

Abstract
Regulation of gene expression ensures an organism responds to stimuli and undergoes proper development. Although the regulatory networks in bacteria have been investigated in model microorganisms, nearly nothing is known about the evolution and plasticity of these networks in obligate, intracellular bacteria. The phylum Chlamydiae contains a vast array of host-associated microbes, including several human pathogens. The Chlamydiae are unique among obligate, intracellular bacteria as they undergo a complex biphasic developmental cycle in which large swaths of genes are temporally regulated. Coupled with the low number of transcription factors, these organisms offer a model to study the evolution of regulatory networks in intracellular organisms. We provide the first comprehensive analysis exploring the diversity and evolution of regulatory networks across the phylum. We utilized a comparative genomics approach to construct predicted coregulatory networks, which unveiled genus- and family-specific regulatory motifs and architectures, most notably those of virulence-associated genes. Surprisingly, our analysis suggests that few regulatory components are conserved across the phylum, and those that are conserved are involved in the exploitation of the intracellular niche. Our study thus lends insight into a component of chlamydial evolution that has otherwise remained largely unexplored.
Keywords: coregulatory networks, transcription factors, intracellular bacteria, Chlamydia
Introduction
All organisms rely on regulatory mechanisms to control the expression of certain genes at certain times or in response to certain stimuli. In bacteria, regulation of gene expression is often carried out by DNA-binding proteins that recognize specific motifs found in promoter regions and serve to either activate or repress transcription through interactions with RNA polymerase. These transcription factors and their target genes thus comprise how a cell may respond to different environmental or developmental signals (Perez and Groisman 2009). These regulatory networks may be highly conserved between related organisms, but growing evidence suggests that many of these networks confer species-specific regulator and target gene associations (Price et al. 2007). The evolution of regulatory networks in a given organism is thus highly reflective of its environment, where free-living bacteria harboring large and diverse gene sets also contain a proportional number of regulatory factors. For instance, the soil bacterium Streptomycetes avermitilis has a genome size of 9.1 Mb (7,582 predicted genes) and is predicted to harbor 623 regulatory proteins (Madan Babu et al. 2006). Contrarily, bacterial symbionts, which experience a stable intracellular environment, have reduced genomes and tend to harbor few regulatory elements, such as the aphid endosymbiont Buchnera aphidicola which is predicted to only encode four regulatory proteins for its 507 predicted genes (Madan Babu et al. 2006). Unique among obligate, intracellular bacteria are the Chlamydiae, which undergo a biphasic developmental cycle, in which hundreds of genes must be temporally regulated. This conserved developmental cycle, taken with the diversity of ecological niches occupied by chlamydiae and their respective hosts, and reduced impact of horizontal gene transfer in the phylum makes the Chlamydiae prime candidates to study the evolution of regulatory networks among intracellular bacteria.
All members of the phylum Chlamydiae are associated with eukaryotic hosts. The family Chlamydiaceae includes many well-known animal and human pathogens, including the largest contributor to bacterial sexually transmitted disease, Chlamydia trachomatis. Outside of this family lies a vast array of chlamydiae that are collectively referred to as “environmental chlamydia.” There are at least eight described families outside of the Chlamydiaceae, whose members are associated with a smorgasbord of eukaryotes, ranging from protists, enigmatic marine worms, arthropods, and fish (Horn 2008; Lagkouvardos et al. 2014; Taylor-Brown et al. 2015). Despite this tremendous diversity in host range, a paramount unifying feature is a shared biphasic developmental cycle in which an infectious, extracellular elementary body (EB) enters a host cell, and transitions into a replicative and fully metabolically active reticulate body (RB). Following replication, the RBs differentiate back to EBs and are subsequently released into the environment, usually as a result of host cell lysis. Several pioneering transcriptomics studies in the human pathogens Chlamydia trachomatis (Belland et al. 2003; Nicholson et al. 2003) and Chlamydia pneumoniae (Mäurer et al. 2007; Albrecht et al. 2011) illustrated that this developmental cycle is marked by differential temporal expression patterns of large sets of genes, which have been broadly characterized as early (EB to RB conversion), mid (RB replication), and late (RB to EB conversion).
Despite the wide importance of these organisms in animal and human health, the infancy of tools for genetic manipulation (Heuer et al. 2007; Nguyen and Valdivia 2013) and the difficulty of intracellular systems have made the elucidation of the major regulatory players arduous (Tan 2012). Only a handful of microarray and RNA sequencing studies are available (Belland et al. 2003; Nicholson et al. 2003; Mäurer et al. 2007; Albrecht et al. 2011) and within this subset even fewer provide the resolution needed to characterize expression profiles over the developmental cycle. Here, we utilize the power of phylogenomics to lend insights into the evolution and diversity of the regulatory proteins and schemes we find distributed throughout the phylum. We systematically predicted transcription factors found in the chlamydial phylum, and we provide the first comprehensive prediction of transcription regulatory networks for various members of the Chlamydiae.
Results and Discussion
Diversity of Regulatory Elements Reflects Host Diversity and Ecology
The genome size of members of the Chlamydiae varies over 2 Mb, from the smallest genome of Chlamydia trachomatis (1.04 Mb) to the largest of Parachlamydia acanthamoeba (3.07 Mb). Of the 9,933 gene families in the phylum, only 409 families are conserved between all Chlamydiae. This indicates a small subset of genes that likely function in core chlamydial biology, such as the developmental cycle, and a large repertoire of genes that likely serve more specific roles tailored to each organism’s environment (Collingro et al. 2011). Diversity and expansion of gene content are often met with the need to regulate these genes (Perez and Groisman 2009). To see whether this large diversity of genes was matched with increased regulatory elements, we exhaustively searched for predicted regulators in the genomes of all sequenced chlamydia by identifying all proteins containing DNA-binding domains and/or sequence homology to known regulators. Combined with the nine previously described regulators in the Chlamydiaceae, we uncovered a striking diversity of 73 putative regulators found with varying frequency throughout the phylum (fig. 1 and supplementary table S1, Supplementary Material online). Indeed, we observe that the more reduced genomes of the Chlamydiaceae harbor relatively few transcription factors (12–15), whereas the larger genomes of the environmental chlamydia harbor an extensive diversity of putative regulators. It is interesting to note, however, that the largest chlamydial genomes do not harbor the largest set of predicted regulatory elements, that honor is bestowed upon Rubidus massiliensis, which harbors 37 predicted regulators. Protochlamydia naegleriophila and Simkania negevensis both contain 31 predicted regulators; however, they vary in genome size by nearly approximately 0.5 Mb (which in real terms is half the size of the genome of C. trachomatis). Simkania negevensis notably harbors the most unique set of predicted regulators, with 11 being unique to only this organism.
Fig. 1.

Distribution and conservation of putative transcriptional regulators in the Chlamydiae. The plot shows gene presence and absence of predicted regulators for members of the phylum Chlamydiae (yellow background indicates members of the family Chlamydiaceae; green background indicates environmental chlamydiae). The species phylogeny shown was calculated from a concatenation of 33 markers genes using PhyloBayes (Lartillot et al. 2013) under the CAT-GTR model. The box inlay displays those genes that are conserved throughout different taxonomic levels. We included AtoC as a globally conserved regulator, as it is only absent in the incomplete genome of Criblamydia (*). pc1704 refers to the Protochlamydia amoebophila UWE25 locus tag of the conserved, yet uncharacterized protein.
