

Background: Satellite cells (SCs) are muscle-specific stem cells that are essential for muscle regeneration.
Results: Conditional abrogation of a disintegrin and metalloprotease 10 (ADAM10) in SCs induces premature differentiation and results in depletion of SCs.
Conclusion: ADAM10 is indispensable to retain quiescence in SCs.
Significance: This study is the first to show the crucial role of ADAM10 as a gatekeeper of SC differentiation.
Keywords: ADAM, muscle regeneration, myogenesis, Notch pathway, skeletal muscle, satellite cells
Abstract
Satellite cells (SCs) are muscle-specific stem cells that are essential for the regeneration of damaged muscles. Although SCs have a robust capacity to regenerate myofibers, the number of SCs decreases with aging, leading to insufficient recovery after muscle injury. We herein show that ADAM10 (a disintegrin and metalloprotease 10), a membrane-bound proteolytic enzyme with a critical role in Notch processing (S2 cleavage), is essential for the maintenance of SC quiescence. We generated mutant mice in which ADAM10 in SCs can be conditionally abrogated by tamoxifen injection. Tamoxifen-treated mutant mice did not show any apparent defects and grew normally under unchallenged conditions. However, these mice showed a nearly complete loss of muscle regeneration after chemically induced muscle injury. In situ hybridization and flow cytometric analyses revealed that the mutant mice had significantly less SCs compared with wild type controls. Of note, we found that inactivation of ADAM10 in SCs severely compromised Notch signaling and led to dysregulated myogenic differentiation, ultimately resulting in deprivation of the SC pool in vivo. Taken together, the present findings underscore the role of ADAM10 as an indispensable component of Notch signaling in SCs and for maintaining the SC pool.
Introduction
Satellite cells (SCs)2 are muscle-specific stem cells that reside between myofibers and the basement membrane with essential roles in repairing damaged muscle (1–3). Under normal conditions, the majority of SCs exist in a quiescent state or G0 phase. However, in response to injury, SCs rapidly proliferate and differentiate to form myofibers and repair damaged muscles. The number of SCs and their regenerative potency decrease with age, leading to insufficient recovery after muscle trauma. Because loss of muscle strength and plasticity can compromise activity of daily living, especially in the elderly, it is mandatory to learn more about the mechanisms underlying the maintenance of the number and the regenerative capacity of SCs.
Past studies have identified various molecules that are potentially involved in the maintenance and aging of SCs, including p38 mitogen-activated protein kinase (4, 5), Wnt (6), TGF-β (7), Jak-Stat (8), p16 (9), and Notch (10–12). Notch receptors and their ligands are highly conserved gene families and are critically involved in various cellular functions, including cell fate decision, cell growth, and differentiation (13, 14). There are four Notch receptors (Notch 1–4) and five ligands (Jagged 1 and 2 and DLL 1, 3, and 4) in mammals. Notch signaling is also involved in the maintenance of stem cells in certain type of tissues, and loss of Notch signaling in the stem cells in these tissues often results in dysregulated differentiation. Accordingly, studies in the past few years have shown that Notch signaling has a crucial role in the maintenance of SCs and that suppression of this signaling pathway results in dysregulated differentiation of SCs and a decrease in the SC population (10–12).
The Notch signaling pathway has a complex and unique mode of action in transmitting cell signaling. Because the receptors and ligands are all membrane-bound, Notch signaling is initiated through cell-cell contact between a receptor-expressing cell and a ligand-expressing cell. Upon activation by ligand binding, Notch receptors undergo sequential proteolytic cleavage by a disintegrin and metalloprotease (ADAM) and the γ-secretase complex (each responsible for S2 and S3 cleavage, respectively), releasing the intracellular domain. The intracellular domain translocates into the nucleus to form a complex with a transcriptional cofactor Rbpj-1 and functions as a transcriptional activator (14, 15).
There are >20 ADAM genes in mammals. Among these, ADAM10 and ADAM17 (also known as TNFα-converting enzyme or TACE) are probably best characterized as enzymes involved in the proteolytic release of membrane-bound proteins, a process also referred to as ectodomain shedding (16, 17). These two genes are closely related to one another and have distinct and overlapping substrates. The identity of the ADAM protease responsible for S2 cleavage of the Notch receptor remains somewhat controversial. Several studies have suggested the potential involvement of ADAM17 in S2 cleavage (18, 19). However, results from studies of ADAM10 mutant mice, which often exhibit Notch-related defects (20–23), suggest that ADAM10 is the principal enzyme responsible for S2 cleavage in vivo.
In the present study we aimed to clarify the potential roles of ADAM10 in SCs and muscle regeneration. We generated a mutant mouse line in which Adam10 can be inactivated specifically in SCs upon tamoxifen injection. The mutant mice did not exhibit any apparent defects under unchallenged conditions; however, these mice almost completely lacked the capacity for muscle generation after muscle injury. Most importantly, we found that the mutant mice are nearly devoid of SCs in skeletal muscle due to defective Notch signaling. Collectively, our data show that ADAM10 is indispensable for maintaining the SC population and for Notch signaling in SCs and further consolidate the notion that ADAM10 as the major sheddase for Notch in vivo.
