

Abstract
Macrophages are a key cell type in the innate immune system, and its proinflammatory (M1) activation in the liver plays a critical role in pathogenesis of alcoholic steatohepatitis. Emerging evidence indicates the involvement of Notch signaling in regulation of innate immune response and cellular metabolism. Metabolic switch to glycolysis characterizes macrophages undergoing M1 activation. It has been proposed that metabolic reprograming in response to extrinsic stimulation, such as bacterial endotoxin, triggers intrinsic signal to dictate cell differentiation. Using an obesity-alcohol synergistic ASH mouse model, we have recently shown that Notch1 pathway promotes M1 activation of hepatic macrophages, through direct upregulation of M1 gene transcription and through reprograming of mitochondrial metabolism to glucose oxidation and subsequent mtROS generation to augment M1 gene expression. Our studies demonstrate a novel mechanism of Notch1 signaling in metabolic reprograming of macrophage for M1 activation in ASH.
Introduction
Alcoholic liver disease encompasses a spectrum of injuries ranging from steatosis, steatohepatitis (ASH), fibrosis, to cirrhosis. M1 activation of hepatic macrophages (HMac) by LPS is a key pathogenic event in ASH (1). Although nuclear factor NF-κB is considered the prototypical proinflammatory signaling pathway, emerging evidence suggests an important role of Notch pathway in M1 activation of macrophages (Mac). In addition to the signaling pathways that regulate Mac activation, metabolic reprograming to glycolysis in M1 Mac and fatty acid oxidation in alternatively activated (M2) Mac suggest a role of metabolism in Mac fate determination (2). We have recently demonstrated that M1 Mac have concomitant increases in aerobic glycolysis and glucose oxidative phosphorylation (OXPHOS), and the latter enhances mitochondrial ROS (mtROS) generation. All of these metabolic events are in a manner dependent on Notch1 pathway. The increased OXPHOS is achieved by Notch1 mediated activation of pyruvate dehydrogenase (PDH) and expression of mitochondrial DNA (mtDNA); and the increased mtROS in turn augments M1 activation. Most importantly, myeloid Notch1 deficiency attenuates transmigration of peripheral blood monocyte/Mac into the liver and subsequent M1 activation in an obesity-alcohol synergistic ASH mouse model (3).
Obesity-alcohol synergism mouse model of steatohepatitis
Alcoholic and non-alcoholic steatohepatitis are the two most common life-style liver diseases caused by excessive intake of alcohol and calories, respectively. Although most alcoholics and obese individuals develop fatty liver, only a fraction of these individuals develop advanced disease such as steatohepatitis, suggesting multifactorial nature of the liver disease (4). Alcohol abuse in obese people, however, causes significantly increases the risk of developing steatohepatitis and cirrhosis (5-7). In experimental animals, earlier study showed that oral gavage of alcohol (4 g/kg) every 12 hours for 3 days caused increased serum ALT and mild ASH in obese rats but not in lean controls (8). In an intragastric feeding mouse model, we previously demonstrated that obesity aggravated alcohol-induced liver injury (9). In this model, mice overfed high fat diet (OF mice) develop moderate obesity and mild steatohepatitis, while mice fed alcohol (Alc mice) developed moderate ASH. When alcohol was co-administrated with the high fat diet, these OF plus alcohol fed (OF+Alc) mice had synergistic increase in serum ALT (393±29 U/L in OF+Alc vs. 240±23 U/L in Alc vs. 28±3 U/L in OF mice), severe steatosis, marked HMac infiltration and M1 activation, pericellular fibrosis, and intensified nitrosative stress induced by a 40-fold induction of nitric oxide synthase (Nos2) (9). These studies clearly show obesity-alcohol synergism in steatohepatitis.
Notch signaling in M1 macrophage activation and ASH
Notch is a single transmembrane receptor with 4 isoforms, Notch1 to 4. Notch signaling is highly conserved pathway and it integrates environmental cues to specify cell fate during development (10). There are five known Notch ligands, Jagged1 and 2, and Delta-like ligand 1, 3, and 4. Notch activation is initiated by ligand binding, which leads to sequential proteolytic cleavage and liberation of Notch intracellular domain (NICD) by γ-secretase. The NICD translocates into the nucleus where it interacts with a DNA binding protein CSL and other nuclear proteins to form a transcriptional co-activator complex (11). It is now clear that Notch functions in cell fate determination extend beyond development. For example, Notch is known to be critical for liver development, but it is also involved in liver regeneration/repair, liver carcinogenesis, and metabolism (12).
