

Abstract
AIM: Proinflammatory cytokines TNF-α and IL-6 play a main role in acute pancreatitis (AP). Cytokine biosynthesis runs through two major signaling pathways at the level of proteins: nuclear transcription factor-κB (NF-κB) and p38 mitogen-activated protein kinase (p38 MAPK). The aim of the study was to investigate the effect of NF-κB and p38 MAPK in activated monocytes/macrophages on cytokines of rats with acute pancreastitis.
METHODS: Taurocholate (3% and 5%) at doses of 1 mL/kg was administered into the biliopancreatic duct of male Sprague-Dawley (SD) rats to reduce acute edematous pancreariris (AEP) and acute necrotizing pancreatitis (ANP). Pancreatic tissues were prepared immediately after death. At this point, blood was obtained for determination of serum amylase and pro-inflammatory TNF-α and IL-6. Activated monocytes/macrophages were captured from blood and so were ascites. NF-κB and p38 MAPK in activated monocytes/macrophages were measured by immunohistochemistry method. Pancreatic tissue samples were prepared for routine light microscopy, using hematoxylin and eosin (HE) staining.
RESULTS: The serum levels of amylase were 3056.00 ± 1232.35 IU/L and 4865.12 ± 890.34 IU/L at 3 and 6 h in ANP group, which were significantly higher than those (3056.00 ± 1232.35 IU/L and 3187.17 ± 821.16 IU/L) (P < 0.05, respectively) in AEP group. In ascites the levels were 3.32 ± 1.01 g and 3.76 ± 1.12 g at 3 and 6 h in ANP group, which were significantly higher than those (1.43 ± 1.02 g and 2.56 ± 1.21 g) (P < 0.05, respectively) in AEP group. The serum levels of TNF-α were 54.27 ± 23.48 pg/mL and 67.83 ± 22.02 pg/mL in AEP group and 64.28 ± 20.79 pg/mL and 106.59 ± 43.71 pg/mL in ANP group, and the serum levels of IL-6 were 428.12 ± 140.30 pg/mL and 420.13 ± 139.40 pg/mL in AEP group and 1600.32 ± 309.78 pg/mL and 2203.76 ± 640.85 pg/mL in ANP group, which were far significantly higher than those in sham group (P < 0.001, respectively). The serum level of TNF-α 6 h after establishment of the studied model and that of IL-6 at 3 and 6 h in ANP group were significantly higher than those in AEP (P < 0.05, P < 0.001, P < 0.05). In ANP group, the levels of serum TNF-α and IL-6 6 h after establishment of the studied model were significantly higher than those 3 h after establishment of studied model (P < 0.05, P < 0.05, respectively). Three and 6 h after establishment of the model, typical pathological changesof AEP and ANP were found, such as large numbers of inflammatory cells, edema, hemorrhage, necrosis, large amount of ascites. In AEP, NF-κB and p38 MAPK in activated monocytes/macrophages were moderately found at 3 and 6 h after introduction of the model. However, in ANP, the expression of NF-κB and p38 MAPK in activated monocytes/macrophages was upregulated evidently at 3 and 6 h after introduction of the model, reaching their highest levels at 6 h after introduction of the model, which were consistent with the levels of TNF-α and IL-6.
CONCLUSION: Cytokine TNF-α and IL-6 play a main role in acute pancreatitis, expression of NF-κB and p38 MAPK in activated monocytes/macrophages might play a major role in cytokine transcription and biosynthesis.
INTRODUCTION
The etiology of acute pancreatitis (AP) is mostly alcoholic or gallstone. Whatever the etiologic factors are, once the disease is initiated by an early molecular mechanism (which is still controversial) in or around acinar cells, a host inflammatory response is evoked in the pancreas. This locally limited inflammation is then manifested and amplified by the actions of diverse inflammatory mediators, such as cytokines [tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1) and interleukin-6 (IL-6)], reactive oxygen species, proteolytic enzymes, lipids, as well as gaseous mediators, resulting in the induction of systemic inflammatory response syndrome (SIRS)[1-3]. Although SIRS is principally considered to be the normal host response to the insults, sustained or exaggerated SIRS, followed by the development of multiple organ dysfunction syndrome (MODS), is ultimately responsible for most pancreatitis- associated mortality and morbidity. Thus, the most distinctive feature of AP could be defined as its propensity to propagate locally limited inflammation into SIRS, and subsequently into MODS[4,5].
In AP, the very early phenomena that appear to occur are monocytes and neutrophils in the circulation migrating into the pancreatic interstitial space, mainly mediated by adhesion molecules on leukocytes[6,7]. These infiltrating cells accelerate further production and secretion of cytokines, as well as other inflammatory mediators. As a result, leukocytes in the circulation, as well as endothelial cells, in the pancreas and specific distant organs such as the lung, liver and spleen, are activated, eliciting further microcirculatory derangements, such as increased vascular permeability and accelerated leukocyte transmigration. The contribution of cytokines during this cascade of events has been increasingly apparent in the past decade, as indicated by several clinical and experimental studies exhibiting increased levels of cytokines in plasma, lymph, and ascites, as well as enhanced activitity of circulating leukocytes. Of the various kinds of inflammatory mediators, TNF-α and IL-1 are regarded as most prominent "first-line" cytokines[8-10]. IL-6 is well known to be the primary inducer of acute-phase response. Therefore, IL-6 was recently reported to be the best prognostic parameter of pulmonary failure[11]. IL-6, IL-8 and TNF-α can be synthesized by monocytes/macrophages, neutrophils, endothelial cells, and even pancreatic duct cells.