Experimental Procedures
Mice
The generation of Adam10flox/flox mice was previously described (20). Adam10flox/flox mice were crossed with Pax7tm2.1(cre/ERT2)Fan/J transgenic mice (24) to specifically abrogate the Adam10 allele from SCs (henceforth referred to as Adam10Pax7 mice). Conditional excision of the floxed allele was achieved by intraperitoneal injection of tamoxifen (75 μg/kg; Toronto Research Chemicals, Toronto, Canada) dissolved in corn oil (20 mg/ml). For fate-mapping experiments, we crossed Adam10Pax7 mice with CAG-CAT-EGFP reporter mice (25) (referred to as Adam10Pax7-EGFP mice), by which deletion of the floxed Adam10 allele and transcriptional activation of EGFP in SCs can be simultaneously achieved. As a control, we also generated mice hemizygous for both the Pax7-Cre and EGFP transgenes by crossing Pax7tm2.1(cre/ERT2)Fan/J transgenic mice and CAG-CAT-EGFP reporter mice (henceforth referred to as WTPax7-EGFP mice). SC-specific expression of Cre recombinase was confirmed by analyzing the expression of EGFP and immunostaining for PAX7 in the muscle fibers collected from the WTPax7-EGFP mice (data not shown). The Adam10flox/flox mice exhibited no apparent defects and were used as control animals in the present study (henceforth referred to as Ctrl mice) (20). All animal experiments were approved by the Institutional Animal Care and Use Committee of the Keio University School of Medicine.
Reagents and Antibodies
All siRNAs were purchased from Sigma. The following antibodies were used: anti-PAX7 (1:100, ab34360; Abcam, Cambridge, England), anti-activated Notch1 (1:400, ab8925; Abcam), anti-MyoD (1:50, sc-32758; Santa Cruz, Dallas, TX), anti-GFP (1:500, GF090R; Nacalai Tesque, Kyoto, Japan), anti-perilipin (1:500, D1D8; Cell Signaling Technology), anti-ADAM10 (1:2000, 422751; EMD Millipore, Germany), and anti-GAPDH (1:5000, G9545; Sigma).
Flow Cytometry
Skeletal muscle from 8–12-week-old mice was harvested for flow cytometric analysis. After visible fat tissues, vessels, nerves, and tendons were removed, the muscles were thoroughly chopped and digested in a mixture of collagenase (Wako Pure Chemical Industries, Osaka, Japan), dispase (Life Technologies), and CaCl2. Digested samples were filtered through cell strainers to remove debris, and red blood cells were removed using Red Blood Cell Lysis Buffer (Roche Diagnostics) before antibody application. The following fluorochrome-conjugated monoclonal antibodies were used: anti-Sca1 (1:200, D7), anti-CD31 (1:80, MEC13.3), and anti-CD45 (1:1333, 30-F11). These antibodies were purchased from Biolegend. The biotinylated-SM/C2.6 monoclonal antibody was generously provided by Dr. S. Fukada (26). The flow cytometric analysis was performed using a Gallios Flow Cytometer (Beckman Coulter, Brea, CA).
In Situ Hybridization
Paraffin-embedded sections of cardiotoxin-treated tibialis anterior (TA) muscles were used for in situ hybridization. The sections were deparaffinized and then probed for Pax7 transcripts using an RNAscope Fluorescent Multiplex Reagent kit (Probe-Mm-Pax7; 314181; Advanced Cell Diagnostics, Hayward, CA) as instructed by the manufacturer. The sections were counterstained with Mayer's hematoxylin.
Muscle-injury Model
The mice were anesthetized with a peritoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg). Muscle injury was induced with an intramuscular injection of 50 μl cardiotoxin/PBS (10 μm) in the TA muscle. The mice were closely monitored until fully recovered from the anesthesia and treatment.
Isolation, Culture, and Staining of SCs
SCs were isolated from myofibers of the extensor digitorum longus muscles digested with type 1 collagenase (Worthington, Lakewood, NJ). The isolated fibers were cultured in DMEM, high glucose, GlutaMax supplement (Life Technologies), 20% FBS, 1% chicken embryo extract (USBiological, Pittsburgh, PA), and antibiotics on dishes coated with Matrigel (Corning, Corning, NY). When necessary, 4-hydroxytamoxifen (4-OHT, Sigma) diluted in ethanol (10 μm) was added to the medium. The SCs that were detached from the fibers and were cultured for 3–4 days. The cells were fixed with 4% paraformaldehyde, permeated with 0.2% Triton X/PBS, and immunostained for analysis. Images were collected using an Olympus FSX100 fluorescence microscope (Olympus, Tokyo, Japan) and Olympus FSX-BSW software, and processed using Adobe Photoshop CS6.
C2C12 Cell Culture
C2C12 cells were maintained in DMEM high glucose (Nacalai Tesque) supplemented with 20% FBS and antibiotics. To induce myogenic differentiation, the medium was replaced with DMEM supplemented with 2% horse serum and antibiotics. Transfection of siRNAs was performed using Lipofectamine RNAiMAX Transfection Reagent (Life Technologies) as instructed by the manufacturer. For the induction of Notch signaling, cells were plated on culture dishes coated with recombinant human soluble DLL4 (1.5 μg/ml; PeproTech, Rocky Hill, NJ).
Quantitative RT-PCR
Total RNA was extracted from freshly isolated muscles and cultured cells using Sepasol-RNA I Super G (Nacalai Tesque) and reverse-transcribed using ReverTra Ace reverse transcriptase (Toyobo Life Science, Osaka, Japan). PCR amplification and quantification were performed using Thunderbird qPCR Mix (Toyobo) and a Light Cycler II (Roche Diagnostics). Gene transcript levels were normalized to the expression levels of Gapdh transcripts. Muscle tissues were collected 3 and 5 days after injury.
Statistical Analysis
GraphPad Prism software (Version 6.05; La Jolla, CA) was used for the statistical analyses. Student's t test (for two samples, assuming equal variances) was used to calculate the p values (two-tailed), and p < 0.05 was considered statistically significant.
