

Abstract
This study explored the association between cerebral metabolic rates of glucose (CMRGlc) and the severity of Vascular Parkinsonism (VP) and Parkinson’s disease (PD). A cross-sectional study was performed to compare CMRGlc in normal subjects vs. VP and PD patients. Twelve normal subjects, 22 VP, and 11 PD patients were evaluated with the H&Y and MMSE, and underwent 18F-FDG measurements. Pearson’s correlations were used to identify potential associations between the severity of VP/PD and CMRGlc. A pronounced reduction of CMRGlc in the frontal lobe and caudate putamen was detected in patients with VP and PD when compared with normal subjects. The VP patients displayed a slight CMRGlc decrease in the caudate putamen and frontal lobe in comparison with PD patients. These decreases in CMRGlc in the frontal lobe and caudate putamen were significantly correlated with the VP patients’ H&Y, UPDRS II, UPDRS III, MMSE, cardiovascular, and attention/memory scores. Similarly, significant correlations were observed in patients with PD. This is the first clinical study finding strong evidence for an association between low cerebral glucose metabolism and the severity of VP and PD. Our findings suggest that these changes in glucose metabolism in the frontal lobe and caudate putamen may underlie the pathophysiological mechanisms of VP and PD. As the scramble to find imaging biomarkers or predictors of the disease intensifies, a better understanding of the roles of cerebral glucose metabolism may give us insight into the pathogenesis of VP and PD.
Keywords: Vascular Parkinsonism, Parkinson’s disease, cerebral glucose metabolism, frontal lobe, caudate putamen, non-motor symptoms
Imaging with 18F-fluorodeoxyglucose (18F-FDG) is commonly used to obtain differential information regarding the patterns of regional cerebral glucose metabolism and abnormal functional connectivity in neurodegenerative diseases using network imaging analysis. By quantifying glucose metabolism in the brain, this imaging approach may provide a quantitative analysis of synaptic activity in the examined brain regions and allow the evaluation of pharmacological effects before or after specific drug treatments. Increasing numbers of studies have shown that changes in cerebral metabolic rates of glucose (CMRGlc) are closely correlated with the pathogenesis of different neurodegenerative disorders, such as Alzheimer’s disease (AD), cerebral ischemia and Parkinson’s disease (PD). In addition, 18F-FDG kinetics or distributions are normally correlated with the clinical severity and progression in PD, while a limited number of studies have reported an association between cerebral glucose metabolic patterns and PD, typically finding a widespread decrease in cerebral oxygen metabolism. However, no study has reported neuronal functional changes and cerebral glucose metabolism in VP.
Although patients with VP show a different mechanism of disease than Parkinson’s disease, they typically present in similar clinical scenarios as PD patients. In addition, VP shares some similar characters with Alzheimer’s disease (AD) in terms of mechanisms and some clinical characteristics. For instance, both of these diseases normally display severe nonmotor dysfunction, including cognitive impairment and depression, as clinical features. Although neurofibrillary tangle and A-beta accumulation are widely prevailing mechanisms in AD, recent studies reveal that disturbances in cerebral glucose metabolism and cerebrovascular dysfunction are invariant pathophysiological features of AD. Similarly, vascular damage is also observed in PD patients. It is this cerebrovascular dysfunction that may contribute to the possible mechanisms underlying changes in CMRGlc in all three diseases. Non-motor symptoms (NMS), such as impaired cognition, progress as the disease develops, although some NMS, such as declined olfactory function, rapid eye movement and behavior disorder, hallucinations, constipation and depression, may predate the motor symptoms and seriously influence the patients quality of life. Clinically, it is very difficult to precisely distinguish VP from other neurodegenerative diseases, such as PD or multiple system atrophy, especially at the early stage. Therefore, a reliable predictor is urgently needed to predict or evaluate the development of VP as well as PD. It would be extremely useful if gross changes in cerebral glucose metabolism are correlated with certain NMS and if these changes could act as a biomarker for the evaluation of VP and PD progression and severity. Quite possibly, these changes could serve as an indicator in disease screening.
In this study, we determined if CMRGlc in patients with VP and PD is associated with disease severity and cognitive dysfunction. The primary aim of this study was to use specific clinical parameters to identify any correlation between symptom scores and CMRGlc levels in patients with VP and PD. A second aim was to quantify the activity of 18FDG data and to evaluate the differential alterations of the CMRGlc in specific brain regions in patients with VP and PD. Lastly, we aimed to assess whether CMRGlc in VP and PD was associated with poor motor function and to identify an association between NMS domains and CMRGlc levels.