Recent experimental studies appeared to have shed some light on the intracellular signaling pathway in the inflammatory cascade in AP. For example, recent evidence strongly suggested the crucial role of NF-κB in the initiation of AP, not only in pancreatic acinar cells and monocytes/macrophages but also in specific distant organs, such as the lung. NF-κB is able to mediate a variety of inflammatory mediators involved in AP, including cytokines and adhesion molecules, as well as specific inducible isoform of nitric oxide synthase enzymes[12-21]. Additionally, recent evidence has also shown the contribution of p38 MAP kinase to the mechanism underlying cytokine expression in pancreatic acinar cells and monocytes/macrophages. An in-vitro experiment showed that cytokine expression of acinar cells was mediated by two separate signal pathways, i.e., NF-κB and p38 MAP kinase. of which p38 MAP kinase is now suggested to play a major role[22-27]. The definitive role of these intracellular signaling cascades may lead to new designs for cytokine modulation therapy.
This study was to investigate the effect of NF-κB and p38 MAPK in activated monocytes/macrophages on pro-inflammatory cytokines of mice with acute pancreatitis and to explore the biosynthesis mechanism of pro-inflammatory cytokines TNF-α and IL-6.
MATERIALS AND METHODS
Materials
Sixty pathogen-free male Sprague-Dawley (SD) rats (Animal Center, Xi’an Jiaotong University), weighing 250-300 g, were used in this experiment. The animals were kept at a constant room temperature of 25 °C with a 12-h light-dark cycle, and were allowed free access to water. The animals were housed in the animal facility for at least seven days prior to use in order to stabilize their intestinal flora. All procedures including the rats receiving a very small amount of methoxyflurane for induction and pentobarbital intra-peritoneal injection (50 mg/kg) for anesthesia were performed under sterile conditions. SD rats were divided into subgroups of pancreatitis and controls. Taurocholate (3% and 5%, Sigma) at doses of 1 mL/kg was administered into the biliopancreatic duct of the rats to establish acute edematous pancreatitis (AEP) and acute necrotizing pancreatitis (ANP). In the sham group, the mice were only operated but not infused anything. All values reported were from animals killed at defined time points. During experiments the rats were closely observed, and pancreas tissues were prepared immediately after death. At this point, blood was obtained for determination of serum amylase and TNF-α and IL-6. The activated monocytes/macrophages were captured from blood and ascites respectively.
Macroscopic assessment of the pancreas
Necrosis, hemorrhage and edema of the pancreas were each graded from 0 to 3 as follows: (a) necrosis: no necrosis = 0, isolated necrotic lesion (< 5% of the pancreas) = 1, necrotic areas 5%-25% = 2, necrotic areas > 25% and partial destruction of the organ = 3; (b) hemorrhage: not evident = 0, single focal hemorrhage = 1, multiple disseminated hemorrhagic dots = 2, massive confluent hemorrhage = 3; (c) edema: normal size = 0, enlargement of the pancreas = 1, recognizable accumulated fluid in the tissue = 2, massive swelling and fluid accumulation = 3. The scores of these parameters were summed, and the highest possible total score for an individual rat was 9[28].
Microscopic assessment of the pancreas
Pancreas tissue samples were prepared for routine light microscopy, using hematoxylin and eosin (HE) staining and examined by a pathologist who was unaware of the source of specimens. HE-stained pancreas sections were observed with a standard light microscope to evaluate morphologic alterations following AP.
Pancreatic water contents
The wet weight-dry weight ratio of the pancreas was obtained after about 200 mg of freshly prepared pancreatic tissue was dried for 20 h at 105 °C.
Amylase assay
Amylase activity in plasma was determined using the a-amylase EPS assay purchased from Roche (Sigma) at 37 °C.
Serum of TNF-α and IL-6
Blood was collected and centrifuged (3000 rpm/min, for 5 min). The serum was captured and stored at -30 °C. The pro- inflammatory TNF-α and IL-6 were measured by enzyme-linked immunosorbent assay (ELISA).
NF-κB and p38 MAPK in activated monocytes/macrophages
The activated monocytes/macrophages were captured from blood and ascites, respectively. They were separated and pasted for 12 h in 10% calf serum of 1640. NF-κB and p38 MAPK in activated monocyte/macrophage were measured by immunohistochemistry method (S-ABC). In brief, endogenous peroxidases were blocked for 15 min with methanol-H2O2, and after several washes unspecific-binding sites were blocked for 2 h with 3% normal horse serum. Samples were then incubated overnight in a humidified chamber at 4 °C with the primary antibody against NF-κB and p38 MAPK (Sigma) diluted 1:200. This was followed by biotinylated goat anti-mouse antibody diluted 1:200 for 1 h, and the avidin-biotin complex (ABC kit, Vector Laboratories) (1:100) for 1 h. The reaction was developed with 0.05% diaminobenzidine and 0.03% H2O2. Negative controls were carried out by omission of the primary antibody in the overnight incubation. Several sections were counterstained with hematoxylin.