This large disparity in the predicted regulatory elements may reflect the differences in host and environmental niches of each organism. All members of the Chlamydiaceae infect higher animal hosts, such as humans, koala, birds, and reptiles (Horn 2008; Lagkouvardos et al. 2014; Taylor-Brown et al. 2015), which suggest that these organisms are well adapted to this particular intracellular environment. This is in contrast with most of the sequenced environmental chlamydia, which primarily have been isolated from free-living amoeba in both soil and aquatic environments (Horn 2008; Lagkouvardos et al. 2014; Taylor-Brown et al. 2015). These environments are much more tumultuous and thus these chlamydial organisms must have a genetic repertoire to compete effectively against other facultative amoeba-associated organisms, such as Legionella species (Moliner et al. 2010), and to survive the harsh conditions while in the extracellular EB stage. From the current fully sequenced environmental chlamydia, only two organisms were not originally isolated from free-living amoeba. Waddlia chondrophila was first isolated from an aborted bovine fetus (Rurangirwa et al. 1999), and Simkania negevensis was originally discovered as a contaminant in human cell culture (Kahane et al. 1995); however, both organisms grow well in Acanthamoeba species (Horn 2008).
Here we find that several of the unique or sparsely distributed transcription factors are putatively involved in regulating operons that function in distinct metabolite metabolism or transport (supplementary table S1, Supplementary Material online), such as arsenic resistance (ArsR), amino acid metabolism (ArgR and TrpR), and carbon storage (CsrA and CsiR). Speculation on the actual roles of regulatory proteins is difficult, as orthologous transcription factors can have vastly different functions, even between closely related bacteria (Price et al. 2007). Most transcription factors acquired through horizontal gene transfer often are local regulators, meaning they only control a small, typically adjacent, subset of genes (Price et al. 2008). Indeed, most of the transcription factors that are species specific are adjacent to other nonconserved genes, although this does not necessarily mean that these genes are under the control of the “local” regulator. However, we do find an apparent cotransfer of the mercury resistance repressor and the corresponding operon into the plasmid of S. negevensis, consisting of the regulator MerR, the mercuric reductase MerA, and one membrane spanning protein MerT (Boyd and Barkay 2012). Evidence for horizontal acquisition is also exemplified with a member of the LysR family of regulators that was transferred into the ancestor of Simkania and Criblamydia from members of the Alphaproteobacteria (supplementary fig. S1, Supplementary Material online). The presence of LysR in only two chlamydiae suggests that multiple losses also occurred, such as in Waddlia and all Parachlamydia, when we reconcile these data with the species tree (shown in fig. 2).
Fig. 2.
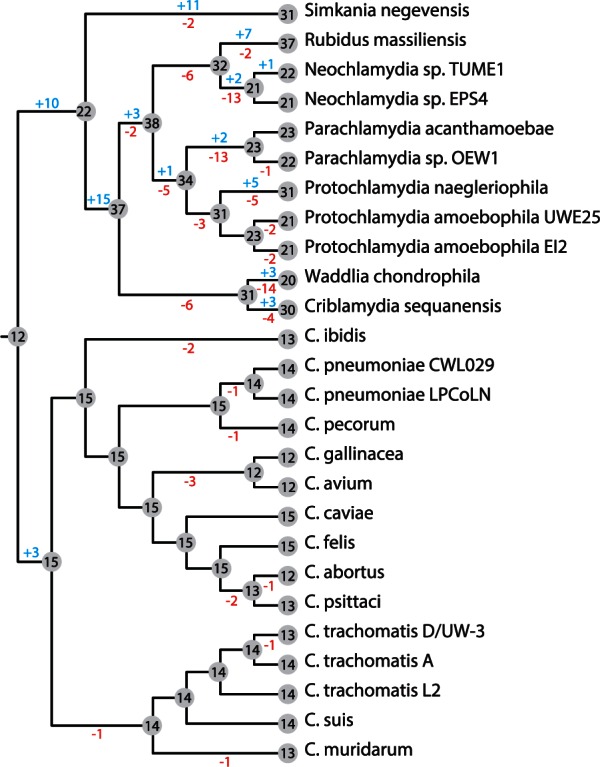
Gain and loss of transcription factor gene families during Chlamydiae evolution. Using the phyletic profile of each gene family containing a predicted regulator, we used the Dollo parsimony approach in COUNT (Csűös 2010) to model the gains and losses of transcription factors along the chlamydial species tree. The environmental chlamydial genomes have undergone extensive gains and losses, whereas the Chlamydiaceae have only three gains at the initial family separation and are then marked by differential losses.
When we modeled the gene family history of all putative transcription factors (Csűös 2010), we find that there has been a veritable mix of gains and losses in every family except for the Chlamydiaceae (fig. 2). The Chlamydiaceae have only acquired three transcription factors when they split from the other families (DcrA, GrgA, and the plasmid regulator pGP4), and all other changes in the transcription factor repertoire have been differential losses. This is in stark contrast to the environmental chlamydia, where a total of 63 regulator genes were acquired in various lineages (fig. 2). Notably, we even find gains and losses among members of the same species, for instance, between the two Protochlamydia species infecting different amoeba hosts, which may indicate that adaptation to novel niches may have been facilitated through changes in gene regulation.
Conserved Regulators Are Essential to Chlamydial Biology
Moving from the surprising diversity of regulators found in the phylum Chlamydiae, those regulators that are conserved are those that likely confer essential and fundamental roles in chlamydial biology. Out of all putative transcription factors, only eight (AtoC, ChxR, DksA, EUO, HrcA, PhoU, YebC, and YtgR) are conserved among all members of the phylum (fig. 1 and supplementary table S1, Supplementary Material online). An additional two (YbjN and NrdR) are nearly conserved (absent in <2 genomes) in all Chlamydiae, and two described additional factors (DcrA and GrgA) are specific for the Chlamydiaceae (Rau et al. 2005; Bao et al. 2012). The role of DcrA as a transcription factor, however, is currently under debate (Kemege et al. 2011). Some of these phylum-wide conserved regulators have been implicated as major players in the chlamydial developmental cycle. For example, EUO has donned the title of the master regulator of late gene expression (RB to EB conversion) in Chlamydiaceae, where previous studies have nicely shown that EUO represses the transcription of late genes (Rosario and Tan 2012; Rosario et al. 2014). Several studies have demonstrated that global regulators, that is those regulators which control large numbers of target genes, evolve more slowly than other regulators, and tend to be vertically inherited (Rajewsky et al. 2002; Price et al. 2008; Perez and Groisman 2009). Thus, the individual gene trees for global regulators tend to be concordant with species trees (Price et al. 2008). As EUO currently represents the only bona fide global regulator in the phylum, we chose to reconstruct the phylogeny of this gene. Indeed, the gene tree for EUO is highly concordant with the chlamydial species tree (supplementary fig. S2, Supplementary Material online), indicative that EUO has been vertically inherited throughout chlamydial evolution, providing further evidence for its role as global regulator in all chlamydiae.
Additionally, ChxR has been implicated as an activator of midcycle genes, where chlamydia are fully metabolically active and dividing as RBs (Koo et al. 2006; Hickey et al. 2011). YtgR has been shown to negatively regulate the ytg operon which is believed to function in metal ion transport (Akers et al. 2011), and likely does not have a major role in developmental cycle regulation. This is likely also the case for the acetate metabolism regulator AtoC and the heat shock response regulator HrcA, which is involved in response to cellular stress and has been experimentally characterized to regulate the dnaK and groE operons in C. trachomatis (Wilson and Tan 2004; Wilson et al. 2005). The putative phosphate regulator PhoU has not been investigated, but likely has a limited role specific to regulating genes under specific environmental stimuli, and has not been implicated as a major player in the developmental cycle. The role of YebC in chlamydia has not yet been investigated, and little can be derived about its function in these organisms. This gene is upregulated late in the Chlamydia developmental cycle (Nicholson et al. 2003), suggesting that it may function in processes involved in the conversion of RB to EB.