MATERIALS AND METHODS
Participants and study design
The participants were chosen and the designed study was performed in the 3rd Hospital of Sun Yat-sen University, Neurological Department, China. The patients presented with VP or PD (Table 1) and were recruited according to criteria defined in our previous study and other diagnostic criteria for VP and PD, as follows: (1) they demonstrated parkinsonism, which is defined as bradykinesia and at least one of rest tremor, rigidity or postural instability; (2) cerebrovascular disease, determined by brain imaging or the presence of focal signs or symptoms that were consistent with stroke; and (3) a combination of (1) and (2) that was defined as an acute or delayed progressive onset of parkinsonism (within 1 year) after stroke, with evidence of infarcts in or near areas that increase basal ganglia motor output or decrease the thalamocortical drive directly, or an insidious onset of parkinsonism with extensive sub-cortical white matter lesions, bilateral symptoms at the onset and the presence of an early shuffling gait or early cognitive dysfunction. MRI scans were obtained with specific sequences as depicted in Fig. 1; the infarct volume for each VP patient was measured using 3D-DOCTOR software and is shown in the Table 1. Among the 22 patients with VP,20 suffered from cerebrovascular-disease-related risks, including hypertension (13), diabetes (7), hyperlipidemias (7), smoking and alcohol drinking (6). A control group (n=12, 6 males and 6 females) was chosen from healthy subjects aged 51 to 81 without hypertension, cerebral ischemia, diabetes, or renal dysfunction. This study has been approved by the local Ethics Committee of the 3rd Hospital of Sun Yat-sen University, and all procedures were set up according to the principles expressed in the Declaration of Helsinki (BMJ 1991; 302: 1194). The patients gave informed consent for the evaluation of 18F-FDG, and they gave their written consent for their measurements on the motor and nonmotor related evaluations. All participants provided their written informed consent to participate in this study, and these consents were documented in the patient files. All of the measurements for the subjects in this study were made blinded and followed the standard assessments: the standard demographic form, the unified Parkinson’s disease rating scale part III (UPDRS I-III), the modified Hoehn and Yahr staging scale (H&Y), and the Mini-Mental State Examination (MMSE). Moreover, all of these scales were available and validated for the Chinese population.
Table 1.
Demographic, motor, and non-motor parameters of patients with VP and PD.
| Clinical parameters | PD | VP | |||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Mean (SD) | Min | Max | Mean (SD) | Min | Max | ||
| Gender (n) | Male n (%) | 6(54.55) | 11(50) | ||||
| Female n (%) | 5(45.55) | 11(50) | |||||
| Age (years) | 59.18(16.29) | 39 | 74 | 65.80(9.83) | 51 | 81 | |
| Disease duration (years) | 4.96(4.43) | 0.6 | 15 | 4.1 (2.47) | 0.1 | 8 | |
| H&Y | 2.36(1.2) | 1 | 5 | 2.55 (1.13) | 1 | 4 | |
| MMSE | 22.54(6.9) | 6 | 30 | 24.21(3.67) | 18 | 30 | |
| Daily L-dopa dosage (mg) | 355.77(166.65) | 125 | 875 | 247.82(131.66) | 125 | 600 | |
| Infarction Volume(mm3) | 0 | 0 | 0 | 16923.27(3683.62) | 12098.09 | 25588.57 | |
| UPDRS | 36.63(14.76) | 11 | 64 | 34.95(14.54) | 13 | 74 | |
| UPRDRS(I) | 3.0(1.31) | 1 | 5 | 3.59(1.94) | 0 | 7 | |
| UPRDRS(II) | 13(6.76) | 4 | 25 | 11.9(6.92) | 5 | 34 | |
| UPRDRS(III) | 20.63(8.15) | 3 | 36 | 19.45(7.86) | 7 | 35 | |
| NMSS (total) | 77.93(48.26) | 6 | 188 | 99.73(5.20) | 18 | 235 | |
| Cardiovascular | 4.27(3.6) | 0 | 12 | 3.81(2.13) | 0 | 6 | |
| Sleep/Fatigue | 16.45(8.73) | 1 | 36 | 19.72(10.26) | 7 | 42 | |
| Mood | 18.63(12.82) | 2 | 55 | 17.77(8.39) | 3 | 32 | |
| Perceptual problem | 4.27(6.06) | 0 | 19 | 1.5(3.16) | 0 | 12 | |
| Attention/memory | 10.0(9.6) | 2 | 26 | 10.81(6.6) | 3 | 21 | |
| Gastrointestina | 12.0(7.32) | 0 | 26 | 9.45(6.69) | 0 | 19 | |
| Urinar | 8.63(6.34) | 0 | 36 | 8.63(5.19) | 0 | 24 | |
| Sexual function | 4.72(5.72) | 0 | 21 | 6.81(6.10) | 0 | 16 | |
| Miscellaneous | 7.81(8.7) | 0 | 36 | 7.95(6.37) | 0 | 38 | |
Figure 1.