Statistical analysis
The values were expressed as the mean ± standard error (SEM). The data were analyzed for statistical significance using Student’s t test. There were six animals in each group at each time point. As the values in some data sets were not normally distributed or exhibited unequal standard deviations, the nonparametric Wilcoxon rank sum test was used to test for statistical differences between groups. In all instances P < 0.05 was considered to be significant.
RESULTS
Amylase assay and ascites
The serum levels of AMS and ascites in AEP and ANP groups were significantly higher than those in sham group (P < 0.01). The serum levels of AMS and ascites at 3 and 6 h after the establishment of the studied model in ANP group were significantly higher than that in AEP (P < 0.05). In ANP group, the levels of serum AMS and ascites 6 h after the establishment of the studied model were not significantly higher than those 3 h after the establishment of the studied model (P > 0.05) (Table 1).
Table 1.
Levels of AMS and ascites of AEP and ANP (¯x ± s)
| Group |
AMS (IU/L) |
Ascites (g) |
||
| 3 h | 6 h | 3 h | 6 h | |
| Sham | 792.80 ± 265.08 | 793.8 0 ± 265.08 | 0 | 0 |
| AEP | 3056.00 ± 1232.35b | 3187.17 ± 821.16b | 1.43 ± 1.02b | 2.56 ± 1.21b |
| ANP | 4345.30 ± 1005.77ab | 4865.12 ± 890.34ab | 3.32 ± 1.01ab | 3.76 ± 1.12ab |
P < 0.01 vs sham,
P < 0.05 vs AEP.
Pancreatic water contents
The extent of pancreatic edema was determined as the wet weight/dry weight ratio of the tissue, and the macroscopic appearance of the pancreas was evaluated by a score (Table 2). In pancreatitis both parameters were significantly increased compared to the control (P < 0.05).
Table 2.
Macrocopic appearance and edema of pancreas in AEP and ANP (at 6 h after of model)
The macroscopic appearance of the pancreas was scored. For an individual rat the highest possible score was 9. Pancreatic water content was estimated as the wet weight/dry weight ratio (ww/dw) of the tissue. N: untreated animals, anesthesia omitted; S: sham operations.
P < 0.05 compared to the untreated group;
P < 0.05 compared to the control group at 6 h after induction of model.
Serum levels of TNF-α and IL-6
The serum levels of TNF-α and IL-6 in AEP and ANP groups were far significantly higher than those in sham group (P < 0.001). The serum levels of TNF-α and IL-6 6 h after establishment of the model in ANP group were significantly higher than those in AEP (P < 0.05, P < 0.001, P < 0.05). In ANP group, the levels of serum TNF-α and IL-6 6 h after establishment of the studied model were significantly higher than those 3 h after establishment of the studied model (P < 0.05) (Table 3).
Table 3.
Serum levels of TNF-α and IL-6 in AEP and ANP (¯x ± s)
| Group |
TNF-α (pg/mL) |
IL-6 (pg/mL) |
||
| 3 h | 6 h | 3 h | 6 h | |
| Sham | 4.13 ± 2.5 | 55.87 ± 3.79 | 100.79 ± 53.89 | 151.04 ± 91.79 |
| AEP | 54.27 ± 23.48b | 67.83 ± 22.02b | 428.12 ± 140.30b | 420.13 ± 139.40b |
| ANP | 64.28 ± 20.79b | 106.59 ± 43.71abc | 1600.32 ± 309.78ab | 2203.76 ± 640.85abc |
P < 0.001 vs sham group;
P < 0.05 or 0.001 vs AEP group;
P < 0.05 vs 3 h group.
Morphological examination
After induction of the AEP and ANP models, the pancreas showed mild edema and congestion. Three and 6 h after introduction of the model, typical pathological changes of AEP and ANP were found, such as large numbers of inflammatory cells, edema, hemorrhage, necrosis, large amount of ascites (Table 1).
NF-κB and p38 MAPK in activated monocytes/macrophages
The expression of NF-κB and p38 MAPK in activated monocytes/macrophages at 3 and 6 h was assessed by immunohistochemistry. In AEP, mild expression of NF-κB and p38 MAPK in activated monocytes/macrophages was found 3 and 6 h after introduction of the model. However, in ANP, the expression of NF-κB and p38 MAPK in activated monocytes/macrophages was upregulated evidently 3 and 6 h after introduction of the model, reaching their highest levels at 6 h after introduction of the model (Figure 1, Figure 2, Figure 3 and Figure 4).
Figure 1.
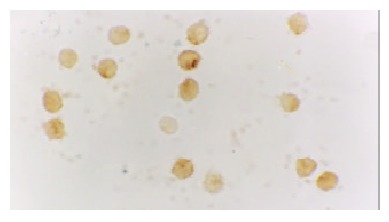
Moderate expression of NF-κB in the cytoplasm/nucleolus of monocytes in AEP (6 h) 10 × 100.