Results
Abrogation of ADAM10 in SCs Did Not Lead to Any Noticeable Defects during Postnatal Growth
To investigate the potential roles of ADAM10 in SCs, we generated a mutant mouse line in which ADAM10 can be specifically abrogated in SCs by tamoxifen treatment (Adam10Pax7 mice) (20, 24). We first isolated SCs from Adam10Pax7 and Ctrl mice and cultured the cells in the presence of 4-OHT for 6 days. As shown in Fig. 1A, we found that Ctrl SCs expressed ADAM10 and that the expression of ADAM10 was markedly reduced in the cells derived from Adam10Pax7 mice, indicating that the floxed allele in Adam10Pax7 mice was successfully removed upon 4-OHT treatment.
FIGURE 1.

Adam10Pax7 mice do not exhibit any apparent growth defects. A, expression of ADAM10 in 4-OHT-treated SCs collected from Ctrl and Adam10Pax7 mice. B, outline of the schedule for tamoxifen treatment. 3-Week-old mice were treated with tamoxifen for five consecutive days and once a week thereafter. The mice were sacrificed (SAC) for analysis when they reached 9 weeks of age. C, H&E-stained sections of the TA muscles from 9-week-old Ctrl and Adam10Pax7 mice. Bars, 1 mm (upper panels) and 100 μm (lower panels). D, frequency distribution of the cross-sectional area (CSA) of the TA muscles collected from 9-week-old Ctrl and Adam10Pax7 mice. The median value of the cross-sectional area of each genotype is presented. The histogram summarizes the data obtained from four mice of each genotype.
3-Week-old Adam10Pax7 and Ctrl littermate mice were treated with tamoxifen consecutively for 5 days and once a week afterward as outlined in Fig. 1B. The Adam10Pax7 mice grew normally and did not exhibit any apparent systemic defects at the time of analysis (9-week-old) compared with their Ctrl littermates (data not shown). Cross-sections of the TA muscle and the evaluation of the myofiber cross sectional area (CSA) showed no noticeable differences between Ctrl and Adam10Pax7 mice (Fig. 1C and D). These observations indicate that, at least under the current unchallenged experimental settings, ADAM10 in SCs is not essential for the postnatal development or homeostasis of skeletal muscle.
Loss of Muscle Regeneration Capacity in Adam10Pax7 Mice
Because SCs play essential roles in muscle regeneration, we next asked how the abrogation of ADAM10 in SCs affects the capacity for regeneration in skeletal muscle after injury. Cardiotoxin was injected into the TA muscle of 9-week-old Adam10Pax7 mice and Ctrl mice to induce extensive muscle injury. The mice were euthanized 7 or 28 days post-injury (DPI) for analysis (Fig. 2A). The size of the cross-sections of the TA muscle, which reflects the degree of regeneration, was significantly smaller in Adam10Pax7 mice compared with Ctrl mice even at 7 DPI (Fig. 2, B and C). A large number of regenerating myofibers, characterized by central nuclei, were found in the TA muscle sections from the Ctrl mice (Fig. 2D). However, these regenerating myofibers were almost completely missing from the TA muscle of the Adam10Pax7 mice, as the damaged region was predominantly filled with immature adipocytes, fibrous tissue, and infiltrating immune cells (Fig. 2E). Consistent with these findings, immunostaining for the adipocyte marker perilipin revealed positive staining in the damaged muscle of the Adam10Pax7 mice but not in that of the Ctrl mice (Fig. 2, F and G). Masson Trichrome staining showed dense collagen fibers surrounding the vessels and very few myofibers in Adam10Pax7 mice (Fig. 2, H and I). At 28 DPI, the damaged muscle was almost fully regenerated in the Ctrl mice with a few myofibers exhibiting central nuclei (Fig. 2, J and L). However, in the Adam10Pax7 mice, the damaged myofibers were replaced by adipocytes with no regenerating myofibers (Fig. 2, K and M). Immunostaining for perilipin clearly demonstrated infiltration of mature adipocytes in the Adam10Pax7 mice but almost none in Ctrl mice (Fig. 2, N and O). In addition, the vessels were densely surrounded with fibroblasts and collagenous fibers in the Adam10Pax7 mice (Fig. 2, P and Q). These observations show that the abrogation of ADAM10 in SCs results in nearly complete loss of the capacity for muscle regeneration after injury and in muscle degeneration, which is accompanied by fat cell infiltration and accumulation of collagen fibers.
FIGURE 2.

Muscle regeneration is severely compromised in Adam10Pax7 mice. A, outline of the schedule for tamoxifen treatment and muscle-injury experiments. Tamoxifen-treated mice were subjected to cardiotoxin injection (CTX) when they reached 9 weeks of age. The mice were sacrificed (SAC) at 7 DPI or 28 DPI for analysis. Sections of the TA muscles at 7 DPI (B–E) and 28 DPI (J–M) were stained with H&E. The inset in D shows a magnified image of myofibers with central nuclei. Bars, 1 mm (B, C, J, and K) and 100 μm (D, E, L, and M). Sections immunostained for perilipin with DAPI counterstaining (F, G, N, and O). The DAPI-stained images were artificially converted to green to enhance the visualization of the nuclei. Bar, 1 mm. The insets in G and O show a magnified image of perilipin-positive cells. Sections were stained by Masson Trichrome (H, I, P, and Q). Bar, 100 μm. Muscle fibers and cytoplasm are stained in red, collagen fibers are in blue, and nucleuses are in dark brown, respectively.