Loss of σ28 Reveals Plasticity in Gene Regulation
Sigma factors allow the differential binding of RNA polymerase to the promoter region of genes and thus are transcriptional regulators. Within the Chlamydiae only three sigma factors have been identified: The primary σ66, the alternative σ54, and a minor σ28 (Tan 2012). Thus far, the role ascribed to σ28 is a temporal regulator of late gene expression (Yu et al. 2006). Intriguingly, only members of the Chlamydiaceae contain the minor σ28. The absence of σ28 in the environmental chlamydia is perplexing as several of the genes shown to be regulated by this protein in the Chlamydiaceae are still present in these organisms.
Several lines of evidence indicate that σ28 was lost in environmental chlamydia rather than acquired by the Chlamydiaceae. Phylogenetic analysis suggests that σ28 in the Chlamydiaceae was vertically inherited from the last common ancestor with the Verrucomicrobia (fig. 3), which is in line with our current understanding of the evolution of the Planctomycetes–Verrucomicrobia–Chlamydiae superphylum (Wagner and Horn 2006; Kamneva et al. 2012). We exhaustively searched in the intergenic regions of the environmental chlamydia genomes to detect any remaining fragments of σ28, but it seems that such an ancient loss has left little remnants. Second, as it has been proposed that σ28 may be subject to regulation through a partner-switching mechanism (Hua et al. 2006), we find several members of this pathway, such as RsbV2, RsbW, and RsbU2, present in environmental chlamydial genomes. The losses of the other members of this regulatory cascade, such as RsbU and RsbV, which are conserved in the Chlamydiaceae (Hua et al. 2006), reflect that these proteins were likely no longer needed once σ28 was lost. The retention of the other Rsb proteins might suggest that these proteins were recruited in a regulatory cascade for one of the two remaining sigma factors present in the environmental chlamydia.
Fig. 3.

Loss of sigma factor σ28 in environmental chlamydia. Phylogenetic analysis under the GTR model in PhyloBayes supports a scenario in which the Chlamydiaceae σ28 was inherited from the Verrucomicrobia (PP = 0.92), and an ancient loss of this protein occurred in the environmental chlamydia. The topology of the gene tree for the Chlamydiaceae is largely congruent with the species phylogeny. Arrow indicates outgroup consisting of various representatives of different bacterial phyla.
The loss of σ28 would have prompted major changes in the transcriptional regulatory network within all environmental chlamydia. At the sequence level, these changes may be borne out as losses of transcriptional regulatory binding sites for genes once under the control of a σ28. As functional elements, such as transcription factor binding sites, tend to be under selective constraint, these elements may be highly conserved throughout evolution (Molina and van Nimwegen 2008). Thus, one can scan the orthologous promoter regions in multiple species to find conserved sequence motifs, an approach called phylogenetic footprinting (Cliften et al. 2003; Katara et al. 2011). In this vein, the tail-specific protease, Tsp, is a σ28-regulated gene expressed late in the Chlamydiaceae (Lad et al. 2007), but is present also in all environmental chlamydia genomes. When we look at the phylogenetic footprint in the promoter region of this gene, there is a significant motif found through all Chlamydiaceae, as would be expected (fig. 4A). There is a notable absence of a motif shared between any members of the environmental chlamydia, suggesting that major independent sequence evolution has occurred in these promoter regions. This is in stark contrast with the promoter region of the globally conserved heat shock response regulator, HrcA, which is known to self-regulate its own expression in addition to the other heat shock response genes in Chlamydiaceae (Wilson and Tan 2004; Wilson et al. 2005). Here, we find conserved motifs found throughout all members of the phylum Chlamydiae (fig. 4B), suggestive that this gene is regulated in the same manner throughout all members. Thus, the key question becomes “how are the σ28-regulated genes in the Chlamydiaceae that are present in the environmental chlamydia regulated?” Given that we cannot detect any significant conserved motifs in the promoters of these genes among the environmental chlamydia, this suggests a loss of strict regulation. Indeed, when we examine the preliminary transcriptome of Protochlamydia amoebophila (König L, personal communication), we find that all of these “σ28-late genes” are now constitutively expressed throughout the developmental cycle, again suggesting a loss of the temporal regulation of these genes as seen in the Chlamydiaceae.
Fig. 4.
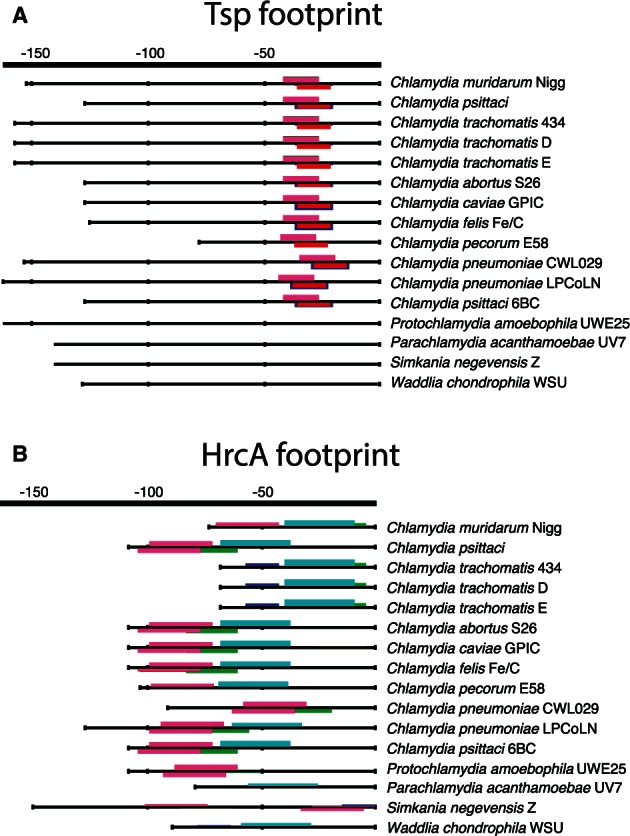
Phylogenetic footprints for tsp and hrcA. The conserved overrepresented DNA motifs detected for (A) tsp and (B) hrcA are shown as the output of the program matrix-scan from the RSAT package (Thomas-Chollier et al. 2008). The colored boxes in the promoter regions indicate discovered motifs, and the height represents the statistical significance score. The promoters for each organism correspond to the direct orthologous promoter regions found in each species for either tsp or hrcA. The hrcA (B) promoter contains a well-conserved motif, which is found throughout the chlamydial phylum. Transcription of tsp in the Chlamydiaceae is mediated by σ28 and we detect a well-defined motif (A) among these organisms. However, the loss of σ28 in environmental chlamydia is matched with the loss of this binding site for these organisms.
Evolutionary Dynamics within Chlamydial Regulatory Networks
To investigate the evolution of regulatory networks within the Chlamydiae, we used a combinatorial approach of comparative genomics and existing transcriptomics data from various chlamydial organisms. Our approach focused around phylogenetic footprinting, and using the approach from Brohee et al. (2011) we linked genes together that share similar footprints to construct predicted coregulatory networks for each of the fully sequenced chlamydial genomes (n = 17). We then used the transcriptomic studies from C. trachomatis (Belland et al. 2003; Nicholson et al. 2003) and C. pneumoniae (Mäurer et al. 2007; Albrecht et al. 2011) to further corroborate predicted regulatory schemes. This sequence-based approach was shown to infer coregulation networks just as well as microarray-derived networks in yeast (Brohee et al. 2011), and thus can serve to elucidate regulatory schemes for nonmodel organisms.