Figure 2.
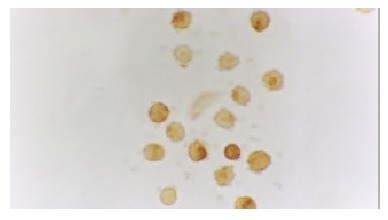
Strong expression of NF-κB in the cytoplasm/nucleolus of monocytes in ANP (6 h) 10 × 100.
Figure 3.
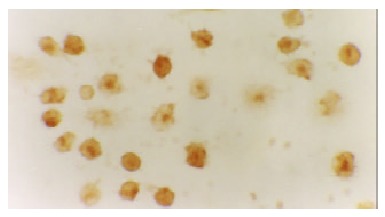
Moderate expression of p38MAPK in the cytoplasm/nucleolus of monocytes in AEP (6 h) 10 × 100.
Figure 4.
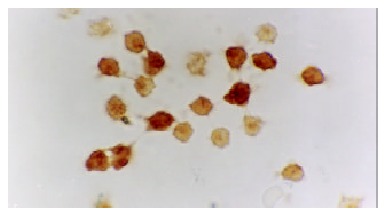
Strong expression of p38MAPK in the cytoplasm/nucleolus of monocytes in ANP (6 h) 10 × 100.
DISCUSSION
The clinical manifestations of AP vary significantly from mild to lethal, severity of the disease is largely determined by the actions of various kinds of inflammatory mediators, including cytokines, reactive oxygen species, proteolytic enzymes, and lipids, as well as gaseous mediators. Despite increasing knowledge implicating the involvement of cytokines in the progression of AP, no clinical trials pertaining to cytokine modulation have been performed so far. Progress in intensive care technologies has contributed to the improvement of mortality and morbidity rates in severe AP in the past decade. However, it appears to be reasonable for clinicians to "line up their sights" on the modulation of cytokines as a direct treatment.
Pro-inflammatory cytokines played a main role in acute pancreatitis and complications due to acute pancreatitis[29,30]. Failure of different organ systems is a frequent problem in severe acute pancreatitis. The majority of fatalities in patients with severe acute pancreatitis are associated with the failure of at least one or more organ systems. Organ failure in severe acute pancreatitis has to be regarded as part of the inflammatory response following the liberation of activated enzymes from the pancreas. The mediators involved in this process include IL-1, TNF-α, IL-6, platelet activating factor, and IL-8. Of the various kinds of inflammatory mediators, TNF-α and IL-1 are regarded as the most prominent "first-line" cytokines. The primary involvement of TNF-α was demonstrated by elevation of TNF-α levels in rat severe AP models. This was confirmed by subsequent TNF-α antagonism experiments, using anti-TNF-α antibodies or a recombinant dimeric form of p55 TNF receptor. Similarly, the concurrent blockade of cytokines showed no additional benefit in the amelioration of pancreatic injury, however, it attenuated the systemic response, as indicated by reduced IL-6 levels and decreased mortality rates. With few exceptions, most studies carried out with the purpose of blocking proximal cytokines, such as TNF-α, IL-1 or IL-6, during the progression of AP showed favorable results in experimental settings. However, neither TNF-α nor IL-1 has a causative role in the initiation of AP. For example, exposure of isolated acinar cells to TNF-α and IL-1 in vitro did not induce either intracellular zymogen activation or release of enzymes. In addition, these cytokines were not able to induce either biological or histological evidence of pancreatitis by in-situ perfusion of isolated human pancreas[31].
As to the primary site of cytokine production, infiltrating macrophages in the pancreas and ascites are suggested to be the initial cellular origins of TNF-α. In a mouse cerulein AP model, the expression of TNF-α messenger RNA within the inflamed pancreas was observed as early as 30 min after the induction of AP, and was followed by elevation of intrapancreatic and serum TNF-α levels. In addition, neutrophils that have migrated into the inflamed pancreas appear to be equally involved in the production of TNF-α, IL-6 and IL-1. The production of these cytokines was subsequently observed in specific distant organs, such as the lungs, liver, and spleen. In particular, production of TNF-α and IL-1 in the lung was known to be closely associated with the development of adult respiratory distress syndrome (ARDS), which was the major cause of early death in patients with SAP[32].
IL-6 is well known to be the primary inducer of acute-phase response, while its level in circulation during AP reaches the maximum value 24-48 h before the maximal C-reactive protein level is reached. Therefore, IL-6 has been recently reported to be the best prognostic parameter of pulmonary failure. IL-6 is synthesized by monocytes/macrophages, neutrophils, endothelial cells, and even pancreatic duct cells. It is able to stimulate neutrophil chemotaxis and release of enzymes, thereby leading to tissue destruction when overproduced. In clinical settings, some authors have reported elevated IL-6 levels in AP patients, particularly in those with complications. This observation has been supported by in-vitro studies, using isolated blood monocytes from patients with AP, in which systemic complications were found to be closely associated with enhanced secretion of IL-6 and IL-8 from the cells. Based on an increasing body of evidence, both IL-6 and IL-8 are now considered to be early and excellent predictors of patient outcome, and are widely used as secondary end points in clinical trails[33].
