

Abstract
AIM: To construct a vacA-knockout Helicobacter pylori mutant strain, whose only difference from the wild strain is its disrupted vacA gene.
METHODS AND RESULTS: A clone containing kanamycin resistance gene used for homologous recombination was constructed in a directional cloning procedure into pBluescript II SK, and then transformed into vacA+ H pylori by electroporation. Colonies growing on the selective media containing kanamycin were harvested for chromosomal DNA extraction, and the allelic exchange was determined by polymerase chain reactions and sequencing. Loss of vacuolating activity of the vacA-knockout strain was confirmed by examining the gastric cells co-cultured with cell-free supernatants from H pylori wild strain or the mutant.
CONCLUSION: We constructed a vacA-knockout strain of H pylori through direct mutagenesis, which creates an important precondition for the future research on virulence comparison with gene expression analysis.
INTRODUCTION
Helicobacter pylori (H pylori) is a Gram-negative bacterium that colonizes the gastric mucosa of humans[1], and plays an important role in pathogenesis of chronic gastritis, peptic ulcers, gastric adenocarcinomas, and gastric mucosa-associated lymphoid tissue lymphomas[2-6]. Leunk et al[7] first reported in 1988 that cell-free supernatants from H pylori broth cultures induced striking vacuolar degeneration when added to cultured eukaryotic cells. Subsequently in 1992, this effect was disclosed to be caused by a secreted toxin VacA[8].
The gene encoding the vacuolating cytotoxin has been cloned from an H pylori isolate, and termed vacA[9]. Analysis of the nucleotide sequence of the vacA open reading frame (ORF) suggested that vacA encoded a 139-kDa protoxin that has three functional domains: a 33-amino-acid N-terminal signal sequence, a mature cytotoxin domain (approximately 87 kDa), and a cleaved C-terminal domain (approximately 50 kDa)[10,11]. VacA could induce vacuole formation from the cell cytosol, as determined by transfection of epithelial cells with a plasmid encoding the complete 95-kDa domain of VacA[12]. These vacuoles are acidic, and their membrane contains the vacuolar ATPase proton pump and the small GTP-binding protein rab7. Therefore, they have been suggested to arise from late compartments of the endocytic pathway[13].
Over the past decade, there has been considerable effort directing toward understanding the molecular mechanisms underlying VacA action. But till now, little is known about the mechanisms of vacuole formation and other effects of VacA. In this study, using the technique of direct mutagenesis to disrupt vacA gene, we constructed a vacA-knockout H pylori mutant strain for the further research on virulence comparison between the H pylori wild strain and the mutant.
MATERIALS AND METHODS
Bacterial strain and growth conditions
H pylori NCTC 11638 as a gift from Dr. Tong Shi (Shanghai Institute of Digestive Diseases) was cultured routinely on brain heart infusion (BHI) agar plates with 5% sheep blood in an environment containing 6% CO2 at 37 °C. For the preparation of cell-free supernatants from H pylori broth cultures, H pylori was cultured in BHI broth +10% fetal bovine serum (FBS) in an environment containing 6% CO2 at 37 °C with agitation (200 rpm) for 48 h. The cultures were centrifuged (15000 g, 30 min, 4 °C) and filtrated with a 0.2 μm syringe filter.
Disruption of vacA gene
The strategy for disruption of vacA gene by direct mutagenesis is shown in Figure 1, and genetic techniques involved were described as follows.
Figure 1.