Genes, that is, nodes, are incorporated into the predicted coregulation network if they have a significant motif shared in the promoter region with other genes or are predicted to be in an operon, where the edges are weighted by the strength of the similarity between these motifs, called the DPbits score (Brohee et al. 2011). Thus connections between nodes suggest that the respective genes are regulated in the same fashion, and large regulons would appear as highly connected subnetworks. Out of the 874 genes present in C. trachomatis 434/Bu, 644, or 76%, are represented in the inferred coregulation network (table 1). Similarly, the C. pneumoniae CWLO29 network comprises 733 genes (70%) out of the total 1,052 total genes (fig. 5A and table 1). With this method, we detect subnetworks of previously well-defined regulons, such as the HrcA regulon. This subnetwork in C. pneumoniae is comprised well-characterized members of the regulon, such as hrcA, dnaK, groEL, groES, and grpE (Wilson and Tan 2004; Wilson et al. 2005) (fig. 5B). Intriguingly, two genes (phoH and CPn0105/CT_016) are strongly predicted to be coregulated within the HrcA regulon, possibly representing additional members within this regulatory network. These novel members are conserved in the predicted networks across the phylum and contain nearly perfect motifs matching the described CIRCE (supplementary fig. S3, Supplementary Material online) element recognized by chlamydial HrcA (Wilson and Tan 2004), strengthening the argument that these are likely part of this regulon. The EUO regulon currently consists of 15 members (Rosario and Tan 2012; Rosario et al. 2014), 10 of which are integrated into the networks (supplementary fig. S4, Supplementary Material online). Six of these members, including ltuB, omcA, hctB, and scc2, have direct links to each other in the network, but all members are part of a tightly linked subnetwork that is strongly indicative of a regulon (supplementary fig. S4, Supplementary Material online). We additionally investigated whether the known five members of the ChxR regulon (Koo et al. 2006; Hickey et al. 2011) were present in the network. Indeed, four members of the ChxR regulon are present, including chxR, tufA, infA, and CT_084 (supplementary fig. S4, Supplementary Material online). Therefore, of the three defined regulons described for chlamydial organisms, we correctly predict that many of the respective members are coregulated with each other.
Table 1.
Properties of Predicted Coregulatory Networks.
| Network | DPbits Score | Nodes | Edges | Average No. of Neighbors |
|---|---|---|---|---|
| Chlamydia trachomatis | 1 | 644 (422) | 3,968 (3,617) | 12.3 (17.1) |
| 5 | 233 (89) | 656 (468) | 5.6 (10.5) | |
| Chlamydia pneumoniae | 1 | 733 (558) | 4,523 (4,238) | 12.3 (15.1) |
| 5 | 286 (86) | 679 (412) | 4.74 (9.6) | |
| Protochlamydia amoebophila | 1 | 710 (450) | 2,261 (1,889) | 6.3 (8.4) |
| 3 | 350 (143) | 627 (368) | 3.6 (5.1) | |
| Simkania negevensis | 1 | 734 (266) | 1,668 (1,060) | 4.6 (7.9) |
| 3 | 314 (100) | 621 (250) | 3.9 (5.0) | |
| Chlamydiaceae | 1 | 443 (134) | 975 (581) | 4.4 (8.7) |
| “Chlamydia” clade | 5 | 194 (71) | 488 (325) | 5.0 (9.2) |
| “Chlamydophila” clade | 5 | 230 (50) | 346 (133) | 3.0 (5.3) |
| Environmental chlamydia | 1 | 165 (NA) | 169 (NA) | 2.1 (NA) |
| Chlamydiae phylum | 1 | 122 (NA) | 115 (NA) | 1.9 (NA) |
Note.—Numbers inside parenthesis refer to counts within the large interconnected subnetwork.
Fig. 5.

Predicted coregulatory network and HrcA regulon for Chlamydia pneumoniae. The predicted coregulatory network (A) for C. pneumonaie CWL029 is shown where nodes in the network represent genes, and edges are predictions of genes to be coregulated based on the similarity of phylogenetic footprints between genes. The color of the edges scale with the strength of a prediction, where dark red represents genes strongly predicted to be coregulated and yellow for weaker predictions. The large interconnected set of genes contains many type III effector proteins and virulence-associated genes. Many of the subnetworks outside of this large “hairball” represent predicted operons. The HrcA regulon (B) is shown as an example of genes that are strongly predicted to be coregulated.
The deep RNA-sequencing study of C. pneumoniae revealed that 70% of all genes detected could be affiliated with an operon (Albrecht et al. 2011), and thus operon prediction should be taken into consideration for network construction. The approach we used to infer operons (Thomas-Chollier et al. 2008) is based on a simple distance metric (default of 55 base pairs) of genes oriented in the same direction, which has been reported to be approximately 80% accurate (Janky and van Helden 2008). To ensure that our operon prediction was reasonable, we compared these predictions with that of the DOOR 2.0 database (Mao et al. 2014). Here operon prediction is based on a sophisticated algorithm considering a number of additional parameters, which correctly predicted 78.6% of the operons identified from the C. pneumoniae RNA-sequencing experiment (Albrecht et al. 2011), and was shown to be the best overall operon prediction software (Brouwer et al. 2008). We find excellent agreement between these two methods, as the percentage of Regulatory Sequence Analysis Tools (RSAT) operon predictions that were also predicted by the DOOR database was 98% for C. trachomatis 434/Bu, 98% for C. pneumoniae CWLO29, and 96% for Protochlamydia amoebophila UWE25 (supplementary table S2, Supplementary Material online). Therefore, the operon prediction applied is quite accurate for the data set and—as it accounts for roughly 50% of the genes present in the networks (supplementary table S2, Supplementary Material online)—important for the correct prediction of coregulated genes.
Untangling the Hairball of Virulence Genes
One of the major goals of chlamydia research, at both a clinical and basic research level, is to identify those proteins translocated into the host cell to manipulate the host and subvert resources to the chlamydia. This is primarily achieved by the type III secretion system (Peters et al. 2007; Beeckman and Vanrompay 2010; Betts-Hampikian and Fields 2010; Mueller et al. 2014). Quite strikingly, when we reduce the individual networks to only those edges that are the strongest predictions for being coregulated (DPbits score ≥ 7), a subnetwork of primarily virulence genes is preserved. In C. trachomatis 434/Bu, this network consists of 27 genes, 16 of which are either known type III secreted effector proteins or membrane proteins (fig. 6). These genes include the actin modulating effector TarP (CT_456) (Clifton et al. 2004); the family of DUF582 proteins recently reported to be effectors (CT_620, CT_711, CT_712) (Muschiol et al. 2011); a protein that interacts with the host cell cycle regulator GCIP (CT_847) (Chellas-Géry et al. 2007); the Pmp-like secreted protein CT_050 (Jorgensen and Valdivia 2008); CT_132 and CT_142-144, all of which either demonstrated translocation by a surrogate type III system or were computationally predicted to be effectors (Da Cunha et al. 2014). In addition to these type III effectors, we find several genes encoding membrane proteins, such as a predicted membrane protein OMC1 (CT_073), the predicted virulence-associated inclusion membrane protein (Inc) CT_837 (Dehoux et al. 2011), and the cysteine-rich outer membrane protein OmcA (Everett and Hatch 1995). Given the inherent difficulties surrounding the prediction of type III effectors, it is rather striking that our approach uncovered such a highly connected network of virulence genes. By relaxing the threshold (DPbits score ≥ 5) this subnetwork expands to 89 nodes (supplementary fig. S5, Supplementary Material online), which includes other known type III secreted effectors such as GlgC (Ball et al. 2013) CT_365, CT_620, and CT_695 (Muschiol et al. 2011), and several other putative Inc proteins (CT_005, CT_814) (Dehoux et al. 2011). These top-scoring predictions connecting virulence genes hold over all organisms investigated (supplementary fig. S6, Supplementary Material online).