Marked Decrease in the Expression of Myogenic Markers in Adam10Pax7 Mice after Injury
To further investigate the defects in Adam10Pax7 mice, we next examined the gene expression profiles of the muscles of Adam10Pax7 mice and Ctrl mice after injury. Because the genes that are involved in muscle regeneration are robustly induced within the first week after injury, we collected muscle tissues 3 and 5 days after cardiotoxin treatment. Consistent with the histology, there was a significant decrease in the transcript levels of myogenic markers, including Pax7, Myh3 (encodes embryonic myosin heavy-chain), Myod1, and Myf5, in the injured TA muscle of Adam10Pax7 mice compared with Ctrl mice (Fig. 3). On the other hand, there was a transient increase in the adipogenic markers Pparg (encodes PPARγ (peroxisome proliferator-activated receptor γ)) and Lep (encodes leptin) after injury in the Ctrl mice; however, these transcripts decreased nearly to basal levels by 5 DPI. By contrast, these adipogenic markers remained highly expressed in the Adam10Pax7 mice, reflecting the adipogenic changes revealed by the histological assays (Fig. 2). In addition, we also found an increase in the expression of Pdgfra, a marker for fibro/adipogenic progenitors (FAPs) or PDGFRα+ mesenchymal cells, which serve as a source for fibroblasts and adipogenic cells in muscle (27–29), in Adam10Pax7 mice compared with Ctrl mice. Taken together, the gene expression analyses suggest that myogenic differentiation was severely hampered in the Adam10Pax7 mice, whereas adipogenic and fibroblastic differentiation were enhanced.
FIGURE 3.

Expression of myogenic transcripts is significantly reduced in Adam10Pax7 mice. Relative expression of Pax7, Myh3, Myod1, Myf5, Pparg, Lep, and Pdgfra in uninjured TA muscles (UI) or in muscles at 3 DPI and 5 DPI collected from Ctrl and Adam10Pax7 mice. n = 4–10 for each genotype. Error bars represent S.D. **, p < 0.005.
SCs Are Depleted in Adam10Pax7 Mice
The decreased expression of myogenic-related genes in Adam10Pax7 mice suggests that SCs lacking ADAM10 failed to differentiate into myogenic lineage or that the number of SCs was decreased in Adam10Pax7 mice. To explore these possibilities, we performed in situ hybridization using a cRNA probe against Pax7 transcripts. As shown in Fig. 4A, there were numerous Pax7-positive cells in the injured muscle sections of Ctrl mice; however, very few cells were positive for Pax7 transcripts in the sections of Adam10Pax7 mice. This result suggests that the SC population was depleted or that the proliferation of SCs was severely hampered in Adam10Pax7 mice. To test these hypotheses, we next performed flow cytometric analysis using a single-cell suspension prepared from uninjured muscles to evaluate the size of the SC pool under unchallenged conditions. We found that the SC population (defined as SM/C2.6+ Sca1− CD45− CD31− cells) (26) was significantly reduced in Adam10Pax7 mice compared with Ctrl mice (Fig. 4, B and C). Although we cannot fully exclude the possibility that abrogation of ADAM10 also exhibits negative effects on myogenic differentiation or proliferation of SCs during muscle regeneration, these observations lean toward the idea that ADAM10 is involved in the maintenance of the SC pool in vivo. We thus concluded that Adam10Pax7 mice are incapable of regenerating damaged muscles most likely due to the loss of SCs.
FIGURE 4.
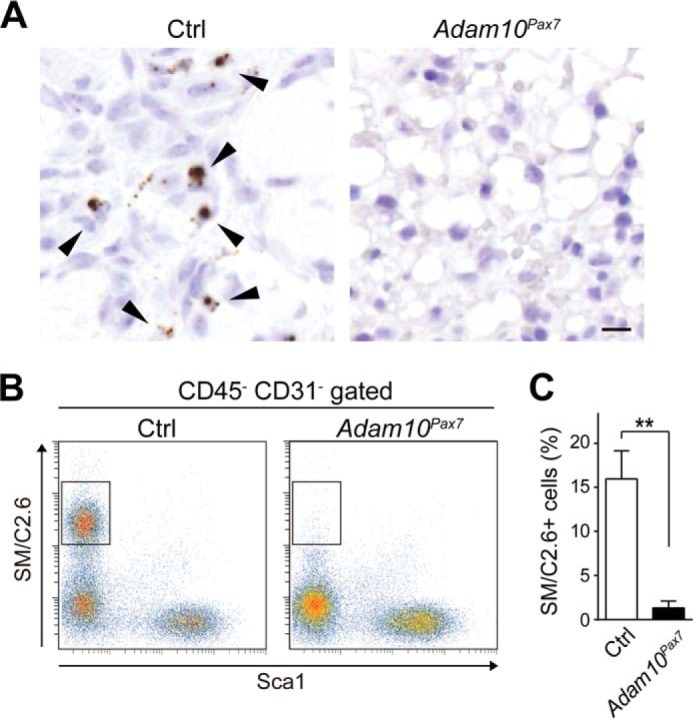
Depletion of SCs in Adam10Pax7 mice. A, sections of the TA muscle taken at 7 DPI from Ctrl and Adam10Pax7 mice probed for Pax7 transcripts and counterstained with Mayer's hematoxylin. The arrowheads indicate Pax7-positive cells. Bar, 10 μm. B, flow cytometric analysis of the SC population using a single-cell suspension prepared from the muscles of uninjured Ctrl and Adam10Pax7 mice (boxed areas). C, percentage of SM/C2.6+ Sca1− cells in the CD45− CD31− gated population of a single-cell suspension collected from the muscles of Ctrl and Adam10Pax7 mice. n = 4 mice for each genotype. Error bars represent S.D. **, p < 0.005.