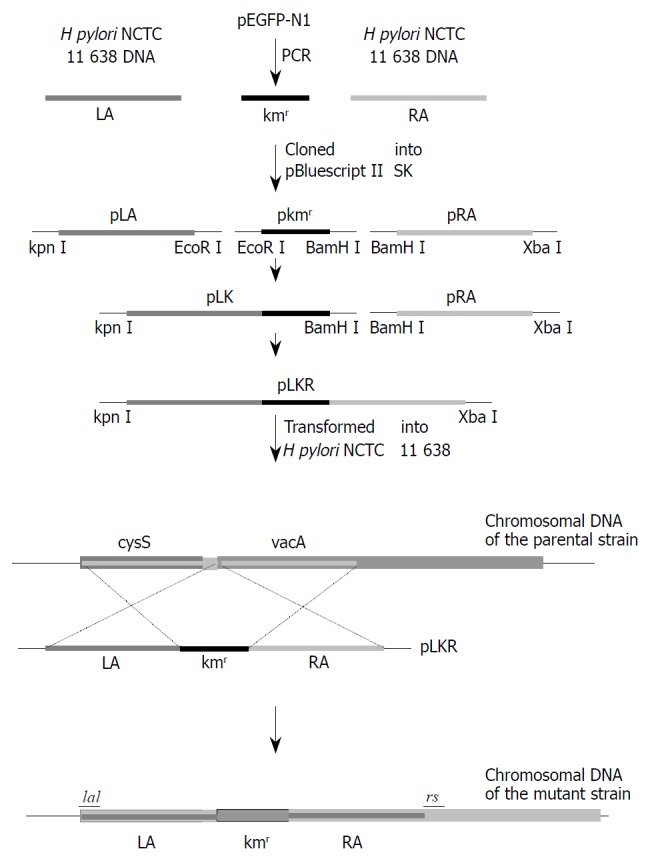
The strategy for disruption of vacA gene by directed mutagenesis. LA and RA were PCR-amplified from H pylori NCTC 11638 genomic DNA, and the kanamycin resistance gene, from pEGFP-N1. PCR products with different restriction sites on both sides were digested with corresponding endonucleases, and then cloned into pBluescript II SK digested with the same enzymes. Because there is an EcoR II site in RA, pLA and pkmr were firstly joined together, resulting in pLK, which then was joined with pRA, resulting in pLKR. The plasmid pLKR was transformed into H. pyloi NCTC 11638, where the Kmr marked mutation was introduced into the genome by homologous recombination, resulting in the vacA-Kmr mutant strain.
DNA isolation To isolate chromosomal DNA, H pylori cells were lysed in lysis buffer (10 mM Tris·HCl, pH8.0, 0.1 M EDTA, pH8.0, 0.5% (w/v) SDS, 20 μg/mL DNase free pancreatic RNase), and then, protease K (Sangon, Shanghai, China) was added in to a final concentration of 100 μg/mL. The lysate was incubated in a water bath at 50 °C for 3 h. Then, the solution was cooled to room temperature, and mixed with an equal volume of phenol equilibrated with 0.1 M Tris·HCl (pH8.0). The two phases were separated by centrifugation at 5000 g for 15 min at room temperature, and the aqueous phase was extracted with phenol twice again. Afterwards, 0.2 volume of 10 M ammonium acetate and 2 volume of ethanol were added to the aqueous phase. The precipitate was collected by centrifugation at 5000 g for 2 min, washed twice with 70% ethanol, and dissolved in an appropriate volume of TE buffer (pH8.0)[14].
Polymerase chain reactions (PCR) PCR was carried out in 100 μL volume containing 100 ng of genomic DNA, 1 U of Ex Taq (Takara), 50 pmol of each primer, and 10 nmol of each deoxynucleoside triphosphate in a standard buffer. Oligonucleotide primers (5’-CGTGGAAATCTTATTACTCTTAGC-3’ and 5’-TGATGCTGACTAATGCTCCT-3’) were used to amplify a 1.7 kb product from H pylori NCTC 11638. Primers for amplifying kanamycin resistance gene (kanR) and two fragments flanking kanR, LA and RA, were designed as shown in Table 1.
Table 1.