Recent experimental studies have shed some light on the intracellular signaling pathway in the inflammatory cascade in AP. For example, recent evidence strongly suggested the crucial role of NF-κB in the initiation of AP, not only in pancreatic acinar cells, monocytes/macrophages and neutrophils, but also in specific distant organs such as the lung. NF-κB is able to mediate a variety of inflammatory mediators involved in AP, including cytokines and adhesion molecules, as well as specific enzymes. NF-κB plays a key role in the transcriptional regulation of adhesion molecules, enzymes and cytokines involved in acute inflammatory diseases. NF-κB is a key transcription factor for the expression of various proinflammatory molecules such as chemokines, cytokines, and adhesion molecules. This notion has recently been verified by adenoviral-mediated gene transfer of an active subunit, RelA/p65, into acinar cells delivered through an injection into the rat pancreatic duct. The overexpressed RelA/p65 protein trans-activated NF-κB and induced pancreatic inflammation, a pathological state very similar to acute pancreatitis. In addition to the importance of NF-κB in the pathogenesis of acute pancreatitis, it was also reported that NF-κB activation in macrophages was a key event both in the development of generalized complications during severe acute pancreatitis and as a cause of the high mortality in this condition. Studies also showed that glucocorticoids were one of the most potent inhibitors of NF-κB, which might be attributable to the inhibition of this key transcription factor both in acinar cells and in inflammatory cells such as monocytes/macrophages. It is known that the mechanism of the inhibition of NF-κB by corticosteroids is multifactorial. One is via the induction of IκBa, the inhibitor molecule of NF-κB, which can form a complex with NF-κB in the cytoplasm, inhibiting the translocation of NF-κB into the nucleus, where it enhances the transcriptional activation of various proinflammatory genes. The other is a cross-coupling mechanism of inhibition between activated glucocorticoid receptors and activated NF-κB. Because the activation of NF-κB is an early event in the pathogenetic mechanism of acute pancreatitis, it is reasonable that in most studies corticosteroids showed beneficial effects on acute pancreatitis[34].
Moreover, inflammatory mediators released during acute diseases activate multiple intracellular signalling cascades including the MAPK signal transduction pathway, which plays a significant role in the recruitment of leukocytes to sites of inflammation. Stimulation of leukocytes by pro-inflammatory cytokines is known to result in activation of MAPK isoform p38. However, the functional consequences of p38 MAPK activation during leukocyte recruitment, including adhesion, migration and effector functions such as oxidative burst and degranulation, are only just beginning to be elucidated. Recently, p38 MAPK has been implicated in the activation of NF-κB. Several groups have demonstrated that p38 MAPK-specific inhibitor SB203580 potently attenuated NF-κB-dependent transcription. However, phosphorylation of NF-κB subunits, nuclear translocation and DNA binding to NF-κB were not affected by this p38 inhibitor. This suggests that NF-κB and p38 MAPK are activated by separate effector pathways, which might converge further downstream in the cell nucleus. So recent experimental studies have shed some light on the intracellular signaling pathway in the inflammatory cascade. Cytokine transcription and biosynthesis bifurcate into two major signaling pathways at the level of proteins. One runs through the NF-κB inducing kinase route, which regulates phosphorylation of inhibitory-kb (IκB) protein, the cytosolic inhibitor of NF-κB which allows NF-κB complex to translocate to the nucleus and promotes gene expression. The other is mediated through p38MAPK pathway and makes p38MAPK phosphorylated and activated[35].
So in recent years, much effort has been focused on delineating intracellular signaling cascades in inflammatory pathways. The intracellular signaling pathways used by leukocytes in response to pro-inflammatory stimuli have only begun to be unravelled. Much attention has been given to MAPK superfamily owing to their activation by pro-inflammatory cytokines IL-6 and TNF-α, which play a central role during inflammatory responses and in inflammatory diseases.
In this study, it showed that retrograde infusion of sodium taurocholate into the pancreatic duct resulted in an increase of amylase activity and ascites 3 and 6 h after pancreatitis. The levels of serum AMS and ascites in AEP and ANP groups were far significantly higher than those in sham group (P < 0.01). The serum levels of TNF-α and IL-6 in AEP and ANP groups were far significantly higher than those in sham group (P < 0.001). The serum level of TNF-α of the studied model and the serum level of IL-6 3 and 6 h in ANP group were significantly higher than that in AEP (P < 0.05, P < 0.001, P < 0.05). In ANP group, the levels of serum TNF-α and IL-6 6 h after establishment of the studied model were significantly higher than those 3 h after establishment of the studied model (P < 0.05, P < 0.05).
After induction of AEP and ANP models, pancreas showed mild edema and congestion. Three and 6 h after introduction of the model, typical pathological changes of AEP and ANP were found, such as large numbers of inflammatory cell, edema, hemorrhage, necrosis, large amount of ascites.