Myogenic Differentiation Is Accelerated in SCs in the Absence of ADAM10
Depletion of the SC population can be explained by either increased apoptosis or a dysregulation of myogenic differentiation in the SCs. We collected myofibers from tamoxifen-treated Adam10Pax7 and Ctrl mice and co-immunostained them with anti-PAX7 and anti-MYOD1 antibodies (the schedule for the tamoxifen treatment is shown in Fig. 5A). Cells that were positive for PAX7 and/or MYOD1 were counted, and the numbers were statistically analyzed. There was a significant reduction in the number of PAX7+/MYOD1− cells (quiescent SCs) and an increase in PAX7−/MYOD1+ cells (committed myoblasts) in Adam10Pax7 mice compared with Ctrl mice (Fig. 5B). On the other hand, no significant difference was found in the number of caspase-3-positive apoptotic cells between Adam10Pax7 and Ctrl mice (data not shown), suggesting that ADAM10 in SCs is not essential for cell survival. These observations indicate that SCs lacking ADAM10 cannot efficiently maintain their quiescent state and consequently undergo myogenic differentiation.
FIGURE 5.

ADAM10 is critically involved in the maintenance of quiescence in SCs mediated by Notch signaling. A, outline of the schedule for tamoxifen treatment and myotube culture. 8–12-Week-old Ctrl and Adam10Pax7 mice were treated with tamoxifen for five consecutive days and then sacrificed (SAC) on day 7 (D7). Myofibers were harvested and incubated in the presence of 4-OHT for 4 days and analyzed on day 11 (D11). B, percentages of PAX7−/MYOD1+, PAX7+/MYOD1+, and PAX7+/MYOD1− cells harvested from the myofibers of Ctrl and Adam10Pax7 mice. n = 3 mice per genotype. *, p < 0.05. NS = not significant. C, immunostaining for N1ICD in SCs isolated from the myofibers of WTPax7-EGFP and Adam10Pax7-EGFP mice. Cells positive for both EGFP and N1ICD (arrowheads) represent quiescent SCs, whereas cells positive for EGFP but negative for N1ICD represent activated or differentiated SCs. D, percentage of N1ICD+/EGFP+ cells harvested from the myofibers of WTPax7-EGFP and Adam10Pax7-EGFP mice. n = 4 mice per genotype. **, p < 0.005. E and F, C2C12 cells transfected with control siRNA or siRNA against Adam10 were incubated on recombinant soluble DLL4 (sDDL4)-coated wells for 4 days. The formation of myotubes and the expression levels of Myog transcripts were evaluated by microscopy (E) and quantitative PCR (F), respectively. Bar, 100 μm. n = 3. **, p < 0.005. G, efficacy of the gene silencing was confirmed by quantitative PCR (left panel) and Western blot (right panel, arrowheads). n = 3. **, p < 0.005.
SCs Lacking ADAM10 Are Defective in Notch Signaling
The phenotype of Adam10Pax7 mice observed in the current study resembles those described for Notch signaling-defective mice and SC-depleted mutant mice (30–34). Because a disruption of Notch signaling results in enhanced differentiation of SCs along the myogenic lineage and because ADAM10 is potentially the most important sheddase for Notch, the defects observed in Adam10Pax7 mice were assumed to arise from aberrant Notch processing. We performed fate-mapping analysis of SCs in Adam10Pax7 mice by crossing these mice with CAG-CAT-EGFP reporter mice (25) (Adam10Pax7-EGFP mice). As a control, we used mice hemizygous for both the Pax7-Cre and EGFP transgenes (WTPax7-EGFP mice). Cells were isolated from the myofibers of these mice and immunostained using a Notch1 intracellular domain (N1ICD)-specific antibody. We found that significantly fewer EGFP-positive cells were also positive for N1ICD in the Adam10Pax7-EGFP mice than in the WTPax7-EGFP mice (Fig. 5, C and D), suggesting that upon the loss of ADAM10, SCs lose the ability to sustain Notch signaling and consequently differentiate along the myogenic lineage.
To test the hypothesis that ADAM10 mediates Notch-dependent inhibition of myogenic differentiation, we next used the myoblast-like cell line C2C12. C2C12 cells differentiate into myoblast-like cells and form myotubes in low serum conditions. In agreement with past studies, we found that the formation of myotubes was significantly inhibited when the cells were plated on Notch ligand DLL4-coated dishes (Fig. 5E) (35). However, the Notch-mediated suppression of the myogenic differentiation and expression of Myog were effectively rescued by gene-silencing of Adam10 (Fig. 5, E and F). Gene silencing and reduced expression of ADAM10 by siRNA were confirmed by quantitative PCR and Western blot (Fig. 5G). These observations support the idea that ADAM10 functions as a negative regulator of SC differentiation.
Discussion
In the present study we showed that abrogation of ADAM10 in SCs results in depletion of the SC pool and loss of the muscle regenerative capacity after injury. The decrease in the SC population was not due to increased apoptosis but due to a dysregulated and accelerated differentiation of SCs toward the myogenic lineage. We also found that SCs lacking ADAM10 are defective in Notch signaling, a critical regulator for maintaining the quiescent state in SCs (10–12). In agreement, suppression of ADAM10 expression in C2C12 cells offsets the inhibitory effect of Notch signaling induced by DLL4. These observations suggest that ADAM10 in SCs negatively regulates the differentiation SCs through activation of Notch signaling (Fig. 6). On the other hand, lack of any apparent defects in Adam10Pax7 mice under unchallenged conditions indicates that ADAM10 in SCs may not be prerequisite for homeostasis or growth of muscle tissues after birth.
FIGURE 6.

The schematic model proposed by the present study. A, under normal conditions, Notch receptors and the expression of their down-stream molecules (such as HES and HEY proteins (30)) are constitutively activated by ADAM10 (and through binding of Notch ligands) and thereby maintain the quiescent state of SCs. B, loss of ADAM10, in turn, suppresses Notch signaling and consequently results in the dysregulated differentiation of SCs. NICD, Notch intracellular domain.