Notch pathway has been shown to control expression of genes involved in Mac polarization (13), thereby modulates inflammatory response. Stimulation of bone marrow derived Mac with LPS or toll-like receptor 4 (TLR4) ligands leads to Notch1 and NF-κB activation, and M1 Mac polarization, which are all abrogated by Notch inhibition with γ-secretase inhibitor DAPT or by Notch1 silencing (14,15). At transcriptional level, ChIP-seq reveals NICD1 binding at regions near the transcription start site of M1 genes such as Nos2, Tnf, and Il-17rc (3,16,17). In consistent with these findings, ChIP-qPCR shows increased enrichment of NICD1 at the proximal promoters of Nos2 and Tnf-α in M1 Mac (3,14). The Nos2 promoter activity and expression are suppressed by DAPT or Notch1 deficiency, but is increased in NICD1 overexpressing Mac (3). At translational level, Notch signaling integrates with TLR4 pathway to induce translation of transcription factors IRF8 (interferon regulatory factor 8), a key factor associated with M1 Mac activation (18). These studies demonstrate that Notch1 activation through its effector NICD1 transactivates M1 genes, most likely underlying a molecular mechanism for the Notch1-dependent Mac M1 activation.
Recent studies have shown that hepatic monocyte/Mac migrated from peripheral blood contribute to liver injury, such as N-acetyl-p-aminophenol induced acute liver injury (19), ASH (20), and NASH (21). In Notch1+/- mice, Mac recruitment and M1 activation are decreased during wound healing (22). In the synergistic ASH model of OF+Alc mice, our studies show that HMac isolated from the wild-type mice have upregulated expression of Notch1 along with M1 genes, but not other Notch isoforms and M2 genes. HMac isolated from the myeloid Notch1 KO mice have diminished expression of M1 genes. More importantly, myeloid Notch1 KO prevents hepatic Mac infiltration and M1 activation in the liver of the ASH model (3). Using a method of ex vivo labeling and in vivo tracking of transplanted monocytes, we show significantly increased migration of transplanted wild-type but not Notch1-KO monocytes into the liver of the ASH mice. Expression of M1 genes in the migrated Notch1-KO HMac is significantly decreased compared to the wild-type HMac. These results suggest Notch1 pathway regulates migration of peripheral blood monocytes into the liver and subsequent M1 activation in ASH (3).
Notch and glucose metabolism in M1 macrophages
To meet with the energy demand, cells constantly utilize glucose, fatty acids, and to some extend the amino acids to produce ATP. Glycolysis converts glucose to pyruvate. Under hypoxic environment, pyruvate is further metabolized in cytoplasm to lactate, a process called aerobic glycolysis. In the presence of oxygen sufficiency, pyruvate is converted to acetyl-CoA in mitochondria, which enters tricarboxylic acid (TCA) cycle to generate reducing equivalent nicotinamide adenine dinucleotide (NADH) for OXPHOS. Comparing to OXPHOS, glycolysis generates ATP at a higher rate and therefore enables cells such as cancer cells to gain competitive advantage for shared energy resources such as glucose. On the other hand, glucose through OXPHOS will generate ∼18 fold more ATP than glycolysis (23), which is important for cells undergoing energy consuming process such as phagocytosis by Mac.
The increased glucose uptake and lactate production in Mac undergoing M1 activation makes it generally accepted that M1 Mac relies mainly on aerobic glycolysis for energy metabolism (24,25). However, metabolic switch to glycolysis is energetically unfavorable for the phagocytic activity of M1 Macs (24-26). In fact, it has been shown that Mac rely a significant degree on OXPHOS during phagocytosis (27) or in response to Listeria monocytogenes challenging (27), whereas neutrophils and monocytes are dependent on glycolysis as a source of metabolic energy (28).