Primers for amplifying kmr, LA and RA
| Primer | Sequence (5’ to 3’) | Site | Coordinates |
| la1 | GGGGTACCCTTTTGAGCCTTTAGTT | Kpn I | cysS bp 36-54 |
| la2 | CGGAATTCTCCTTTCTTTTTGTAAAAC | EcoR I | HPU07145 bp 382-400 |
| kmr1 | CGGAATTCATGATTGAACAAGATGGATTG | EcoR I | pEGFP-N1 bp 2629-2649 |
| kmr2 | CGGGATCCTCAGAAGAACTCGTCAAG | BamH I | pEGFP-N1 bp 3406-3423 |
| ra1 | CGGGATCCATCGCCCTCTGGTTTCTC | BamH I | HPU07145 bp 437-454 |
| ra2 | GCTCTAGACACCCACTTGATTATTCACTCT | Xba I | HPU07145 bp 1786-1807 |
Gel purification and enzyme digestion PCR products were electrophoresed and excised from a 1% agarose gel, purified using a Qiaquick gel extraction kit (Qiagen, Hilden, Germany), and digested with corresponding restriction enzymes (Promega, Madison, USA) depending on different restriction sites.
Cloning of different DNA fragments Purified PCR products for sequencing were cloned into pGEM-T vector (Promega). Fragments kanR, LA, and RA with different restriction sites on both sides were digested with corresponding endonucleases (Promega), and then cloned into pBluescript SK II digested with the same enzymes.
Sequencing Every clone was sequenced with the ABI DNA sequencer (Bioasia Biotechnology Company, Shanghai, China).
H pylori DNA transformation by electroporation H pylori NCTC 11638 cells were transformed with plasmid pLKR by electroporation, and kanamycin-resistant (Kmr) transformants were selected by a method similar to that described by Clayton et al[15]. Briefly, H pylori cultured on plates were scraped and suspended in 30 mL cold double-distilled water. Cells were harvested by centrifugation at 4360 g at 4 °C for 5 min, and the pellet was suspended in 20 mL of cold 10% glycerol. The cells were centrifuged once, and resuspended in 2 mL ice-cold 10% glycerol. Plasmid DNA (1 μg in 5 μL TE buffer) was mixed with 0.2 mL cell suspension. The mixture was added to a prechilled (-20 °C) 0.2 cm electroporation cuvet (Bio-Rad, Hercules, USA), and subjected to single-pulse electroporation of initial voltage 2.5 kV, 25 μF and 600Ω in parallel. The sample was transferred onto a cold plate and incubated for 12 h at 37 °C. Then the cells were inoculated onto selective media with 30 μg/mL kanamycin, followed by incubation for 4 d to allow the growth of transformants.
Cell culture and detection of vacuole formation
Cells of gastric cancer cell line SGC7901 as a gift from Jie Yang (Department of Cell Biology, Shanghai Second Medical Univ ersity) were grown in DMEM (GIBCO-BRL, Gaithersburg, USA) supplemented with 10% FBS, 2 mmol/L glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin in a humidified 5% CO2 atmosphere. SGC7901 cells were co-cultured with cell-free supernatants from H pylori NCTC11638 wild strain or mutant strain for 12 h, and then observed by contrast microscopy.
RESULTS
Upstream sequence close to vacA gene
Genome of NCTC 11638 was not completely sequenced, and the upstream portion close to vacA gene going to be used in the mutagenesis technique was not published in GeneBank. Therefore, the upstream sequences close to vacA gene of 26695 and J99, whose genomes were completely sequenced and published, were aligned and searched for conservative sequences. Then a 1.7-kb product was PCR-amplified from H pylori NCTC11638 DNA and sequenced. The sequencing result showed the complete cysS (cysteinyl-tRNA synthetase) gene of NCTC 11638 (Figure 2).
Figure 2.

Nucleotide sequence of cysS gene and the downstream sequence amplified from the vacA-Kmr mutant H pylori. The 1398 bp cysS ORF and the 795 bp kmr ORF are shown. Primers la1, la2, ra1, ra2, and rs for amplifying LA, RA, and ASm are indicated. -35 signal, -10 signal, and rbs of vacA gene serving km r gene in the mutant strain are also shown.