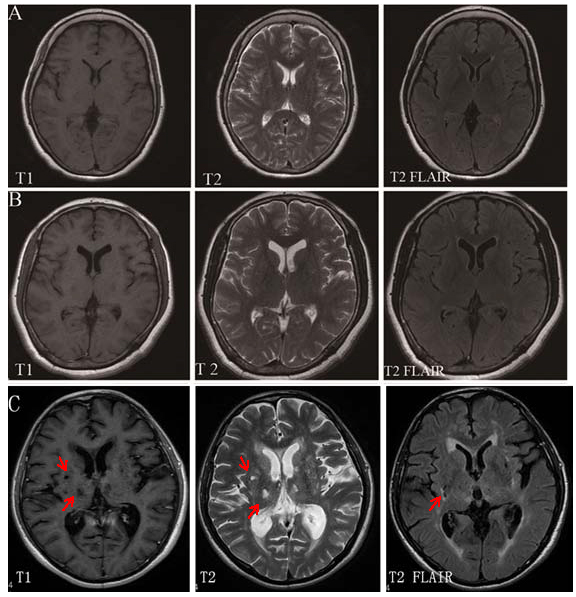
MRI Imaging scans for VP and PD patients and normal subjects. (A) Normal subjects, (B) PD patients, (C) VP patients. The extent of white matter hyperintensities and multiple infarctions in the basal ganglia in the VP patients are shown in T2-weighted and FLAIR images. Arrows indicate the infarction.
18F FDG imaging
The 18F FDG cerebral metabolic imaging procedure was performed with modification as previously described. Briefly, all of the subjects fasted for more than 6 hours and were taken off their anti-parkinsonian medications for at least 12 h prior to imaging. A 185 MBq injection of 18F-FDG (Atom High-tech Isotope Pharmaceutical Company, Guangzhou, China; > 95% pure) was intravenously administered to the patients with their eyes covered in a quiet and poorly lit room. An 18F-FDG is commonly used to assess the levels of resting CMRGlc. The imaging was performed using dual headed gamma cameras with coincidence circuit system (Discovery VH, GE Company, USA) in 2D mode and at 40 min post-injection. During the acquisition, a head-holder was applied to the patient to immobilize the head. The X-ray CT transmission tomography scans were first performed with a 10-min CT continuous acquisition, and the FDG imaging was performed then continuously for 18 min. After the X-ray attenuation correction and coincidence-ordered subsets expectation maximization (COSEM) reconstruction, the transverse, sagittal, and coronal section and the fusion images were obtained. After sequential analysis of the transverse and coronal section images, the regions of interest (ROI) in the bilateral frontal lobe and caudate putamen were delineated to quantify the radioactivity in examined areas. All regional radioactivity counts in the examined brain areas were compared with that in the cerebellum and was indicated as a semi-ratio value.
Statistical analysis
All of the data for the continuous variables (age, disease duration, daily levodopa dosage, the UPDRS, MMSE, and NMSS) are shown as the means ± SD, and the categorical variables (gender) are shown as percentages. One-Way ANOVA (Statistical Product and Service Solutions 19.0 program, Chicago, IL) followed by Tukey’s post hoc analysis was used to assess the presence of differences in CMRGlc between groups. A Spearman’s rank correlation coefficient was used to evaluate the association between CMRGlc and the different clinical parameters. P-values of less than 0.05 were regarded as statistically significant.