Notch has been shown to enhance glycolysis via PI3-K/Akt pathway (29) and upregulation of HIF-1α (30,31). In HMac, our studies show glycolysis is indeed increase. However, these cells have a concomitant ∼23% increase in glucose flux to TCA cycle, higher oxygen consumption rate, and enhanced mtROS generation. The 23 % increase in glucose flux to TCA cycle seems to be trivial, but it would produce 4.2-fold more of ATP than that of glycolysis based on the presumption that 2 ATP is generated through glycolysis and 36 ATP through oxidation per glucose molecule. More importantly, these metabolic events are abrogated by the DAPT treatment, Notch1 silencing, or Notch1 KO, suggesting Notch dependent glucose oxidation and subsequent mtROS in M1 Mac (3). The importance of mtROS in mediating M1 Mac activation and function has recently been shown (32), and will be further discussed below.
Notch1 reprograms mitochondrial metabolism in M1 macrophages
The oxidation of acetyl-CoA to CO2 by the TCA cycle is the central process in mitochondria for energy metabolism. Two enzymes control glucose flux to TCA cycle for oxidation: pyruvate dehydrogenase (PDH) that converts pyruvate to acetyl-CoA as the primary pathway, and pyruvate carboxylase (PC) that converts pyruvate to oxaloacetate as an alternate pathway. The activity of PDH is inhibited by PDH-kinase (PDK) mediated phosphorylation of three serine residues on PDH-E1α catalytic subunit (Figure 1). On the other hand, PDP1 dephosphorylates these residues and thereby activates PDH (33,34). In M1 Mac, our studies show increased NICD1 binding to Pdp1 promoter and expression of Pdp1. This is associated with decreased phosphor-/total- PDH-E1α protein ratio, and increased PDH activity. Notch1 silencing or KO abrogates the induction of Pdp1 and decreases PDH activity (3). These results suggest that Notch1 upregulates PDP1 and subsequent activation of PDH to enhance glucose flux to the TCA cycle for OXPHOS (Figure 1).
Figure 1.
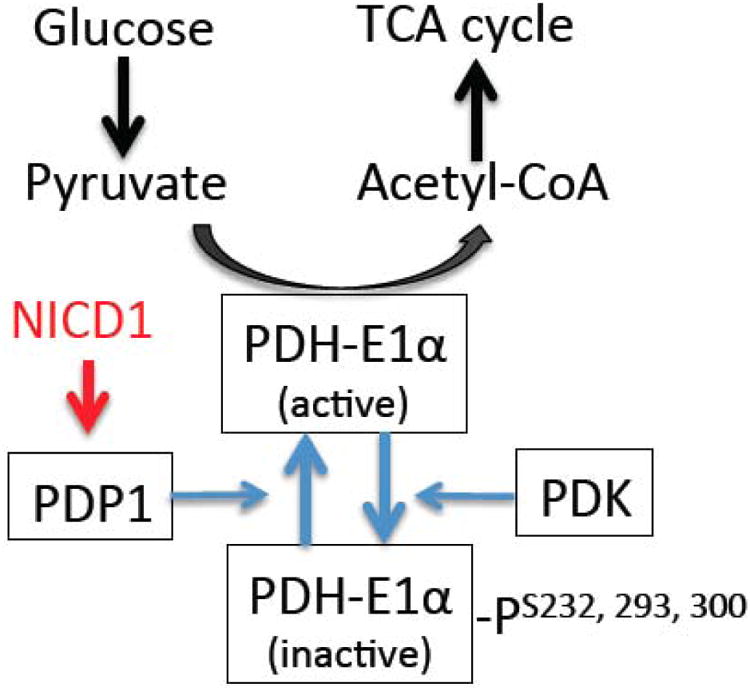
A schematic diagram of Notch1-dependent upregulation of PDP1, which dephosphorylates and activates PDH-E1α to enhance glucose flux to mitochondrial TCA cycle metabolism.
Mitochondrial OXPHOS produces ATP through sequential reductions of respired oxygen via the electron transport chain (ETC) complexes. The components of ETC are encoded by both nuclear DNA and mtDNA, but all proteins encoded by mtDNA are ETC subunits. We and other laboratories have shown translocation of NICD1 to mitochondria (3,35), suggesting a role of Notch1 in mitochondrial function. In fact, mutations in Notch are associated with impaired mitochondrial respiration in CADASIL patients with neurological defects (36,37) and in Drosophila (38). Using ChIP-seq and ChIP-qPCR, we show enrichment of NICD1 in the promoter region of mtDNA displacement loop (D-loop). The expression of mtDNA-encoded ETC components (NADH dehydrogenase, cytochrome b, cytochrome c oxidase, and ATP synthases) is upregulated in a manner dependent on Notch in M1 Mac. These results demonstrate Notch reprograming mitochondrial metabolism to glucose oxidation through upregulation of PDP1 and mtDNA expression (Figure 2).