Fig. 6.
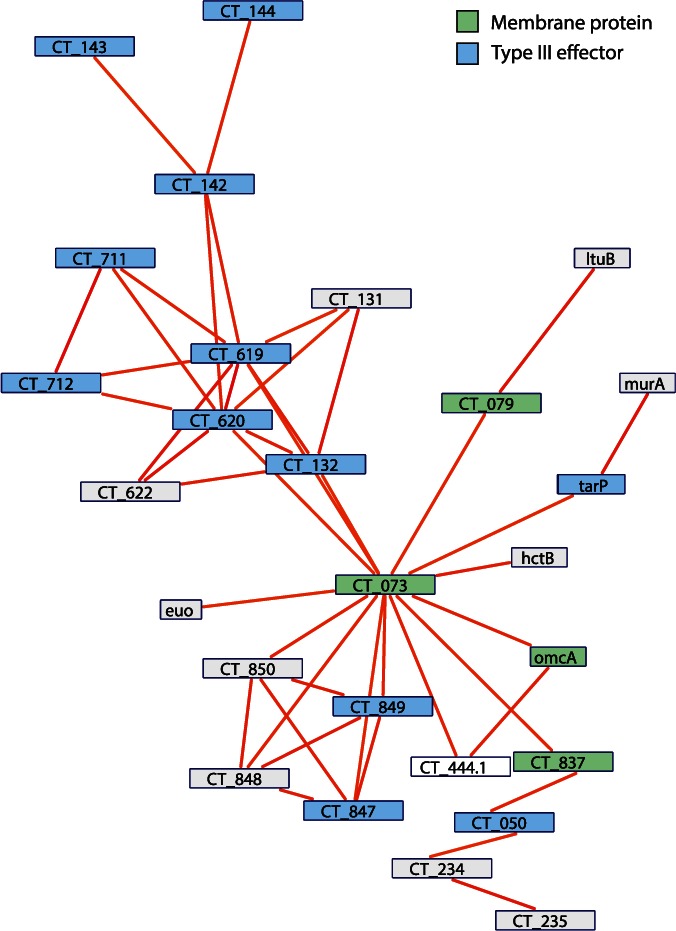
Putative virulence regulon of Chlamydia trachomatis. When we filter the predicted coregulation network to only include those edges that are the most strongly predicted (DPbits score ≥ 7), a tightly connected subnetwork appears, consisting mainly of virulence-associated genes. Of the 29 nodes in the subnetwork, 10 have been described as type III effector proteins and 5 described as membrane proteins. Nodes within this subnetwork likely represent strong candidates for a role in facilitating host–microbe interactions.
The topologies in these networks may indicate differential regulation between effector protein sets. For example, in the C. trachomatis subnetwork, there appear to be two main cliques: One containing the membrane proteins and those proteins connected to CT_073, and the other with those proteins clustering around CT_619 and CT_620 (fig. 6). These two cliques may represent two different sets of effectors, regulated at different times, by different factors, or for different functions. For instance, TarP and CT_849 represent effector proteins that are involved in initial inclusion formation and modification (Valdivia 2008), whereas the family of DUF582 proteins (CT_620, CT_712) in the other clique have been proposed to function in mid/late stages of the developmental cycle with a possible role facilitating exit from the host cells (Muschiol et al. 2011).
Conservation of Coregulatory Networks across the Phylum
Key questions we can ask using the individual predicted coregulatory networks are how similar are they across differing taxonomic levels. If a particular prediction was conserved throughout these organisms, we would consistently recover these edges in the individual networks, and thus they would be present in a consensus network. Indeed, within-family comparisons revealed many shared coregulated genes for the Chlamydiaceae, for instance, C. trachomatis and C. pneumoniae share 556 nodes of 820 orthologs (table 1). Among 6 members of the Chlamydiaceae, 443 nodes are present in the consensus network (table 1 and supplementary fig. S7, Supplementary Material online). If we construct a consensus network where we only consider edges from individual networks if they are top predictions (DPbits ≥ 5), we uncover certain network properties that are different between two groups within the Chlamydiaceae, represented by C. trachomatis, C. muridarum, and C. suis (“Chlamydia” clade), and the group containing C. pneumoniae and relatives (“Chlamydophila” clade, previously classified as a separate genus; Stephens et al. 2009). Despite the “Chlamydophila” clade having more total nodes in the networks (230–194), the large putative virulence regulon (i.e., the main network) is comprised fewer members (50–71) and has far fewer edges than that of the “Chlamydia” clade network (133–325). Another parameter we can assess between these networks is the average number of neighbors a node has, which is 5.3 in the “Chlamydophila” and 9.15 for the “Chlamydia,” again confirming a higher degree of conservation for the “Chlamydia” clade. This disparity suggests that the regulatory network controlling the genes in the “Chlamydia” clade is more conserved than that of the former “Chlamydophila” clade. The members of the “Chlamydophila” have a wider breadth of hosts than that of the “Chlamydia” clade, and thus this difference might be borne out here in that certain members of the “Chlamydophila” clade have more specialized network architecture. In summary, the consensus network of the Chlamydiaceae is still similar to the individual organisms’ networks. The presence and retention of many of the genes of the “virulence”-subnetwork indicate family- and genus-specific regulons involved in host manipulation.
When we ask which predicted interactions are conserved across the whole phylum (C. trachomatis, C. pneumoniae, and all environmental chlamydia; n = 6), 122 genes remain in the network. Most of these represent genes that are in predicted operons whose gene order has been preserved, such as the ribosomal proteins, the ATP-synthase subunits, cell wall components, and type III secretion machinery. The highly conserved HrcA regulon, whose various members are not in an operon, was recovered, serving almost as an internal positive control for this analysis. If we relax our stringency on conservation to an edge being present in five of the six genomes analyzed, the consensus network doubles in size, to 240 genes (fig. 7A). Here, we recover a tightly connected subnetwork of 49 genes primarily involved in host manipulation and acquisition/processing of nutrients (fig. 7B). This includes the ATP/ADP translocase 1 (tlcA), adenylate kinase (adk), the well-studied type II effector CPAF (Zhong et al. 2001), glycogen metabolism genes (glgC and glgX), and several other genes involved in nucleotide metabolism (dut, surE, and pyrG). This consensus network is notably void of the virulence-associated genes found in the stringently filtered individual networks. The phylum-wide subnetwork (fig. 7B) is enriched in eggNOG functional categories in metabolism compared with the equivalent (i.e., the “hairball”) in C. trachomatis (44–26%, respectively; supplementary fig. S8, Supplementary Material online). This conserved subnetwork thus seems to comprise genes needed for exploitation of the intracellular niche and nutrient metabolism. Unlike the establishment of infection, which may require highly specialized effectors for distinct host species, once inside a eukaryotic cell it seems the ways to exploit this niche by chlamydial species are rather conserved.
Fig. 7.

Phylum-wide conservation of predicted coregulations in the Chlamydiae. The consensus network was created by comparison of individual organism’s predicted coregulation networks (n = 6). An edge was kept if it was present in five or more networks. The 240 genes present in this network (A) mainly represent conserved operons, with the exception of a putative regulon of 41 genes (B) that mainly have function in exploitation of the intracellular niche. The activator of this subnetwork may be ChxR, which is indicated. Notably, the virulence genes detected in individual organism’s networks are absent in this consensus network.
