
Figure 1.
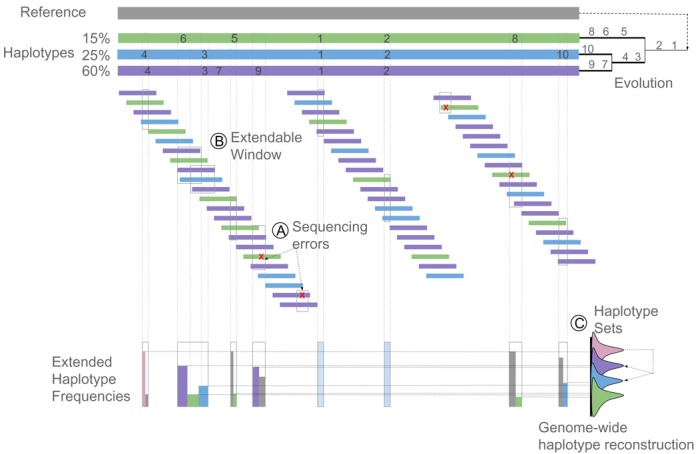
Method Overview. A clonal community where three descendant haplotypes evolved from a reference genome is depicted. The haplotypes are present at respectively 60%, 25% and 15% in the population. A total of 10 different mutations accumulated in the evolving population. Mutations ‘1’ and ‘2’ were acquired by the last common ancestor of extant haplotypes and are therefore shared by all haplotypes in the population. Mutations ‘3’ and ‘4’ were acquired before the origin of the blue and purple haplotypes, whereas the remaining mutations are unique to one of the haplotypes. Small colored horizontal bars represent the reads obtained by deep sequencing the aforementioned population. The coloring of the reads corresponds to the colors of the haplotypes from which they originated. Reads are mapped to the ancestral reference genome. Step 1: Local haplotype reconstruction and window extension. (A) Haplotype templates and their frequencies are first inferred per window. Windows are represented by gray rectangles. A window is defined as a genomic region that is covered by a sufficient number of reads and that contains a set of one or more consecutive tentative polymorphic sites. Tentative polymorphisms can refer to both sequencing errors (red crosses) and true polymorphisms. Per window, accepted template windows will be defined by performing a local haplotype reconstruction simultaneously with the error correction (see Supplementary Figure S1). (B) Template haplotypes are extended over flanking windows with overlapping polymorphic sites based on the consistency in the polymorphisms present in the non-empty read overlap of these flanking windows and in the frequencies of the extended template haplotypes. Two windows for which the ‘window extension’ will be performed are indicated by gray rectangles. In the example the template haplotypes within these flanking windows will be merged into 3 extended haplotypes (the purple, green and blue one, see Supplementary Figure S2). (C) Step 2: Genome-wide haplotype reconstruction. Extended haplotypes from the different concatenated windows will be merged into genome-wide haplotypes based on the frequency information. To this end, we use a mixture model approach in which extended haplotypes occurring at similar frequencies in the population, referred to as haplotype sets are represented by a different mixture component in the model (distributions drawn at the right of the picture). Haplotype sets containing polymorphisms unique for the haplotype will occur at a lower frequency than haplotype sets that contain polymorphisms shared by several genome-wide haplotypes (indicated in pink, see also Supplementary Figure S3). The genome-wide haplotype reconstruction searches for combinations of haplotype sets that provide the best explanation for the observed frequencies of all haplotype sets.