The expression of NF-κB and p38 MAPK in activated monocytes/macrophages at 3 and 6 h after pancreatitis was assessed by immunohistochemistry. In AEP, moderate expression of NF-κB in activated monocytes/macrophages was found 3 and 6 h after introduction of the model. However, in ANP, the expression of NF-κB and p38 MAPK in activated monocytes/macrophages were upregulated evidently 3 and 6 h after introduction of the model, reaching their highest levels 6 h after introduction of the model. So the expression of NF-κB and p38 MAPK in activated monocytes/macrophages at 3 and 6 h assessed by immunohistochemistry was consistent with the increase of pro-inflammatory cytokines TNF-α and IL-6.
Thus, increased understanding of the signal transduction pathways involved in the regulation of cytokine production and cytokine signaling in inflammatory cells has opened the door for the discovery of novel therapeutics for treating a variety of inflammatory diseases in which cytokine production or signaling is implicated. The availability of potent and selective inhibitors of such signaling pathways also provides a means to further dissect the pathways to increase our understanding about the complex events underlying a particular immune mediated event at the cellular and molecular level. NF-κB and p38 MAPK are the most studied signaling molecules in this regard and their relevance to such disease states has been established primarily through the use of inhibitors. Indeed, selective inhibition of NF-κB and p38 MAP kinase inhibitors has demonstrated efficacy in a variety of animal models[36-40]. Specific p38 inhibitors and selective inhibition of NF-κB aimed at reducing the production of inflammatory mediators are now being developed, and might in the future provide more effective treatment for inflammatory diseases. At present, it may be too early to take an optimistic view of therapeutic cytokine modulation in AP. There are many obstacles to overcome. However, direct therapy by cytokine modulation sounds very attractive to clinicians.
Footnotes
Edited by Zhu LH and Wang XL
References
- 1.Morrison CP, Teague BD, Court FG, Wemyss-Holden SA, Metcalfe MS, Dennison AR, Maddern GJ. Experimental studies of serum cytokine concentration following pancreatic electrolytic ablation. Med Sci Monit. 2003;9:BR43–BR46. [PubMed] [Google Scholar]
- 2.Zhang Q, Ni Q, Cai D, Zhang Y, Zhang N, Hou L. Mechanisms of multiple organ damages in acute necrotizing pancreatitis. Chin Med J ( Engl) 2001;114:738–742. [PubMed] [Google Scholar]
- 3.Makhija R, Kingsnorth AN. Cytokine storm in acute pancreatitis. J Hepatobiliary Pancreat Surg. 2002;9:401–410. doi: 10.1007/s005340200049. [DOI] [PubMed] [Google Scholar]
- 4.Bhatia M, Neoptolemos JP, Slavin J. Inflammatory mediators as therapeutic targets in acute pancreatitis. Curr Opin Investig Drugs. 2001;2:496–501. [PubMed] [Google Scholar]
- 5.Wereszczynska-Siemiatkowska U, Dabrowski A, Siemiatkowski A, Mroczko B, Laszewicz W, Gabryelewicz A. Serum profiles of E-selectin, interleukin-10, and interleukin-6 and oxidative stress parameters in patients with acute pancreatitis and nonpancreatic acute abdominal pain. Pancreas. 2003;26:144–152. doi: 10.1097/00006676-200303000-00010. [DOI] [PubMed] [Google Scholar]
- 6.Lundberg AH, Granger DN, Russell J, Sabek O, Henry J, Gaber L, Kotb M, Gaber AO. Quantitative measurement of P- and E-selectin adhesion molecules in acute pancreatitis: correlation with distant organ injury. Ann Surg. 2000;231:213–222. doi: 10.1097/00000658-200002000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qin RY, Zou SQ, Wu ZD, Qiu FZ. Experimental research on production and uptake sites of TNFalpha in rats with acute hemorrhagic necrotic pancreatitis. World J Gastroenterol. 1998;4:144–146. doi: 10.3748/wjg.v4.i2.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shimada M, Andoh A, Hata K, Tasaki K, Araki Y, Fujiyama Y, Bamba T. IL-6 secretion by human pancreatic periacinar myofibroblasts in response to inflammatory mediators. J immunol. 2002;168:861–868. doi: 10.4049/jimmunol.168.2.861. [DOI] [PubMed] [Google Scholar]
- 9.Riché FC, Cholley BP, Laisné MJ, Vicaut E, Panis YH, Lajeunie EJ, Boudiaf M, Valleur PD. Inflammatory cytokines, C reactive protein, and procalcitonin as early predictors of necrosis infection in acute necrotizing pancreatitis. Surgery. 2003;133:257–262. doi: 10.1067/msy.2003.70. [DOI] [PubMed] [Google Scholar]