Cloning of pLKR for transforming H pylori
As shown in Figure 1, LA which contains the H pylori vacA promoter and RA were amplified from genomic DNA of NCTC11638, while kanR gene which has no promoter was amplified from the plasmid pEGFP-N1 (Clontech, Palo Alto, USA). PCR-products LA, kanR and RA, with restriction sites incorporated at the termini, were joined together in a directional cloning procedure into pBluescript II SK, resulting in pLKR.
Construction of vacA-knockout H pylori mutant strain
pLKR which is unable to replicate in H pylori, was introduced into H pylori NCTC11638 by electroporation. After 4 d of growth, five Kmr single colonies were isolated. To determine whether vacA had been disrupted in the transformed strains through allelic exchange, DNA isolated from H pylori NCTC11638 wild-type strain and the Kmr mutant strain were PCR-amplified with the primers la1 and rs (5’-GCGCTTGATCCTGCCAAAGCATAGC-3’) annealing to H pylori NCTC 11638 vacA at bp 1808 to 1832 flanking ra2 (Figure 1 and Figure 2). A 3.8-kb product consistent with the expected size was PCR-amplified from Kmr mutant strain, as compared with a 3.0-kb product amplified from wild strain (Figure 3), suggesting a substitution of Kmr gene for a short fragment of vacA gene by homologous recombination between plasmid and chromosomal sequences. The sequencing result of the 3.8-kb product confirmed the occurrence of allelic exchange (Figure 2).
Figure 3.
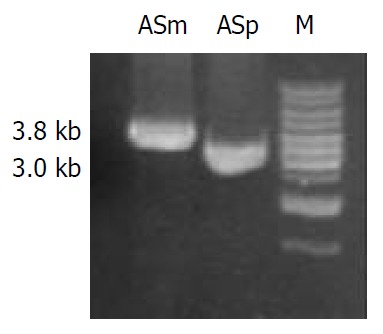
PCR amplification for the determination of homolo-gous recombination in Kmr mutant strain. Genomic DNA of NCTC 11638 wild strain and Kmr mutant strain were respec-tively PCR-amplified for the fragments ASm and ASp using the primers ra1 and rs. A single 3.8 kb product ASm was am-plified from Kmr mutant strain as compared with the 3.0 kb product ASp amplified from the wild strain.
Characterization of vacA-knockout H pylori mutant
To determine the loss of vacuolating activity of the mutant strain, gastric cells SGC7901 were co-cultured with cell-free supernatants from H pylori NCTC11638 wild strain or mutant strain for 12 h, and then observed by contrast microscopy. Intracellular vacuoles developed in cells co-cultured with supernatant from the wild strain, while no vacuoles developed in cells co-cultured with supernatant from vacA-Kmr mutant strain (Figure 4).
Figure 4.