Figure 2.
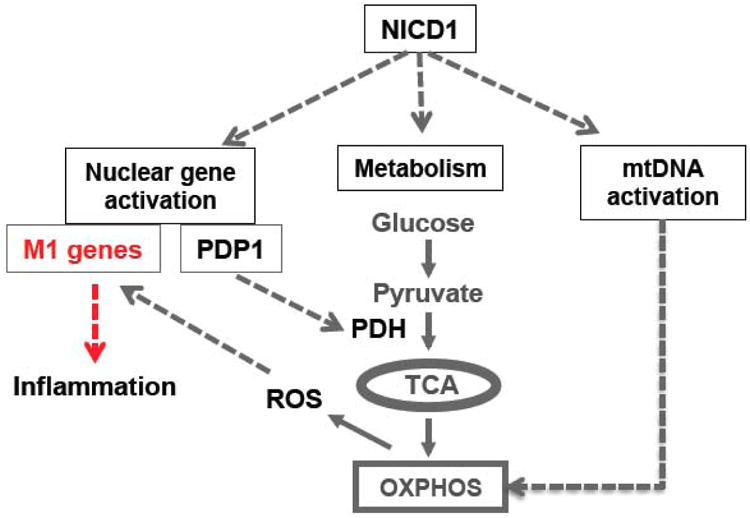
A schematic diagram of Notch1-dependent reprogramming of mitochondrial metabolism in M1 HMacs through upregulation of PDH activity and mtDNA expressions. The metabolic reprogramming provides a positive regulatory loop via enhanced OXPHOS and mtROS to augment NICD1-driven M1 gene expression.
Notch1 through mtROS links metabolic reprograming to M1 macrophage activation
As discussed above, coupled with the increased glucose flux to TCA, Notch may drive mitochondrial respiration to generate ATP for M1 Mac activity. The mitochondrial ETC is also a major source for cellular ROS, although ROS generated by NADPH oxidase is consider for phagocytes against bacteria invasion (39). Recent studies demonstrate significant contribution of mtROS to the bactericidal activity and M1 activation of Mac (32,40,41). In fact, mtROS is well documented to activate NF-κB pathway and stabilize HIF-1α (42-44), the two transcriptional factors implicated in M1 activation. Treatment of M1 Mac with mitochondrial specific superoxide scavenger MitoQ significantly decreases expression of M1 genes. More importantly, glycolytic inhibitor 2-deoxyglucose, Notch1 KO, or Pdp1 silencing reduces mtROS and M1 gene expression in M1 Mac. Myeloid Notch1 KO also reduces hepatic ROS by ∼40% in the ASH mice (3). These results establish a metabolic axis of glucose-pyruvate-OXPHOS and subsequent generation of mtROS, which links Notch dependent glucose metabolism to M1 Mac activation in ASH (Figure 2).
Concluding remarks
Metabolic reprogramming is increasingly recognized to dictate cell fate determination (2). This concept has also been explored in M1 vs. M2 Mac activation, revealing contrasted metabolic shifts of increased glycolysis in M1 Macs vs. increased fatty acid oxidation in M2 Macs (45,46). Our studies provide evidence suggesting Notch pathway regulates M1 activation through two mechanisms: 1) Notch1 directly activates transcription of M1 genes; and 2) Notch1 reprograms Mac metabolism to mitochondrial glucose oxidation, resulting in increased mtROS, which augments M1 gene expression (Figure 2). The Notch-dependent upregulation of mtDNA expression may also sustain the M1 Mac activity through improving mitochondrial function in the setting of chronic ASH. Lastly, recent studies have demonstrated glutamine metabolism through TCA cycle is important for M1 Mac (47). The effect of glutamine metabolism in M1 activation, however, is attenuated by glycolytic inhibitor 2-deoxyglucose (48), indicating interaction of glycolytic and glutamine fluxes in TCA cycle metabolism. Thus, further studies focusing on the regulation of different metabolic fluxes that fuel TCA cycle and their impact on Mac cell fate will provide new insight into the mechanisms of M1 activation in ASH.