Intriguingly, ChxR and EUO, two of the conserved and previously described chlamydial transcriptional regulators, are highly integrated in this network (fig. 7B). Given that ChxR is known to autoregulate its own expression, it is tempting to suggest that those promoters linked with ChxR in this regulon may be under its control. Of the conserved predictions of coregulation with ChxR we find several genes associated with type II secretion, including the type II secreted effector CPAF and the type II secretion machinery operon (gspDEF and the conserved hypothetical protein CT_573). Although there is debate as to the biological function CPAF serves in chlamydial infections (Chen et al. 2012), it is intriguing that we uncover the conservation of predictions involving ChxR with a type II secreted substrate and the type II secretion system, both considered midcycle genes. ChxR is also connected to ATP/ADP translocase (tlcA) and the adenylate kinase (adk), both of which are involved in nucleotide metabolism and appear to be midcycle genes (Belland et al. 2003). The interconnectedness of all nodes in the conserved phylum-wide network suggests that, indeed, these genes all have shared motifs and may be under the control of the same regulator. The presence of one of the experimentally demonstrated targets of ChxR, CT_084 (Koo et al. 2006), in the network offers more evidence that this may represent genes under the control of this activator. As it has been proposed that ChxR may function as the activator of midcycle genes (Koo et al. 2006), our networks support this notion and suggest that ChxR may be an even more important regulator of global gene expression than previously thought.
Conclusions
Here, we have investigated the evolution and diversity of the transcriptional regulatory architecture at a phylum-wide level. We systematically identified putative transcription factors and demonstrated that there have been extensive gains and losses of these factors during chlamydial evolution. The conserved regulatory players, especially EUO and ChxR, likely play fundamental roles in regulating gene expression, as demonstrated by their conservation across the phylum and their central placement within our regulatory networks. Further investigations, for instance, by CHiP-Seq of various transcription factors, within and between chlamydial organisms, will allow us to fully characterize the regulatory schemes present in the phylum. As we work toward this goal, the comparative genomics approach we implemented here remains a powerful tool to explore this component of evolution that would otherwise remain vastly unexplored. In this vein, we provide the first description of regulatory networks for members of the Chlamydiae, including those with direct relevance for human health. Our analysis revealed that major players involved in host-cell manipulation and virulence are coregulated and are largely genus and family specific in their network organization. Additionally, we uncovered that the regulatory network architecture is not well conserved throughout the phylum, but those connections that are conserved are primarily involved in the exploitation of the intracellular niche, such as nucleotide and ATP scavenging. An invaluable corollary of this network approach is that genes integrated into these networks represent prime candidates as novel virulence-associated genes, and provide the chlamydial research community a solid starting point for investigating the roles of hypothetical proteins. This approach can easily be expanded to other nonmodel systems to elucidate putative functions for hypothetical proteins and determination of virulence factors (Brohee et al. 2011).
Materials and Methods
Identification of Conserved Transcription Factors
The proteome of each organism was scanned using InterProScan v5 (Jones et al. 2014), and hits matching the DNA-binding domain families from the curated DBD database (Wilson et al. 2008) to PFAM (Finn et al. 2013) and SUPERFAMILY (Gough et al. 2001) were extracted and further curated to remove false-positive matches. We additionally searched for Gene Ontology terms associated with gene regulation and those previously described in the literature. Orthologous groups of proteins were determined by OrthoMCL (Li et al. 2003) using default parameters. We inferred the evolutionary history of the transcription factors along the species tree using COUNT with the Dollo parsimony option (Csűös 2010).
Chlamydial Species Phylogeny
Using AMPHORA2 (Wu and Scott 2012) we extracted 31 phylogenetic marker genes from each chlamydial proteome. Each gene family was aligned with MAFFT (Katoh and Standley 2013) using the LINSI algorithm, followed by removal of poorly aligned sites using BMGE (Criscuolo and Gribaldo 2010). The individual alignments were concatenated together using SCaFoS (Roure et al. 2007). Phylogenetic analysis was performed using the CAT-GTR (general time reversible) model in PhyloBayes-MPI (Lartillot et al. 2013) running two independent chains. We determined that the chains had converged when the maximum discrepancies in bipartition frequencies (bpcomp) dropped below 0.1 and effective sampling size of parameters (tracecomp) was at least 100 between the chains, as per the recommendation in the PhyloBayes manual (Lartillot et al. 2009). We additionally performed a maximum-likelihood analysis using RAxML (Stamatakis 2006) under the “PROTGAMMALGF” model with 1,000 bootstraps. The PhyloBayes and RAxML tree topologies were nearly congruent.
Gene Family Phylogenies
EUO protein sequences were obtained from the OrthoMCL data. Protein sequences for the lysR (pecT) and σ28 analysis were obtained through Basic Local Alignment Search Tool (BLAST) against the UniRef 90 database (Suzek et al. 2007). To account for compositional heterogeneity between species in the σ28 analysis, we recoded the alignment into the six DayHoff categories. All alignments were performed with MAFFT (Katoh and Standley 2013) using the LINSI algorithm, followed by removal of poorly aligned sites using BMGE (Criscuolo and Gribaldo 2010). We reconstructed the EUO gene family phylogenetic tree using RAxML (Stamatakis 2006) under the “PROTGAMMALGF” model with 1,000 bootstraps. Phylogenetic trees for LysR and σ28 were calculated with PhyloBayes under the GTR model and convergence checks were performed the same as in the species tree analysis.
Phylogenetic Footprinting and Predicted Coregulatory Networks
We used the RSAT (Thomas-Chollier et al. 2008) suite to construct both the phylogenetic footprints and predicted coregulatory networks. Briefly, for each organisms considered, we determined whether there was a significant phylogenetic footprint for each gene in that genome by detecting overrepresented motifs in promoter regions through the program “dyad-analysis” (Defrance et al. 2008; Janky and van Helden 2008). We then created the coregulation networks in RSAT by linking similar phylogenetic footprints together as previously described (Brohee et al. 2011) through the “footprint-discovery” program within RSAT with the following default parameters: “-lth occ 1 -lth occ_sig 0 -uth rank 50 -bg_model taxfreq -all_genes -sep_genes -filter -infer_operons -task all.” Networks were also constructed without predicting operons by omitting the “-infer_operons” option. Consensus networks were created using a custom Python script using the “NetworkX” package. Networks were viewed and processed using Cytoscape v.3.2.1 (Shannon et al. 2003). Sequence logos were created using WebLogo 3 (Crooks et al. 2004). The MEME software (Bailey et al. 2009) was also used to scan sequence groups, such as the putative EUO regulon for overrepresented motifs. Operon predictions were downloaded from the DOOR 2.0 database (Mao et al. 2014) for comparison against the RSAT operon predictions. Functional categories were assigned to genes through BLAST of the eggNOG 4.0 database (Powell et al. 2014). All networks may be downloaded as GML files from figshare.com/s/0b0b4ebe046a11e59e9c06ec4bbcf141.
Supplementary Material
Supplementary figures S1–S8 and tables S1 and S2 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/)
Acknowledgments
This work was supported by the Austrian Science Fund FWF grant I1628-B22, the European Research Council StG EVOCHLAMY (281633), and the Marie Curie Initial Training Network - SYMBIOMICS.