RESULTS
In the cross-sectional pilot study, 22 VP (11 males, 50% and 11 females, 50%) and 11 PD (6 males, 55% and 5 females, 45%) patients were enrolled. The mean ages of the VP and PD patients were 65.80 ± 9.83 years (range: 51 to 81) and 59.18 ± 16.29 years (range: 39 to 74), respectively, and that of the control group was 68.80 ± 9.83 years (range: 47 to 83). The mean durations of VP and PD symptoms were 4.1 ± 2.47 years (range: 0.1 to 8) and 4.96±4.43 years (range: 0.6 to 15), respectively. The H&Y stages ranged from 1 to 4 for patients with VP and PD. In terms of the levodopa (L-dopa) medication, the average doses were 247.82 mg daily and 355.77 mg daily for VP and PD, respectively. The demographic details of the patients are listed in Table 1.
MRI and 18F FDG imaging
Brain MRI scanning showed no T2 flair hyperintensities or T1 hypointensities in any brain regions in the control subjects. However, many T2 flair hyperintensities and T1 hypointensities in the caudate putamen and subcortical white matter were found in VP patients (Fig. 1). The average infarct volume for VP patients was more than 16923.27±3683.62 mm3/subject based on measurements from the 3D-DOCTOR software; However, the idiopathic PD patients showed no structural infarcts in the basal ganglia (Table 1 and Fig. 1). This result is consistent with previous studies.
A significant difference in terms of hypometabolism in VP and PD patients was detected in the frontal lobe and in the caudate putamen compared to normal subjects (Table 2). The VP and PD patients displayed a significant CMRGlc decrease in the frontal cortex and caudate putamen when compared with normal subjects (Table 2). Moreover, in comparison with PD patients, the VP patients displayed a slight CMRGlc decrease in the caudate putamen and frontal lobe, without significant differences (Table 2). Typical 18F FDG images on normal subjects, VP, and PD patients are shown in Fig. 2. The CMRGlc was evaluated using 3D-SSP analyses in all subjects. We have quantified the correlations between the CMERGlc and infarction volume in the frontal lobe and caudate putamen MRI imaging, showing a 0.031 and 0.045.
Table 2.
The difference in CMRGlc in the frontal lobes and caudate putamen between normal subjects vs. VP and PD patients (Mean ± SD).
| Group | N | frontal lobe /cerebellum | Cpu/cerebellum |
|---|---|---|---|
| Normal | 12 | 1.26±0.1 | 1.29±0.09 |
| PD | 11 | 1.11±0.11 | 1.09±0.17 |
| VP | 22 | 1.06±0.14 | 1.07±0.11 |
| P value | VP vs. normal | 0.000*** | 0.000*** |
| PD vs. normal | 0.007♀♀ | 0.001♀♀ | |
| PD vs VP | 0.272 | 0.6 |
VP vs. normal., p<0.001;
PD vs. normal, p<0.01
Figure 2.
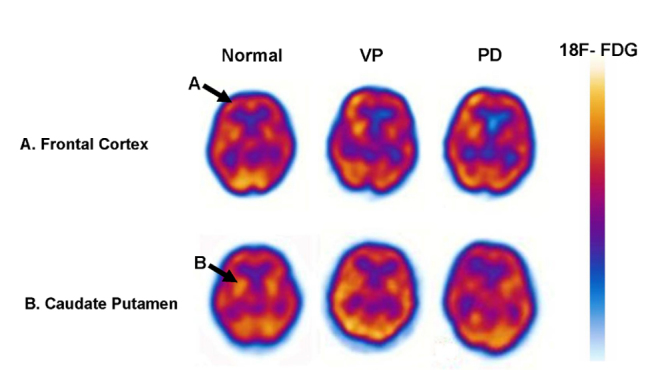
Measurements of CMRGlc in various brain regions in VP, PD, and normal subjects. Significant relative hypometabolism in patients with VP and PD was detected in the frontal lobe and caudate putamen compared to normal subjects (A and B). The images are horizontal brain sections. The changes in glucose metabolism are indicated by high (red) to low (blue) tracer uptake or binding in the scale.
In addition, all subjects were analyzed using the semi-quantitative ROIs/cerebellum ratio.