Although there are studies showing that ADAM17 is responsible for the cleavage of Notch and thus for the activation of Notch signaling (18, 19, 36, 37), several lines of evidence argue against this notion (please also refer to Groot et al. (38) for a detailed discussion on this issue). Most importantly, most of the ADAM10 loss-of-function mutant mice models examined to date show phenotypes that are highly related to Notch loss-of-function (20–22, 39, 40). These observations also imply that ADAM17 is not capable of compensating the loss of ADAM10 in processing Notch in vivo. Furthermore, most ADAM17 mutant mice show defects that are closely related to the loss of EGFR signaling (and of TNFα) but do not exhibit any defects suggestive of defective Notch signaling (41–44). It is possible that ADAM17 cleaves Notch under non-physiological conditions in vitro, but emerging data are in favor of the idea that ADAM10 is solely responsible for Notch processing in vivo. Accordingly, the phenotype of Adam10Pax7 mice showed striking similarities with different mutant mice with defective Notch-signaling in SCs (30–34). Furthermore, we also observed that mutant mice lacking ADAM17 in SCs did not exhibit any defects that are related to defective Notch signaling.3 Taken together, our observations further consolidate ADAM10 as a unique and indispensable enzyme for mediating Notch cleavage.
Muscle injury experiments showed that fat infiltration and accumulation of fibrous tissue in damaged muscle occur in Adam10Pax7 mice. This may appear on the surface that SCs lacking ADAM10 have altered their cell fate so as to differentiate into adipocytes and fibroblasts; however, the data of the present and past studies confute this assumption. First of all, this phenomenon is not unique to Adam10Pax7 mice but is also observed in the other mutant mice that are depleted of SCs (45, 46). Most importantly, recent studies have shown that SCs are highly specific progenitors for myofibers and are not capable of differentiating into other lineages (27, 45–47). Of note, the progenitors for adipocytes and fibroblasts in muscles, which were not related to SCs, have also been identified and were named FAPs/PDGFRα+ mesenchymal cells (although not proven, it is conceivable that these cells are closely related, if not identical) (27–29, 48).
Interestingly, gene expression analysis revealed a transient increase in the transcripts for Pgfra, a marker for the aforementioned FAPs/PDGFRα+ mesenchymal cells, and adipocyte-related genes (Pparg and Lep) after injury in Ctrl mice, indicating that adipocyte differentiation is induced after muscle injury. However, the induction toward adipocyte differentiation subsides in Ctrl mice as the myofibers start to regenerate. On the contrary, the expression of these genes was not suppressed in Adam10Pax7 mice, resulting in severe infiltration of fat tissue in damaged muscles. These observations suggest that both SCs and FAPs/PDGFRα+ mesenchymal cells are activated upon muscle injury; however, the activation of FAPs/PDGFRα+ mesenchymal cells are effectively suppressed as the SCs cells differentiate and regenerate damaged muscle fibers. Given these observations, it is tempting to speculate that there is a potential interaction between these two cell types, by which the balance of myogenesis and adipocyte/fibroblast differentiation is determined.
In summary, our data show that conditional ablation of ADAM10 in Pax7-expressing cells results in a depletion of the SC population and failure to regenerate damaged muscles. Notch processing was hampered in SCs lacking ADAM10, further corroborating the notion that ADAM10 is the major sheddase for Notch. Conclusively, the present study demonstrates that ADAM10 functions as a gatekeeper of SCs by preventing premature differentiation through processing and activation of Notch (Fig. 6).
Author Contributions
S. M. designed, performed, and analyzed most of the experiments. M. Y. designed, performed, and analyzed the experiments shown in Fig. 4, B and C, and provided technical assistance. M. S. and Y. O. performed the histological analysis shown in Fig. 2. T. T., S. T., Y. T., M. N., and M. M. provided technical assistance and contributed to the preparation of the figures. K. H. conceived and coordinated the study and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Yuka Sunaga, Kaori Sue, and Mayumi Ito for excellent technical support, So-ichiro Fukada (Osaka University, Osaka, Japan) for generously providing the SM/C2.6 antibody, and Nozomi Hayashiji (Keio University, Tokyo, Japan) and Naoki Ito (National Center of Neurology and Psychiatry, Tokyo, Japan) for technical advice.
This work was supported in part MEXT (Ministry of Education, Culture, Sports, Science, and Technology) KAKENHI (24390358; to K. H.). The authors declare that they have no conflicts of interest with the contents of this article.
S. Mizuno and K. Horiuchi, unpublished observation.
- SC
- satellite cell
- ADAM
- a disintegrin and metalloprotease
- DPI
- day(s) post-injury
- N1ICD
- Notch1 intracellular domain
- FAP
- fibro-adipose progenitor
- TA
- tibialis anterior
- 4-OHT
- 4-hydroxytamoxifen
- EGFP
- enhanced GFP.