References
- Akers JC, HoDac H, Lathrop RH, Tan M. 2011. Identification and functional analysis of CT069 as a novel transcriptional regulator in Chlamydia. J Bacteriol. 193:6123–6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht M, Sharma CM, Dittrich MT, Müller T, Reinhardt R, Vogel J, Rudel T. 2011. The transcriptional landscape of Chlamydia pneumoniae. Genome Biol. 12:R98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. 2009. MEME Suite: tools for motif discovery and searching. Nucleic Acids Res. 37:W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball SG, Subtil A, Bhattacharya D, Moustafa A, Weber APM, Gehre L, Colleoni C, Arias M-C, Cenci U, Dauvillée D. 2013. Metabolic effectors secreted by bacterial pathogens: essential facilitators of plastid endosymbiosis? Plant Cell Online 25:7–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X, Nickels BE, Fan H. 2012. Chlamydia trachomatis protein GrgA activates transcription by contacting the nonconserved region of σ66. Proc Natl Acad Sci U S A. 109:16870–16875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeckman DSA, Vanrompay DCG. 2010. Bacterial secretion systems with an emphasis on the chlamydial Type III secretion system. Curr Issues Mol Biol. 12:17–41. [PubMed] [Google Scholar]
- Belland RJ, Zhong G, Crane DD, Hogan D, Sturdevant D, Sharma J, Beatty WL, Caldwell HD. 2003. Genomic transcriptional profiling of the developmental cycle of Chlamydia trachomatis. Proc Natl Acad Sci U S A. 100:8478–8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betts-Hampikian HJ, Fields KA. 2010. The chlamydial Type III secretion mechanism: revealing cracks in a tough nut. Front Microbiol. 1:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd ES, Barkay T. 2012. The mercury resistance operon: from an origin in a geothermal environment to an efficient detoxification machine. Front. Microbiol. 3:349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brohee S, Janky R, Abdel-Sater F, Vanderstocken G, Andre B, van Helden J. 2011. Unraveling networks of co-regulated genes on the sole basis of genome sequences. Nucleic Acids Res. 39:6340–6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer RWW, Kuipers OP, van Hijum SAFT. 2008. The relative value of operon predictions. Brief Bioinform. 9:367–375. [DOI] [PubMed] [Google Scholar]
- Chellas-Géry B, Linton CN, Fields KA. 2007. Human GCIP interacts with CT847, a novel Chlamydia trachomatis type III secretion substrate, and is degraded in a tissue-culture infection model. Cell Microbiol. 9:2417–2430. [DOI] [PubMed] [Google Scholar]
- Chen AL, Johnson KA, Lee JK, Sütterlin C, Tan M. 2012. CPAF: a chlamydial protease in search of an authentic substrate. PLoS Pathog. 8:e1002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliften P, Sudarsanam P, Desikan A, Fulton L, Fulton B, Majors J, Waterston R, Cohen BA, Johnston M. 2003. Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science 301:71–76. [DOI] [PubMed] [Google Scholar]
- Clifton DR, Fields KA, Grieshaber SS, Dooley CA, Fischer ER, Mead DJ, Carabeo RA, Hackstadt T. 2004. A chlamydial type III translocated protein is tyrosine-phosphorylated at the site of entry and associated with recruitment of actin. Proc Natl Acad Sci U S A. 101:10166–10171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingro A, Tischler P, Weinmaier T, Penz T, Heinz E, Brunham RC, Read TD, Bavoil PM, Sachse K, Kahane S, et al. 2011. Unity in variety—the pan-genome of the Chlamydiae. Mol Biol Evol. 28:3253–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criscuolo A, Gribaldo S. 2010. BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol Biol. 10:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia J-M, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res. 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csűös M. 2010. Count: evolutionary analysis of phylogenetic profiles with parsimony and likelihood. Bioinformatics 26:1910–1912. [DOI] [PubMed] [Google Scholar]
- Da Cunha M, Milho C, Almeida F, Pais SV, Borges V, Maurício R, Borrego MJ, Gomes JP, Mota LJ. 2014. Identification of type III secretion substrates of Chlamydia trachomatis using Yersinia enterocolitica as a heterologous system. BMC Microbiol. 14:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defrance M, Janky R, Sand O, van Helden J. 2008. Using RSAT oligo-analysis and dyad-analysis tools to discover regulatory signals in nucleic sequences. Nat Protoc. 3:1589–1603. [DOI] [PubMed] [Google Scholar]
- Dehoux P, Flores R, Dauga C, Zhong G, Subtil A. 2011. Multi-genome identification and characterization of chlamydiae-specific type III secretion substrates: the Inc proteins. BMC Genomics 12:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett KD, Hatch TP. 1995. Architecture of the cell envelope of Chlamydia psittaci 6BC. J Bacteriol. 177:877–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, et al. 2013. Pfam: the protein families database. Nucleic Acids Res. 42:D222–D230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough J, Karplus K, Hughey R, Chothia C. 2001. Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J Mol Biol. 313:903–919. [DOI] [PubMed] [Google Scholar]
- Heuer D, Kneip C, Mäurer AP, Meyer TF. 2007. Tackling the intractable—approaching the genetics of Chlamydiales. Int J Med Microbiol. 297:569–576. [DOI] [PubMed] [Google Scholar]
- Hickey JM, Lovell S, Battaile KP, Hu L, Middaugh CR, Hefty PS. 2011. The atypical response regulator protein ChxR has structural characteristics and dimer interface interactions that are unique within the OmpR/PhoB subfamily. J Biol Chem. 286:32606–32616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn M. 2008. Chlamydiae as symbionts in eukaryotes. Annu Rev Microbiol. 62:113–131. [DOI] [PubMed] [Google Scholar]
- Hua L, Hefty PS, Lee YJ, Lee YM, Stephens RS, Price CW. 2006. Core of the partner switching signalling mechanism is conserved in the obligate intracellular pathogen Chlamydia trachomatis. Mol Microbiol. 59:623–636. [DOI] [PubMed] [Google Scholar]
- Janky R, van Helden J. 2008. Evaluation of phylogenetic footprint discovery for predicting bacterial cis-regulatory elements and revealing their evolution. BMC Bioinformatics 9:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P, Binns D, Chang H-Y, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, et al. 2014. InterProScan 5: genome-scale protein function classification. Bioinformatics 30:1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen I, Valdivia RH. 2008. Pmp-like proteins Pls1 and Pls2 are secreted into the lumen of the Chlamydia trachomatis inclusion. Infect Immun. 76:3940–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahane S, Metzer E, Friedman MG. 1995. Evidence that the novel microorganism “Z” may belong to a new genus in the family Chlamydiaceae. FEMS Microbiol Lett. 126:203–207. [DOI] [PubMed] [Google Scholar]
- Kamneva OK, Knight SJ, Liberles DA, Ward NL. 2012. Analysis of genome content evolution in PVC bacterial super-phylum: assessment of candidate genes associated with cellular organization and lifestyle. Genome Biol Evol. 4:1375–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katara P, Grover A, Sharma V. 2011. Phylogenetic footprinting: a boost for microbial regulatory genomics. Protoplasma 249:901–907. [DOI] [PubMed] [Google Scholar]