- 10.Coelho AM, Machado MC, Cunha JE, Sampietre SN, Abdo EE. Influence of pancreatic enzyme content on experimental acute pancreatitis. Pancreas. 2003;26:230–234. doi: 10.1097/00006676-200304000-00004. [DOI] [PubMed] [Google Scholar]
- 11.Mayer J, Rau B, Gansauge F, Beger HG. Inflammatory mediators in human acute pancreatitis: clinical and pathophysiological implications. Gut. 2000;47:546–552. doi: 10.1136/gut.47.4.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jaffray C, Yang J, Carter G, Mendez C, Norman J. Pancreatic elastase activates pulmonary nuclear factor kappa B and inhibitory kappa B, mimicking pancreatitis-associated adult respiratory distress syndrome. Surgery. 2000;128:225–231. doi: 10.1067/msy.2000.107419. [DOI] [PubMed] [Google Scholar]
- 13.Kim H, Seo JY, Kim KH. NF-kappaB and cytokines in pancreatic acinar cells. J Korean Med Sci. 2000;15 Suppl:S53–S54. doi: 10.3346/jkms.2000.15.S.S53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blanchard JA, Barve S, Joshi-Barve S, Talwalker R, Gates LK. Cytokine production by CAPAN-1 and CAPAN-2 cell lines. Dig Dis Sci. 2000;45:927–932. doi: 10.1023/a:1005573024448. [DOI] [PubMed] [Google Scholar]
- 15.Frossard JL, Pastor CM, Hadengue A. Effect of hyperthermia on NF-kappaB binding activity in cerulein-induced acute pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2001;280:G1157–G1162. doi: 10.1152/ajpgi.2001.280.6.G1157. [DOI] [PubMed] [Google Scholar]
- 16.Blanchard JA, Barve S, Joshi-Barve S, Talwalker R, Gates LK. Antioxidants inhibit cytokine production and suppress NF-kappaB activation in CAPAN-1 and CAPAN-2 cell lines. Dig Dis Sci. 2001;46:2768–2772. doi: 10.1023/a:1012795900871. [DOI] [PubMed] [Google Scholar]
- 17.Tando Y, Algül H, Schneider G, Weber CK, Weidenbach H, Adler G, Schmid RM. Induction of IkappaB-kinase by cholecystokinin is mediated by trypsinogen activation in rat pancreatic lobules. Digestion. 2002;66:237–245. doi: 10.1159/000068364. [DOI] [PubMed] [Google Scholar]
- 18.Algül H, Tando Y, Schneider G, Weidenbach H, Adler G, Schmid RM. Acute experimental pancreatitis and NF-kappaB/Rel activation. Pancreatology. 2002;2:503–509. doi: 10.1159/000066090. [DOI] [PubMed] [Google Scholar]
- 19.Murr MM, Yang J, Fier A, Kaylor P, Mastorides S, Norman JG. Pancreatic elastase induces liver injury by activating cytokine production within Kupffer cells via nuclear factor-Kappa B. J Gastrointest Surg. 2002;6:474–480. doi: 10.1016/s1091-255x(01)00071-3. [DOI] [PubMed] [Google Scholar]
- 20.Rakonczay Z, Jármay K, Kaszaki J, Mándi Y, Duda E, Hegyi P, Boros I, Lonovics J, Takács T. NF-kappaB activation is detrimental in arginine-induced acute pancreatitis. Free Radic Biol Med. 2003;34:696–709. doi: 10.1016/s0891-5849(02)01373-4. [DOI] [PubMed] [Google Scholar]
- 21.Altavilla D, Famulari C, Passaniti M, Campo GM, Macrì A, Seminara P, Marini H, Calò M, Santamaria LB, Bono D, et al. Lipid peroxidation inhibition reduces NF-kappaB activation and attenuates cerulein-induced pancreatitis. Free Radic Res. 2003;37:425–435. doi: 10.1080/1071576031000070093. [DOI] [PubMed] [Google Scholar]
- 22.Blinman TA, Gukovsky I, Mouria M, Zaninovic V, Livingston E, Pandol SJ, Gukovskaya AS. Activation of pancreatic acinar cells on isolation from tissue: cytokine upregulation via p38 MAP kinase. Am J Physiol Cell Physiol. 2000;279:C1993–C2003. doi: 10.1152/ajpcell.2000.279.6.C1993. [DOI] [PubMed] [Google Scholar]
- 23.Chen X, Ji B, Han B, Ernst SA, Simeone D, Logsdon CD. NF-kappaB activation in pancreas induces pancreatic and systemic inflammatory response. Gastroenterology. 2002;122:448–457. doi: 10.1053/gast.2002.31060. [DOI] [PubMed] [Google Scholar]
- 24.Murr MM, Yang J, Fier A, Gallagher SF, Carter G, Gower WR, Norman JG. Regulation of Kupffer cell TNF gene expression during experimental acute pancreatitis: the role of p38-MAPK, ERK1/2, SAPK/JNK, and NF-kappaB. J Gastrointest Surg. 2003;7:20–25. doi: 10.1016/s1091-255x(02)00053-7. [DOI] [PubMed] [Google Scholar]
- 25.Masamune A, Kikuta K, Satoh M, Satoh A, Shimosegawa T. Alcohol activates activator protein-1 and mitogen-activated protein kinases in rat pancreatic stellate cells. J Pharmacol Exp Ther. 2002;302:36–42. doi: 10.1124/jpet.302.1.36. [DOI] [PubMed] [Google Scholar]