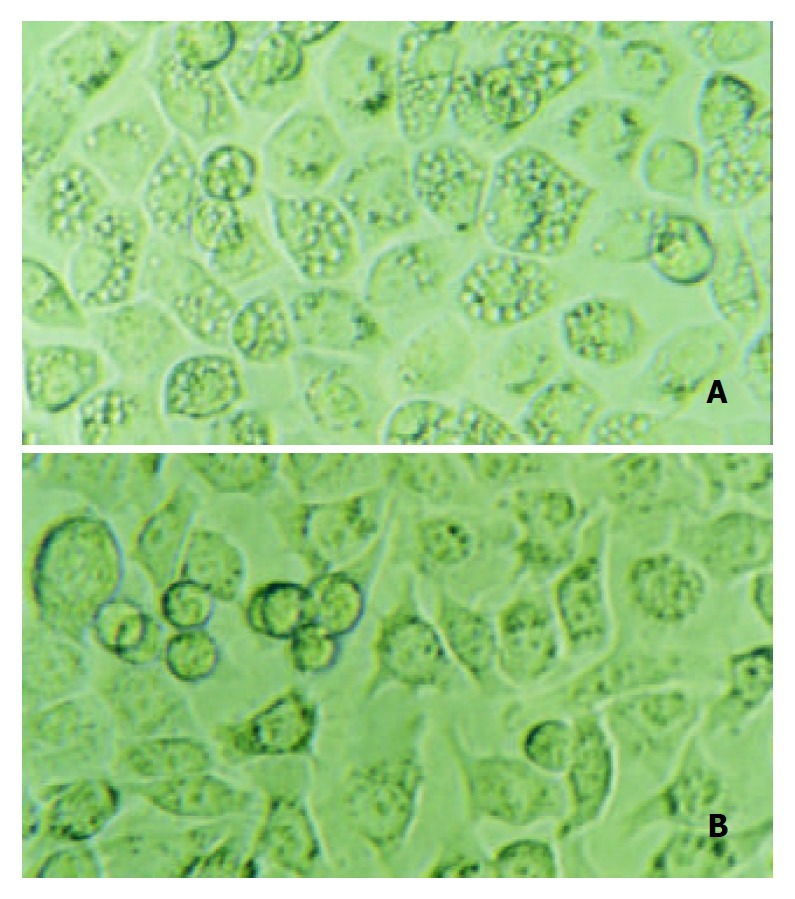
Gastric cancer cells SGC-7901 were co-cultured with the supernatant either from Helicobacter pylori NCTC 11638 or its vacA-knockout mutant strain. 12 h after the incubation, A: cells co-cultured with NCTC 11638 developed vacuoles in the cytosol; B: cells co-cultured with vacA- mutant strain developed no vacuoles at all.
DISCUSSION
VacA, produced by pathogenic strains of H pylori, was a major virulence factor in pathogenesis of gastroduodenal ulcers[8,16]. VacA induced the formation of membrane-delimited vacuoles in intoxicated cells[7], and showed many other effects on cellular functions and viability, such as causing mitochondrial depolarization[17], inducing apoptosis in gastric cells[18], affecting or interacting with various components of cytoskeleton to cause actin rearrangements[19], and even disorganizing microtubular network[20]. To study VacA machanism of action, we have tried to get purified VacA used as single virulence determinant, to study its effect on the expression profile of eukaryocyte. However, like Manetti et al[21], we did not successfully get the expressed VacA as a functional recombinant protein in E. coli, probably due to its incorrect fold. We have also considered letting VacA directly expressed in the cytosol to induce vacuole formation. In our experiments, vacuoles were induced in only 10% of cells transfected with plasmids expressing VacA, because the efficiency of the transfection method was relatively low. In addition, when VacA acts outside the cells, the pathway by which it interacts with the cells is quite different from that when the protein is produced in the cytosol. Under natural conditions, association of VacA with the eukaryotic cell surface was the first step in the intoxication of cells[22]. The initial interaction of VacA with target cells was through high-affinity cell surface receptors, and this interaction was necessary for its biologic activity[23,24]. A 250 kDa receptor protein tyrosine phosphatase (RPTP) β served as a receptor for VacA on AZ521 cells, and another protein, p140, was also commonly detected in VacA-susceptible cells[25,26]. Increased binding of acid- or alkali-activated VacA to RPTPβ may alter its activity and possibly accelerates or inhibits dephosphorylation of tyrosine on cytosolic proteins. Moreover, VacA acting outside the cells is a kind of exogenous antigen, having different pathways of processing and presentation from that of VacA expressed in the cytosol as an endogenous antigen. All of these processes may affect gene expression of the host cells.