References
- 1.Dhawan J., and Rando T. A. (2005) Stem cells in postnatal myogenesis: molecular mechanisms of satellite cell quiescence, activation, and replenishment. Trends Cell Biol. 15, 666–673 [DOI] [PubMed] [Google Scholar]
- 2.Fukada S., Ma Y., Ohtani T., Watanabe Y., Murakami S., and Yamaguchi M. (2013) Isolation, characterization, and molecular regulation of muscle stem cells. Front Physiol 4, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yin H., Price F., and Rudnicki M. A. (2013) Satellite cells and the muscle stem cell niche. Physiol. Rev. 93, 23–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernet J. D., Doles J. D., Hall J. K., Kelly Tanaka K., Carter T. A., and Olwin B. B. (2014) p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat. Med. 20, 265–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cosgrove B. D., Gilbert P. M., Porpiglia E., Mourkioti F., Lee S. P., Corbel S. Y., Llewellyn M. E., Delp S. L., and Blau H. M. (2014) Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat. Med. 20, 255–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brack A. S., Conboy M. J., Roy S., Lee M., Kuo C. J., Keller C., and Rando T. A. (2007) Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science 317, 807–810 [DOI] [PubMed] [Google Scholar]
- 7.Carlson M. E., Hsu M., and Conboy I. M. (2008) Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature 454, 528–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Price F. D., von Maltzahn J., Bentzinger C. F., Dumont N. A., Yin H., Chang N. C., Wilson D. H., Frenette J., and Rudnicki M. A. (2014) Inhibition of JAK-STAT signaling stimulates adult satellite cell function. Nat. Med. 20, 1174–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sousa-Victor P., Gutarra S., García-Prat L., Rodriguez-Ubreva J., Ortet L., Ruiz-Bonilla V., Jardí M., Ballestar E., González S., Serrano A. L., Perdiguero E., and Muñoz-Cánoves P. (2014) Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 506, 316–321 [DOI] [PubMed] [Google Scholar]
- 10.Conboy I. M., and Rando T. A. (2002) The regulation of Notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Dev Cell 3, 397–409 [DOI] [PubMed] [Google Scholar]
- 11.Buas M. F., and Kadesch T. (2010) Regulation of skeletal myogenesis by Notch. Exp. Cell Res. 316, 3028–3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mourikis P., and Tajbakhsh S. (2014) Distinct contextual roles for Notch signalling in skeletal muscle stem cells. BMC Dev. Biol. 14, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koch U., Lehal R., and Radtke F. (2013) Stem cells living with a Notch. Development 140, 689–704 [DOI] [PubMed] [Google Scholar]
- 14.Andersson E. R., Sandberg R., and Lendahl U. (2011) Notch signaling: simplicity in design, versatility in function. Development 138, 3593–3612 [DOI] [PubMed] [Google Scholar]
- 15.Kopan R., and Ilagan M. X. (2009) The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137, 216–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pruessmeyer J., and Ludwig A. (2009) The good, the bad and the ugly substrates for ADAM10 and ADAM17 in brain pathology, inflammation and cancer. Semin. Cell Dev. Biol. 20, 164–174 [DOI] [PubMed] [Google Scholar]
- 17.Reiss K., and Saftig P. (2009) The “a disintegrin and metalloprotease” (ADAM) family of sheddases: physiological and cellular functions. Semin. Cell Dev. Biol. 20, 126–137 [DOI] [PubMed] [Google Scholar]
- 18.Brou C., Logeat F., Gupta N., Bessia C., LeBail O., Doedens J. R., Cumano A., Roux P., Black R. A., and Israël A. (2000) A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Mol Cell 5, 207–216 [DOI] [PubMed] [Google Scholar]
- 19.Murthy A., Shao Y. W., Narala S. R., Molyneux S. D., Zúñiga-Pflücker J. C., and Khokha R. (2012) Notch activation by the metalloproteinase ADAM17 regulates myeloproliferation and atopic barrier immunity by suppressing epithelial cytokine synthesis. Immunity 36, 105–119 [DOI] [PubMed] [Google Scholar]
- 20.Yoda M., Kimura T., Tohmonda T., Uchikawa S., Koba T., Takito J., Morioka H., Matsumoto M., Link D. C., Chiba K., Okada Y., Toyama Y., and Horiuchi K. (2011) Dual functions of cell-autonomous and non-cell-autonomous ADAM10 activity in granulopoiesis. Blood 118, 6939–6942 [DOI] [PubMed] [Google Scholar]
- 21.Hartmann D., de Strooper B., Serneels L., Craessaerts K., Herreman A., Annaert W., Umans L., Lübke T., Lena Illert A., von Figura K., and Saftig P. (2002) The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for α-secretase activity in fibroblasts. Hum. Mol. Genet. 11, 2615–2624 [DOI] [PubMed] [Google Scholar]
- 22.Weber S., Niessen M. T., Prox J., Lüllmann-Rauch R., Schmitz A., Schwanbeck R., Blobel C. P., Jorissen E., de Strooper B., Niessen C. M., and Saftig P. (2011) The disintegrin/metalloproteinase Adam10 is essential for epidermal integrity and Notch-mediated signaling. Development 138, 495–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weber S., and Saftig P. (2012) Ectodomain shedding and ADAMs in development. Development 139, 3693–3709 [DOI] [PubMed] [Google Scholar]
- 24.Lepper C., and Fan C. M. (2010) Inducible lineage tracing of Pax7-descendant cells reveals embryonic origin of adult satellite cells. Genesis 48, 424–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kawamoto S., Niwa H., Tashiro F., Sano S., Kondoh G., Takeda J., Tabayashi K., and Miyazaki J. (2000) A novel reporter mouse strain that expresses enhanced green fluorescent protein upon Cre-mediated recombination. FEBS Lett. 470, 263–268 [DOI] [PubMed] [Google Scholar]
- 26.Fukada S., Higuchi S., Segawa M., Koda K., Yamamoto Y., Tsujikawa K., Kohama Y., Uezumi A., Imamura M., Miyagoe-Suzuki Y., Takeda S., and Yamamoto H. (2004) Purification and cell-surface marker characterization of quiescent satellite cells from murine skeletal muscle by a novel monoclonal antibody. Exp. Cell Res. 296, 245–255 [DOI] [PubMed] [Google Scholar]