Acknowledgments
This review article is based on the work in part supported by NIH grants P50AA011999 (H.T.), R24AA12885 (H.T.), K01AA020524 (J.X.) and DK048522-19 (pilot project to J.X.), and by the Medical Research Service of Department of Veterans Affairs grant 5IO BX001991-02F (H.T.) and ABMRF/The Foundation for Alcohol Research (J.X.),
Footnotes
Note de titre : This article is part of the special issue “Alcohol, Virus and Steatosis evolving to cancer” featuring the conference papers of the 10th International Symposium organized by the Brazilian Society of Hepatology in São Paulo, Brazil, September 30th-October 1st, 2015.
The authors have declared that no conflict of interest exists.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wheeler MD. Endotoxin and Kupffer cell activation in alcoholic liver disease. Alcohol Res Health. 2003;27(4):300–306. [PMC free article] [PubMed] [Google Scholar]
- 2.Agathocleous M, Harris WA. Metabolism in physiological cell proliferation and differentiation. Trends in cell biology. 2013 doi: 10.1016/j.tcb.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 3.Xu J, Chi F, Guo T, Punj V, Lee WNP, French SW, et al. Notch reprograms mitochondrial metabolism for proinflammatory macrophage activation. J Clin Invest. 2015 doi: 10.1172/JCI76468.. Published online ahead of print. http://www.jci.org/articles/view/76468. [DOI] [PMC free article] [PubMed]
- 4.Tsukamoto H, Machida K, Dynnyk A, Mkrtchyan H. “Second hit” models of alcoholic liver disease. Semin Liver Dis. 2009;29(2):178–187. doi: 10.1055/s-0029-1214373. PMC4197797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Naveau S, Giraud V, Borotto E, Aubert A, Capron F, Chaput JC. Excess weight risk factor for alcoholic liver disease. Hepatology. 1997;25(1):108–111. doi: 10.1002/hep.510250120. [DOI] [PubMed] [Google Scholar]
- 6.Ruhl CE, Everhart JE. Joint effects of body weight and alcohol on elevated serum alanine aminotransferase in the United States population. Clin Gastroenterol Hepatol. 2005;3(12):1260–1268. doi: 10.1016/s1542-3565(05)00743-3. [DOI] [PubMed] [Google Scholar]
- 7.Alatalo PI, Koivisto HM, Hietala JP, Puukka KS, Bloigu R, Niemela OJ. Effect of moderate alcohol consumption on liver enzymes increases with increasing body mass index. Am J Clin Nutr. 2008;88(4):1097–1103. doi: 10.1093/ajcn/88.4.1097. [DOI] [PubMed] [Google Scholar]
- 8.Carmiel-Haggai M, Cederbaum AI, Nieto N. Binge ethanol exposure increases liver injury in obese rats. Gastroenterology. 2003;125(6):1818–1833. doi: 10.1053/j.gastro.2003.09.019. [DOI] [PubMed] [Google Scholar]
- 9.Xu J, Lai KK, Verlinsky A, Lugea A, French SW, Cooper MP, et al. Synergistic steatohepatitis by moderate obesity and alcohol in mice despite increased adiponectin and p-AMPK. J Hepatol. 2011;55(3):673–682. doi: 10.1016/j.jhep.2010.12.034. PMC3094601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284(5415):770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 11.Mumm JS, Kopan R. Notch signaling: from the outside. Dev Biol. 2000;228(2):151–165. doi: 10.1006/dbio.2000.9960. [DOI] [PubMed] [Google Scholar]
- 12.Geisler F, Strazzabosco M. Emerging roles of Notch signaling in liver disease. Hepatology. 2015;61(1):382–392. doi: 10.1002/hep.27268. PMC4268103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang YC, He F, Feng F, Liu XW, Dong GY, Qin HY, et al. Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses. Cancer Res. 2010;70(12):4840–4849. doi: 10.1158/0008-5472.CAN-10-0269. [DOI] [PubMed] [Google Scholar]
- 14.Monsalve E, Ruiz-Garcia A, Baladron V, Ruiz-Hidalgo MJ, Sanchez-Solana B, Rivero S, et al. Notch1 upregulates LPS-induced macrophage activation by increasing NF-kappaB activity. Eur J Immunol. 2009;39(9):2556–2570. doi: 10.1002/eji.200838722. [DOI] [PubMed] [Google Scholar]