Direct mutagenesis was probably the most useful technique for assessing the contribution to virulence of specific bacterial gene products[27]. In our study, vacA gene encoding vacuolating cytotoxin that has been identified by conventional biochemical means was disrupted by gene replacement. This technique requires a means for introducing DNA into the pathogen, as well as suitable selective markers and an inherent capacity for homologous recombination. In previous studies, the Kmr determinant often came from Campylobacter coli[28]. Here we introduced a simple method to get the Kmr gene from commercialized plasmids such as pEGFP-N1. Coding sequence of the gene without a promotor was PCR-amplified from pEGFP-N1 and ligated downstream with the promotor of H pylori vacA gene. Upon insertion into chromosomal DNA of H pylori through homologous recombination, this gene could be efficiently transcribed because the vacA promoter was recognized by H pylori transcriptional machinery, introducing kanamycin resistance characteristics into H pylori. Due to the stop coden of Kmr, vacA would not be translated at all although most of the sequences still existed. The results of PCR and sequencing confirmed the occurrence of allelic exchange. Therefore, using the direct mutagenesis technique, we obtained the isogenic mutant strain of H pylori, which differed from the wild strain only in that the vacA gene was knocked out. Through co-culture of cell-free supernatants from the wild or mutant H pylori strain with gastric cells, loss of vacuolating activity of the vacA-knockout strain was confirmed. These results clearly show that VacA is an indispensable toxin secreted by H pylori for the induction of vacuole formation.
Such kind of technique has been used to yield vacA- mutant H pylori[9,28,29]. But no further experiment has been done to compare the virulence between the mutant and the parental strain. On the other hand, microarray analysis has been used in several studies to screen gene expression profiles in gastric epithelial cells induced by H pylori[30-32]. Our group has also analyzed different expression profiles of gastric cancer cells co-cultured with supernatants of VacA+ or VacA- H pylori isolates. However, VacA has not been used as a single virulence determinant to stimulate host cells, thus one can not determine which virulent factors result in the alteration of the expression. In this study, we successfully constructed the vacA- mutant strain, using the direct mutagenesis technique, which creates an important precondition for the further research on virulence comparison with gene expression analysis.
Footnotes
Supported by Ministry of Education Research Foundation for Returned Overseas Chinese Scholars Abroad (2001) 498
Edited by Xia HHX
References
- 1.Dunn BE, Cohen H, Blaser MJ. Helicobacter pylori. Clin Microbiol Rev. 1997;10:720–741. doi: 10.1128/cmr.10.4.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984;1:1311–1315. doi: 10.1016/s0140-6736(84)91816-6. [DOI] [PubMed] [Google Scholar]
- 3.Graham DY, Lew GM, Klein PD, Evans DG, Evans DJ, Saeed ZA, Malaty HM. Effect of treatment of Helicobacter pylori infection on the long-term recurrence of gastric or duodenal ulcer. A randomized, controlled study. Ann Intern Med. 1992;116:705–708. doi: 10.7326/0003-4819-116-9-705. [DOI] [PubMed] [Google Scholar]
- 4.Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, Sibley RK. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med. 1991;325:1127–1131. doi: 10.1056/NEJM199110173251603. [DOI] [PubMed] [Google Scholar]