- Katoh K, Standley DM. 2013. MAFFT Multiple Sequence Alignment Software Version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemege KE, Hickey JM, Lovell S, Battaile KP, Zhang Y, Hefty PS. 2011. Ab initio structural modeling of and experimental validation for Chlamydia trachomatis protein CT296 reveal structural similarity to Fe(II) 2-oxoglutarate-dependent enzymes. J Bacteriol. 193:6517–6528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo IC, Walthers D, Hefty PS, Kenney LJ, Stephens RS. 2006. ChxR is a transcriptional activator in Chlamydia. Proc Natl Acad Sci U S A. 103:750–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lad SP, Yang G, Scott DA, Wang G, Nair P, Mathison J, Reddy VS, Li E. 2007. Chlamydial CT441 is a PDZ domain-containing tail-specific protease that interferes with the NF-κB pathway of immune response. J Bacteriol. 189:6619–6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagkouvardos I, Weinmaier T, Lauro FM, Cavicchioli R, Rattei T, Horn M. 2014. Integrating metagenomic and amplicon databases to resolve the phylogenetic and ecological diversity of the Chlamydiae. ISME J. 8:115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lartillot N, Lepage T, Blanquart S. 2009. PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 25:2286–2288. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Rodrigue N, Stubbs D, Richer J. 2013. PhyloBayes MPI: phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst Biol. 62:611–615. [DOI] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ, Jr, Roos DS. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13:2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan Babu M, Teichmann SA, Aravind L. 2006. Evolutionary dynamics of prokaryotic transcriptional regulatory networks. J Mol Biol. 358:614–633. [DOI] [PubMed] [Google Scholar]
- Mao X, Ma Q, Zhou C, Chen X, Zhang H, Yang J, Mao F, Lai W, Xu Y. 2014. DOOR 2.0: presenting operons and their functions through dynamic and integrated views. Nucleic Acids Res. 42:D654–D659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäurer AP, Mehlitz A, Mollenkopf HJ, Meyer TF. 2007. Gene expression profiles of Chlamydophila pneumoniae during the developmental cycle and iron depletion–mediated persistence. PLoS Pathog. 3:e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina N, van Nimwegen E. 2008. Universal patterns of purifying selection at noncoding positions in bacteria. Genome Res. 18:148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moliner C, Fournier P-E, Raoult D. 2010. Genome analysis of microorganisms living in amoebae reveals a melting pot of evolution. FEMS Microbiol Rev. 34:281–294. [DOI] [PubMed] [Google Scholar]
- Mueller KE, Plano GV, Fields KA. 2014. New frontiers in Type III secretion biology: the Chlamydia perspective. Infect Immun. 82:2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muschiol S, Boncompain G, Vromman F, Dehoux P, Normark S, Henriques-Normark B, Subtil A. 2011. Identification of a family of effectors secreted by the Type III secretion system that are conserved in pathogenic Chlamydiae. Infect Immun. 79:571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen BD, Valdivia RH. 2013. Forward genetic approaches in Chlamydia trachomatis. J Vis Exp. 80:e50636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson TL, Olinger L, Chong K, Schoolnik G, Stephens RS. 2003. Global stage-specific gene regulation during the developmental cycle of Chlamydia trachomatis. J Bacteriol. 185:3179–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez JC, Groisman EA. 2009. Evolution of transcriptional regulatory circuits in bacteria. Cell 138:233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J, Wilson DP, Myers G, Timms P, Bavoil PM. 2007. Type III secretion à la Chlamydia. Trends Microbiol. 15:241–251. [DOI] [PubMed] [Google Scholar]
- Powell S, Forslund K, Szklarczyk D, Trachana K, Roth A, Huerta-Cepas J, Gabaldón T, Rattei T, Creevey C, Kuhn M, et al. 2014. eggNOG v4.0: nested orthology inference across 3686 organisms. Nucleic Acids Res. 42:D231–D239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M, Dehal P, Arkin A. 2008. Horizontal gene transfer and the evolution of transcriptional regulation in Escherichia coli. Genome Biol. 9:R4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP. 2007. Orthologous transcription factors in bacteria have different functions and regulate different genes. PLoS Comput Biol. 3:e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajewsky N, Socci ND, Zapotocky M, Siggia ED. 2002. The evolution of DNA regulatory regions for Proteo-gamma bacteria by interspecies comparisons. Genome Res. 12:298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau A, Wyllie S, Whittimore J, Raulston JE. 2005. Identification of Chlamydia trachomatis genomic sequences recognized by chlamydial divalent cation-dependent regulator A (DcrA). J Bacteriol. 187:443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario CJ, Hanson BR, Tan M. 2014. The transcriptional repressor EUO regulates both subsets of Chlamydia late genes. Mol Microbiol. 94:888–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario CJ, Tan M. 2012. The early gene product EUO is a transcriptional repressor that selectively regulates promoters of Chlamydia late genes. Mol Microbiol. 84:1097–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roure B, Rodriguez-Ezpeleta N, Philippe H. 2007. SCaFoS: a tool for selection, concatenation and fusion of sequences for phylogenomics. BMC Evol Biol. 7:S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rurangirwa FR, Dilbeck PM, Crawford TB, McGuire TC, McElwain TF. 1999. Analysis of the 16S rRNA gene of micro-organism WSU 86-1044 from an aborted bovine foetus reveals that it is a member of the order Chlamydiales: proposal of Waddliaceae fam. nov., Waddlia chondrophila gen. nov., sp. nov. Int J Syst Bacteriol. 49(Pt 2):577–581. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. [DOI] [PubMed] [Google Scholar]
- Stephens RS, Myers G, Eppinger M, Bavoil PM. 2009. Divergence without difference: phylogenetics and taxonomy of Chlamydia resolved. FEMS Immunol Med Microbiol. 55:115–119. [DOI] [PubMed] [Google Scholar]
- Suzek BE, Huang H, McGarvey P, Mazumder R, Wu CH. 2007. UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics 23:1282–1288. [DOI] [PubMed] [Google Scholar]
- Tan M. 2012. Temporal gene regulation during the chlamydial developmental cycle. In: Tan M, Bavoil PM, editors. Intracellular pathogens I: Chlamydiales. Washington (DC): ASM Press; p. 149–169. [Google Scholar]
- Taylor-Brown A, Vaughan L, Greub G, Timms P, Polkinghorne A. 2015. Twenty years of research into Chlamydia-like organisms: a revolution in our understanding of the biology and pathogenicity of members of the phylum Chlamydiae. Pathog Dis. 73:1–15. [DOI] [PubMed] [Google Scholar]
- Thomas-Chollier M, Sand O, Turatsinze J-V, Janky R, Defrance M, Vervisch E, Brohée S, van Helden J. 2008. RSAT: regulatory sequence analysis tools. Nucleic Acids Res. 36:W119–W127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdivia RH. 2008. Chlamydia effector proteins and new insights into chlamydial cellular microbiology. Curr Opin Microbiol. 11:53–59. [DOI] [PubMed] [Google Scholar]
- Wagner M, Horn M. 2006. The Planctomycetes, Verrucomicrobia, Chlamydiae and sister phyla comprise a superphylum with biotechnological and medical relevance. Curr Opin Biotechnol. 17:241–249. [DOI] [PubMed] [Google Scholar]
- Wilson AC, Tan M. 2004. Stress response gene regulation in Chlamydia is dependent on HrcA-CIRCE interactions. J Bacteriol. 186:3384–3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AC, Wu CC, Yates JR, Tan M. 2005. Chlamydial GroEL autoregulates its own expression through direct interactions with the HrcA repressor protein. J Bacteriol. 187:7535–7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson D, Charoensawan V, Kummerfeld SK, Teichmann SA. 2008. DBD––taxonomically broad transcription factor predictions: new content and functionality. Nucleic Acids Res. 36:D88–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Scott AJ. 2012. Phylogenomic analysis of bacterial and archaeal sequences with AMPHORA2. Bioinformatics 28:1033–1034. [DOI] [PubMed] [Google Scholar]
- Yu HHY, Kibler D, Tan M. 2006. In silico prediction and functional validation of σ28-regulated genes in Chlamydia and Escherichia coli. J Bacteriol. 188:8206–8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong G, Fan P, Ji H, Dong F, Huang Y. 2001. Identification of a chlamydial protease-like activity factor responsible for the degradation of host transcription factors. J Exp Med. 193:935–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.