- 15.Palaga T, Buranaruk C, Rengpipat S, Fauq AH, Golde TE, Kaufmann SH, et al. Notch signaling is activated by TLR stimulation and regulates macrophage functions. Eur J Immunol. 2008;38(1):174–183. doi: 10.1002/eji.200636999. [DOI] [PubMed] [Google Scholar]
- 16.Zhao B, Zou J, Wang H, Johannsen E, Peng CW, Quackenbush J, et al. Epstein-Barr virus exploits intrinsic B-lymphocyte transcription programs to achieve immortal cell growth. Proc Natl Acad Sci U S A. 2011;108(36):14902–14907. doi: 10.1073/pnas.1108892108. PMC3169132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang H, Zou J, Zhao B, Johannsen E, Ashworth T, Wong H, et al. Genome-wide analysis reveals conserved and divergent features of Notch1/RBPJ binding in human and murine T-lymphoblastic leukemia cells. Proc Natl Acad Sci U S A. 2011;108(36):14908–14913. doi: 10.1073/pnas.1109023108. PMC3169118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu H, Zhu J, Smith S, Foldi J, Zhao B, Chung AY, et al. Notch-RBP-J signaling regulates the transcription factor IRF8 to promote inflammatory macrophage polarization. Nat Immunol. 2012;13(7):642–650. doi: 10.1038/ni.2304. PMC3513378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zigmond E, Samia-Grinberg S, Pasmanik-Chor M, Brazowski E, Shibolet O, Halpern Z, et al. Infiltrating monocyte-derived macrophages and resident kupffer cells display different ontogeny and functions in acute liver injury. J Immunol. 2014;193(1):344–353. doi: 10.4049/jimmunol.1400574. [DOI] [PubMed] [Google Scholar]
- 20.Wang M, You Q, Lor K, Chen F, Gao B, Ju C. Chronic alcohol ingestion modulates hepatic macrophage populations and functions in mice. J Leukoc Biol. 2014;96(4):657–665. doi: 10.1189/jlb.6A0114-004RR. PMC4163632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morinaga H, Mayoral R, Heinrichsdorff J, Osborn O, Franck N, Hah N, et al. Characterization of Distinct Subpopulations of Hepatic Macrophages in HFD/Obese Mice. Diabetes. 2015;64(4):1120–1130. doi: 10.2337/db14-1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Outtz HH, Wu JK, Wang X, Kitajewski J. Notch1 deficiency results in decreased inflammation during wound healing and regulates vascular endothelial growth factor receptor-1 and inflammatory cytokine expression in macrophages. J Immunol. 2010;185(7):4363–4373. doi: 10.4049/jimmunol.1000720. PMC3887523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Voet D, Voet JG. Biochemistry. Second. John Wiley & Sons, Inc; 1995. [Google Scholar]
- 24.Tavakoli S, Zamora D, Ullevig S, Asmis R. Bioenergetic profiles diverge during macrophage polarization: implications for the interpretation of 18F-FDG PET imaging of atherosclerosis. J Nucl Med. 2013;54(9):1661–1667. doi: 10.2967/jnumed.112.119099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115(23):4742–4749. doi: 10.1182/blood-2009-10-249540. PMC2890190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leeper-Woodford SK, Mills JW. Phagocytosis and ATP levels in alveolar macrophages during acute hypoxia. Am J Respir Cell Mol Biol. 1992;6(3):326–334. doi: 10.1165/ajrcmb/6.3.326. [DOI] [PubMed] [Google Scholar]
- 27.Karnovsky ML, Lazdins J, Simmons SR. Metabolism of activated mononuclear phagocytes at rest and during pahgocytosis. In: R VF, editor. Mononuclear Phagocytes in Immunity, Infection, and Pathology. Blackwell Scientific Publications; Oxford, UK: [Google Scholar]
- 28.Oren R, Farnham AE, Saito K, Milofsky E, Karnovsky ML. Metabolic patterns in three types of phagocytizing cells. J Cell Biol. 1963;17:487–501. doi: 10.1083/jcb.17.3.487. PMC2106210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Landor SK, Mutvei AP, Mamaeva V, Jin S, Busk M, Borra R, et al. Hypo-and hyperactivated Notch signaling induce a glycolytic switch through distinct mechanisms. Proc Natl Acad Sci U S A. 2011;108(46):18814–18819. doi: 10.1073/pnas.1104943108. PMC3219154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nishi K, Oda T, Takabuchi S, Oda S, Fukuda K, Adachi T, et al. LPS induces hypoxia-inducible factor 1 activation in macrophage-differentiated cells in a reactive oxygen species-dependent manner. Antioxid Redox Signal. 2008;10(5):983–995. doi: 10.1089/ars.2007.1825. [DOI] [PubMed] [Google Scholar]