- 5.Forman D, Newell DG, Fullerton F, Yarnell JW, Stacey AR, Wald N, Sitas F. Association between infection with Helicobacter pylori and risk of gastric cancer: evidence from a prospective investigation. BMJ. 1991;302:1302–1305. doi: 10.1136/bmj.302.6788.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xue FB, Xu YY, Wan Y, Pan BR, Ren J, Fan DM. Association of H. pylori infection with gastric carcinoma: a Meta analysis. World J Gastroenterol. 2001;7:801–804. doi: 10.3748/wjg.v7.i6.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leunk RD, Johnson PT, David BC, Kraft WG, Morgan DR. Cytotoxic activity in broth-culture filtrates of Campylobacter pylori. J Med Microbiol. 1988;26:93–99. doi: 10.1099/00222615-26-2-93. [DOI] [PubMed] [Google Scholar]
- 8.Cover TL, Blaser MJ. Purification and characterization of the vacuolating toxin from Helicobacter pylori. J Biol Chem. 1992;267:10570–10575. [PubMed] [Google Scholar]
- 9.Cover TL, Tummuru MK, Cao P, Thompson SA, Blaser MJ. Divergence of genetic sequences for the vacuolating cytotoxin among Helicobacter pylori strains. J Biol Chem. 1994;269:10566–10573. [PubMed] [Google Scholar]
- 10.Schmitt W, Haas R. Genetic analysis of the Helicobacter pylori vacuolating cytotoxin: structural similarities with the IgA protease type of exported protein. Mol Microbiol. 1994;12:307–319. doi: 10.1111/j.1365-2958.1994.tb01019.x. [DOI] [PubMed] [Google Scholar]
- 11.Hou P, Tu ZX, Xu GM, Gong YF, Ji XH, Li ZS. Helicobacter pylori vacA genotypes and cagA status and their relationship to associated diseases. World J Gastroenterol. 2000;6:605–607. doi: 10.3748/wjg.v6.i4.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Bernard M, Arico B, Papini E, Rizzuto R, Grandi G, Rappuoli R, Montecucco C. Helicobacter pylori toxin VacA induces vacuole formation by acting in the cell cytosol. Mol Microbiol. 1997;26:665–674. doi: 10.1046/j.1365-2958.1997.5881952.x. [DOI] [PubMed] [Google Scholar]
- 13.Papini E, de Bernard M, Milia E, Bugnoli M, Zerial M, Rappuoli R, Montecucco C. Cellular vacuoles induced by Helicobacter pylori originate from late endosomal compartments. Proc Natl Acad Sci USA. 1994;91:9720–9724. doi: 10.1073/pnas.91.21.9720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sambrook J, Russell DW. Molecular Cloning-A Laboratory Manual. Vol 1. 3thed, Cold Spring Harbor, New York: Cold Spring Har-bor Laboratory Press; 2001. pp. 6.4–6.11. [Google Scholar]
- 15.Clayton CL, Mobley HLT. Methods in Molecular Medicine, Helicobacter pylori Protocols. 1sted. Totowa: Humana Press Inc; 1997. pp. 145–152. [Google Scholar]
- 16.Cover TL. The vacuolating cytotoxin of Helicobacter pylori. Mol Microbiol. 1996;20:241–246. doi: 10.1111/j.1365-2958.1996.tb02612.x. [DOI] [PubMed] [Google Scholar]
- 17.Kimura M, Goto S, Wada A, Yahiro K, Niidome T, Hatakeyama T, Aoyagi H, Hirayama T, Kondo T. Vacuolating cytotoxin purified from Helicobacter pylori causes mitochondrial damage in human gastric cells. Microb Pathog. 1999;26:45–52. doi: 10.1006/mpat.1998.0241. [DOI] [PubMed] [Google Scholar]
- 18.Kuck D, Kolmerer B, Iking-Konert C, Krammer PH, Stremmel W, Rudi J. Vacuolating cytotoxin of Helicobacter pylori induces apoptosis in the human gastric epithelial cell line AGS. Infect Immun. 2001;69:5080–5087. doi: 10.1128/IAI.69.8.5080-5087.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ashorn M, Cantet F, Mayo K, Megraud F. Cytoskeletal rearrangements induced by Helicobacter pylori strains in epithelial cell culture: possible role of the cytotoxin. Dig Dis Sci. 2000;45:1774–1780. doi: 10.1023/a:1005578110764. [DOI] [PubMed] [Google Scholar]