- 26.Fleischer F, Dabew R, Göke B, Wagner AC. Stress kinase inhibition modulates acute experimental pancreatitis. World J Gastroenterol. 2001;7:259–265. doi: 10.3748/wjg.v7.i2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dabrowski A, Boguslowicz C, Dabrowska M, Tribillo I, Gabryelewicz A. Reactive oxygen species activate mitogen-activated protein kinases in pancreatic acinar cells. Pancreas. 2000;21:376–384. doi: 10.1097/00006676-200011000-00008. [DOI] [PubMed] [Google Scholar]
- 28.Gilgenast O, Brandt-Nedelev B, Wiswedel I, Lippert H, Halangk W, Reinheckel T. Differential oxidative injury in extrapancreatic tissues during experimental pancreatitis: modification of lung proteins by 4-hydroxynonenal. Dig Dis Sci. 2001;46:932–937. doi: 10.1023/a:1010745610954. [DOI] [PubMed] [Google Scholar]
- 29.Osman MO, Gesser B, Mortensen JT, Matsushima K, Jensen SL, Larsen CG. Profiles of pro-inflammatory cytokines in the serum of rabbits after experimentally induced acute pancreatitis. Cytokine. 2002;17:53–59. doi: 10.1006/cyto.2001.0977. [DOI] [PubMed] [Google Scholar]
- 30.Bidarkundi GK, Wig JD, Bhatnagar A, Majumdar S. Clinical relevance of intracellular cytokines IL-6 and IL-12 in acute pancreatitis, and correlation with APACHE III score. Br J Biomed Sci. 2002;59:85–89. doi: 10.1080/09674845.2002.11783640. [DOI] [PubMed] [Google Scholar]
- 31.Yamauchi J, Shibuya K, Sunamura M, Arai K, Shimamura H, Motoi F, Takeda K, Matsuno S. Cytokine modulation in acute pancreatitis. J Hepatobiliary Pancreat Surg. 2001;8:195–203. doi: 10.1007/s005340170016. [DOI] [PubMed] [Google Scholar]
- 32.Denham W, Yang J, Norman J. Evidence for an unknown component of pancreatic ascites that induces adult respiratory distress syndrome through an interleukin-1 and tumor necrosis factor-dependent mechanism. Surgery. 1997;122:295–301; discussion 301-2. doi: 10.1016/s0039-6060(97)90021-0. [DOI] [PubMed] [Google Scholar]
- 33.Powell JJ, Murchison JT, Fearon KC, Ross JA, Siriwardena AK. Randomized controlled trial of the effect of early enteral nutrition on markers of the inflammatory response in predicted severe acute pancreatitis. Br J Surg. 2000;87:1375–1381. doi: 10.1046/j.1365-2168.2000.01558.x. [DOI] [PubMed] [Google Scholar]
- 34.Takaoka K, Kataoka K, Sakagami J. The effect of steroid pulse therapy on the development of acute pancreatitis induced by closed duodenal loop in rats. J Gastroenterol. 2002;37:537–542. doi: 10.1007/s005350200083. [DOI] [PubMed] [Google Scholar]
- 35.Haddad JJ. The involvement of L-gamma-glutamyl-L-cysteinyl-glycine (glutathione/GSH) in the mechanism of redox signaling mediating MAPK (p38)-dependent regulation of pro-inflammatory cytokine production. Biochem Pharmacol. 2002;63:305–320. doi: 10.1016/s0006-2952(01)00870-x. [DOI] [PubMed] [Google Scholar]
- 36.Ethridge RT, Hashimoto K, Chung DH, Ehlers RA, Rajaraman S, Evers BM. Selective inhibition of NF-kappaB attenuates the severity of cerulein-induced acute pancreatitis. J Am Coll Surg. 2002;195:497–505. doi: 10.1016/s1072-7515(02)01222-x. [DOI] [PubMed] [Google Scholar]
- 37.Legos JJ, McLaughlin B, Skaper SD, Strijbos PJ, Parsons AA, Aizenman E, Herin GA, Barone FC, Erhardt JA. The selective p38 inhibitor SB-239063 protects primary neurons from mild to moderate excitotoxic injury. Eur J Pharmacol. 2002;447:37–42. doi: 10.1016/s0014-2999(02)01890-3. [DOI] [PubMed] [Google Scholar]
- 38.Elenitoba-Johnson KS, Jenson SD, Abbott RT, Palais RA, Bohling SD, Lin Z, Tripp S, Shami PJ, Wang LY, Coupland RW, et al. Involvement of multiple signaling pathways in follicular lymphoma transformation: p38-mitogen-activated protein kinase as a target for therapy. Proc Natl Acad Sci USA. 2003;100:7259–7264. doi: 10.1073/pnas.1137463100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.El Bekay R, Alvarez M, Monteseirín J, Alba G, Chacón P, Vega A, Martin-Nieto J, Jiménez J, Pintado E, Bedoya FJ, et al. Oxidative stress is a critical mediator of the angiotensin II signal in human neutrophils: involvement of mitogen-activated protein kinase, calcineurin, and the transcription factor NF-kappaB. Blood. 2003;102:662–671. doi: 10.1182/blood-2002-09-2785. [DOI] [PubMed] [Google Scholar]
- 40.Torres M. Mitogen-activated protein kinase pathways in redox signaling. Front Biosci. 2003;8:d369–d391. doi: 10.2741/999. [DOI] [PubMed] [Google Scholar]