- 31.Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE. 2005;2005(306):re12. doi: 10.1126/stke.3062005re12. [DOI] [PubMed] [Google Scholar]
- 32.West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472(7344):476–480. doi: 10.1038/nature09973. PMC3460538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harris RA, Bowker-Kinley MM, Huang B, Wu P. Regulation of the activity of the pyruvate dehydrogenase complex. Adv Enzyme Regul. 2002;42:249–259. doi: 10.1016/s0065-2571(01)00061-9. [DOI] [PubMed] [Google Scholar]
- 34.Patel MS, Korotchkina LG. Regulation of the pyruvate dehydrogenase complex. Biochem Soc Trans. 2006;34(Pt 2):217–222. doi: 10.1042/BST20060217. [DOI] [PubMed] [Google Scholar]
- 35.Perumalsamy LR, Nagala M, Sarin A. Notch-activated signaling cascade interacts with mitochondrial remodeling proteins to regulate cell survival. Proc Natl Acad Sci U S A. 2010;107(15):6882–6887. doi: 10.1073/pnas.0910060107. PMC2872423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De la Pena P, Bornstein B, Del Hoyo P, Fernández–Moreno MA, Martin MA, Campos Y, et al. Mitochondrial dysfunction associated with a mutation in the Notch3 gene in a CADASIL family. Neurology. 2001;57(7):1235–1238. doi: 10.1212/wnl.57.7.1235. [DOI] [PubMed] [Google Scholar]
- 37.Dotti MT, De Stefano N, Bianchi S, Malandrini A, Battisti C, Cardaioli E, et al. A novel NOTCH3 frameshift deletion and mitochondrial abnormalities in a patient with CADASIL. Archives of neurology. 2004;61(6):942. doi: 10.1001/archneur.61.6.942. [DOI] [PubMed] [Google Scholar]
- 38.Thorig GE, Heinstra PW, Scharloo W. The action of the notch locus in Drosophila melanogaster. I. Effects of the notch8 deficiency on mitochondrial enzymes. Mol Gen Genet. 1981;182(1):31–38. doi: 10.1007/BF00422763. [DOI] [PubMed] [Google Scholar]
- 39.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 40.Rousset S, Emre Y, Join-Lambert O, Hurtaud C, Ricquier D, Cassard-Doulcier AM. The uncoupling protein 2 modulates the cytokine balance in innate immunity. Cytokine. 2006;35(3-4):135–142. doi: 10.1016/j.cyto.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 41.Sonoda J, Laganière J, Mehl IR, Barish GD, Chong LW, Li X, et al. Nuclear receptor ERRa and coactivator PGC-1(3 are effectors of IFN-v-induced host defense. Genes & development. 2007;21(15):1909–1920. doi: 10.1101/gad.1553007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011;21(1):103–115. doi: 10.1038/cr.2010.178. PMC3193400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brunelle JK, Bell EL, Quesada NM, Vercauteren K, Tiranti V, Zeviani M, et al. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005;1(6):409–414. doi: 10.1016/j.cmet.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 44.Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1(6):401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 45.Lacy-Hulbert A, Moore KJ. Designer macrophages: oxidative metabolism fuels inflammation repair. Cell metabolism. 2006;4(1):7–8. doi: 10.1016/j.cmet.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 46.O'Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature. 2013;493(7432):346–355. doi: 10.1038/nature11862. [DOI] [PubMed] [Google Scholar]
- 47.Newsholme P. Why is L-glutamine metabolism important to cells of the immune system in health, postinjury, surgery or infection? J Nutr. 2001;131(9 Suppl):2515S–22S. doi: 10.1093/jn/131.9.2515S. discussion 2523S-4S. [DOI] [PubMed] [Google Scholar]
- 48.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. 2013;496(7444):238–242. doi: 10.1038/nature11986. PMC4031686. [DOI] [PMC free article] [PubMed] [Google Scholar]