- 20.Pai R, Cover TL, Tarnawski AS. Helicobacter pylori vacuolating cytotoxin (VacA) disorganizes the cytoskeletal architecture of gastric epithelial cells. Biochem Biophys Res Commun. 1999;262:245–250. doi: 10.1006/bbrc.1999.1194. [DOI] [PubMed] [Google Scholar]
- 21.Manetti R, Massari P, Burroni D, de Bernard M, Marchini A, Olivieri R, Papini E, Montecucco C, Rappuoli R, Telford JL. Helicobacter pylori cytotoxin: importance of native conformation for induction of neutralizing antibodies. Infect Immun. 1995;63:4476–4480. doi: 10.1128/iai.63.11.4476-4480.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Papini E, Zoratti M, Cover TL. In search of the Helicobacter pylori VacA mechanism of action. Toxicon. 2001;39:1757–1767. doi: 10.1016/s0041-0101(01)00162-3. [DOI] [PubMed] [Google Scholar]
- 23.Garner JA, Cover TL. Binding and internalization of the Helicobacter pylori vacuolating cytotoxin by epithelial cells. Infect Immun. 1996;64:4197–4203. doi: 10.1128/iai.64.10.4197-4203.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Massari P, Manetti R, Burroni D, Nuti S, Norais N, Rappuoli R, Telford JL. Binding of the Helicobacter pylori vacuolating cytotoxin to target cells. Infect Immun. 1998;66:3981–3984. doi: 10.1128/iai.66.8.3981-3984.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yahiro K, Niidome T, Kimura M, Hatakeyama T, Aoyagi H, Kurazono H, Imagawa Ki, Wada A, Moss J, Hirayama T. Activation of Helicobacter pylori VacA toxin by alkaline or acid conditions increases its binding to a 250-kDa receptor protein-tyrosine phosphatase beta. J Biol Chem. 1999;274:36693–36699. doi: 10.1074/jbc.274.51.36693. [DOI] [PubMed] [Google Scholar]
- 26.Yahiro K, Niidome T, Hatakeyama T, Aoyagi H, Kurazono H, Padilla PI, Wada A, Hirayama T. Helicobacter pylori vacuolating cytotoxin binds to the 140-kDa protein in human gastric cancer cell lines, AZ-521 and AGS. Biochem Biophys Res Commun. 1997;238:629–632. doi: 10.1006/bbrc.1997.7345. [DOI] [PubMed] [Google Scholar]
- 27.Henderson B, Wilson M, McNab R, Lax AJ. Cellular Microbiology: Bacteria-host interactions in health and disease. Hoboken: John Wiley Sons Ltd; 1999. pp. 163–188. [Google Scholar]
- 28.Copass M, Grandi G, Rappuoli R. Introduction of unmarked mutations in the Helicobacter pylori vacA gene with a sucrose sensitivity marker. Infect Immun. 1997;65:1949–1952. doi: 10.1128/iai.65.5.1949-1952.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burroni D, Lupetti P, Pagliaccia C, Reyrat JM, Dallai R, Rappuoli R, Telford JL. Deletion of the major proteolytic site of the Helicobacter pylori cytotoxin does not influence toxin activity but favors assembly of the toxin into hexameric structures. Infect Immun. 1998;66:5547–5550. doi: 10.1128/iai.66.11.5547-5550.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sepulveda AR, Tao H, Carloni E, Sepulveda J, Graham DY, Peterson LE. Screening of gene expression profiles in gastric epithelial cells induced by Helicobacter pylori using microarray analysis. Aliment Pharmacol Ther. 2002;16 Suppl 2:145–157. doi: 10.1046/j.1365-2036.16.s2.4.x. [DOI] [PubMed] [Google Scholar]
- 31.Cox JM, Clayton CL, Tomita T, Wallace DM, Robinson PA, Crabtree JE. cDNA array analysis of cag pathogenicity island-associated Helicobacter pylori epithelial cell response genes. Infect Immun. 2001;69:6970–6980. doi: 10.1128/IAI.69.11.6970-6980.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maeda S, Otsuka M, Hirata Y, Mitsuno Y, Yoshida H, Shiratori Y, Masuho Y, Muramatsu M, Seki N, Omata M. cDNA microarray analysis of Helicobacter pylori-mediated alteration of gene expression in gastric cancer cells. Biochem Biophys Res Commun. 2001;284:443–449. doi: 10.1006/bbrc.2001.5006. [DOI] [PubMed] [Google Scholar]