- 27.Uezumi A., Fukada S., Yamamoto N., Takeda S., and Tsuchida K. (2010) Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat Cell Biol. 12, 143–152 [DOI] [PubMed] [Google Scholar]
- 28.Uezumi A., Ito T., Morikawa D., Shimizu N., Yoneda T., Segawa M., Yamaguchi M., Ogawa R., Matev M. M., Miyagoe-Suzuki Y., Takeda S., Tsujikawa K., Tsuchida K., Yamamoto H., and Fukada S. (2011) Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. J. Cell Sci. 124, 3654–3664 [DOI] [PubMed] [Google Scholar]
- 29.Joe A. W., Yi L., Natarajan A., Le Grand F., So L., Wang J., Rudnicki M. A., and Rossi F. M. (2010) Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol. 12, 153–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fukada S., Yamaguchi M., Kokubo H., Ogawa R., Uezumi A., Yoneda T., Matev M. M., Motohashi N., Ito T., Zolkiewska A., Johnson R. L., Saga Y., Miyagoe-Suzuki Y., Tsujikawa K., Takeda S., and Yamamoto H. (2011) Hesr1 and Hesr3 are essential to generate undifferentiated quiescent satellite cells and to maintain satellite cell numbers. Development 138, 4609–4619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bjornson C. R., Cheung T. H., Liu L., Tripathi P. V., Steeper K. M., and Rando T. A. (2012) Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem Cells 30, 232–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lepper C., Partridge T. A., and Fan C. M. (2011) An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development 138, 3639–3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin S., Shen H., Jin B., Gu Y., Chen Z., Cao C., Hu C., Keller C., Pear W. S., and Wu L. (2013) Brief report: Blockade of Notch signaling in muscle stem cells causes muscular dystrophic phenotype and impaired muscle regeneration. Stem Cells 31, 823–828 [DOI] [PubMed] [Google Scholar]
- 34.Mourikis P., Sambasivan R., Castel D., Rocheteau P., Bizzarro V., and Tajbakhsh S. (2012) A critical requirement for notch signaling in maintenance of the quiescent skeletal muscle stem cell state. Stem Cells 30, 243–252 [DOI] [PubMed] [Google Scholar]
- 35.Buas M. F., Kabak S., and Kadesch T. (2009) Inhibition of myogenesis by Notch: evidence for multiple pathways. J. Cell. Physiol. 218, 84–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bozkulak E. C., and Weinmaster G. (2009) Selective use of ADAM10 and ADAM17 in activation of Notch1 signaling. Mol. Cell. Biol. 29, 5679–5695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pisconti A., Cornelison D. D., Olguín H. C., Antwine T. L., and Olwin B. B. (2010) Syndecan-3 and Notch cooperate in regulating adult myogenesis. J. Cell Biol. 190, 427–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Groot A. J., Cobzaru C., Weber S., Saftig P., Blobel C. P., Kopan R., Vooijs M., and Franzke C. W. (2013) Epidermal ADAM17 is dispensable for notch activation. J. Invest. Dermatol. 133, 2286–2288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jorissen E., Prox J., Bernreuther C., Weber S., Schwanbeck R., Serneels L., Snellinx A., Craessaerts K., Thathiah A., Tesseur I., Bartsch U., Weskamp G., Blobel C. P., Glatzel M., De Strooper B., and Saftig P. (2010) The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J. Neurosci. 30, 4833–4844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu H., Zhu J., Smith S., Foldi J., Zhao B., Chung A. Y., Outtz H., Kitajewski J., Shi C., Weber S., Saftig P., Li Y., Ozato K., Blobel C. P., Ivashkiv L. B., and Hu X. (2012) Notch-RBP-J signaling regulates the transcription factor IRF8 to promote inflammatory macrophage polarization. Nat. Immunol. 13, 642–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Horiuchi K., Kimura T., Miyamoto T., Takaishi H., Okada Y., Toyama Y., and Blobel C. P. (2007) TNFα-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J. Immunol. 179, 2686–2689 [DOI] [PubMed] [Google Scholar]
- 42.Franzke C. W., Cobzaru C., Triantafyllopoulou A., Löffek S., Horiuchi K., Threadgill D. W., Kurz T., van Rooijen N., Bruckner-Tuderman L., and Blobel C. P. (2012) Epidermal ADAM17 maintains the skin barrier by regulating EGFR ligand-dependent terminal keratinocyte differentiation. J. Exp. Med. 209, 1105–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saito K., Horiuchi K., Kimura T., Mizuno S., Yoda M., Morioka H., Akiyama H., Threadgill D., Okada Y., Toyama Y., and Sato K. (2013) Conditional inactivation of TNFα-converting enzyme in chondrocytes results in an elongated growth plate and shorter long bones. PLoS ONE 8, e54853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagao K., Kobayashi T., Ohyama M., Akiyama H., Horiuchi K., and Amagai M. (2012) Brief report: requirement of TACE/ADAM17 for hair follicle bulge niche establishment. Stem Cells 30, 1781–1785 [DOI] [PubMed] [Google Scholar]
- 45.Murphy M. M., Lawson J. A., Mathew S. J., Hutcheson D. A., and Kardon G. (2011) Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development 138, 3625–3637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sambasivan R., Yao R., Kissenpfennig A., Van Wittenberghe L., Paldi A., Gayraud-Morel B., Guenou H., Malissen B., Tajbakhsh S., and Galy A. (2011) Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development 138, 3647–3656 [DOI] [PubMed] [Google Scholar]
- 47.Relaix F., and Zammit P. S. (2012) Satellite cells are essential for skeletal muscle regeneration: the cell on the edge returns centre stage. Development 139, 2845–2856 [DOI] [PubMed] [Google Scholar]
- 48.Mitchell K. J., Pannérec A., Cadot B., Parlakian A., Besson V., Gomes E. R., Marazzi G., and Sassoon D. A. (2010) Identification and characterization of a non-satellite cell muscle resident progenitor during postnatal development. Nat. Cell Biol. 12, 257–266 [DOI] [PubMed] [Google Scholar]