In the VP patients, significant correlations were observed between the H&Y and CMRGlc in frontal lobe/C and Cpu/C (r= -0.734, p = 0.001 for frontal lobe;r=-0.701,p = 0.001 for caudate putamen; Table 3); between the UPDRS and CMRGlc in frontal lobe/C and Cpu/C (rS= -0.596, p = 0.03,;rs=-0.625,p = 0.002); between the UPDRS (II) and CMRGlc in frontal lobe/C and Cpu/C (rS = -0.479, p = 0.024;rS = -604, p = 0.003); between the UPDRS (III) and CMRGlc in frontal lobe/C and CPU/C (rS = -0.585, p = 0.004;rS = -0.565, p = 0.006); between CMRGlc in frontal lobe/C and Cpu/C and the NMSS (rS = -0.47,2 p = 0.027;rS = -0.451, p = 0.035); and between CMRGlc in frontal lobe/C and the NMSS domains Mood and Attention/memory (rS = 0.490/0.533, p = 0.021 / 0.011); and between CMRGlc in Cpu/C and the NMSS domains Cardiovascular, Attention/memory, and Gastrointestina (rS = -0.482/-0.605/-0.506, p = 0.023 / 0.003/0.016). There were no significant correlations between CMRGlc and other clinical parameters such as L-dopa dosage in the patients with VP, as described above. Similarly, in the PD patients, the decreases in CMRGlc in the frontal lobe and caudate putamen were significantly correlated with the H&Y (r=-0.894, p=0.001 for frontal lobe;r=-0.668,p=0.02 for caudate putamen; Table 4), UPDRS II (r=-0.729,p=0.01; r=-0.727, p=0.01; Table 4), UPDRS III (r=-0.46,p=0.15;r=-0.831, p=0.001), MMSE (r=0.701,p=.02; r=0.38, p=0.25), cardiovascular (r=-0.649, p=0.03;r=-0.51, p=0.11) and attention/memory scores (r=-0.742, p=0.01; r=-0.664,p=0.03).
Table 3.
Spearman’s rank correlation coefficient (rs) between CMRGlc and clinical parameters in VP patients.
| frontal lobe/cerebella | Cpu/cerebella | |||
|---|---|---|---|---|
| r | p | r | p | |
| UP1 | -.387 | .075 | -.236 | .290 |
| UP2 | -.479* | .024 | -.604** | .003 |
| UP3 | -.585** | .004 | -.565** | .006 |
| UPDRS | -.596** | .003 | -.625** | .002 |
| H&Y | -.734** | .000 | -.701** | .000 |
| MMSE | .632** | .002 | .421 | .051 |
| Cardiovascular | -.245 | .271 | -.482* | .023 |
| Sleep/Fatigue | -.366 | .094 | -.380 | .081 |
| Mood | -.490* | .021 | -.351 | .110 |
| Perceptual problem | -.149 | .508 | -.117 | .603 |
| Attention/memory | -.533* | .011 | -.605** | .003 |
| Gastrointestina | -.170 | .450 | -.506* | .016 |
| Urinar | -.501* | .018 | -.460* | .031 |
| Sexual function | -.420 | .052 | -.226 | .312 |
| Miscellaneous | -.359 | .101 | -.227 | .310 |
| NMSS | -.472* | .027 | -.451* | .035 |
| Disease duration (years) | .219 | .328 | -.047 | .837 |
| Daily L-dopa dosage (mg) | -.130 | .565 | .142 | .528 |
| infarction volume | -.460* | 0.031 | -.565** | 0.003 |
Table 4.
Spearman’s rank correlation coefficient (rs) between CMRGlc and clinical parameters in PD patients.
| Frontal lobe/cerebella | Cpu/cerebella | |||
|---|---|---|---|---|
| r | p | r | p | |
| UP1 | -0.45 | 0.17 | -0.46 | 0.16 |
| UP2 | -.729* | 0.01 | -.727* | 0.01 |
| UP3 | -0.46 | 0.15 | -.831** | 0.00 |
| UPDRS | -.630* | 0.04 | -.834** | 0.00 |
| H&Y | -.894** | 0.00 | -.668* | 0.02 |
| MMSE | .701* | 0.02 | 0.38 | 0.25 |
| Cardiovascular | -.649* | 0.03 | -0.51 | 0.11 |
| Sleep/Fatigue | -.886** | 0.00 | -.634* | 0.04 |
| Mood | -.803** | 0.00 | -.634* | 0.04 |
| Perceptual problem | -0.12 | 0.72 | -0.20 | 0.57 |
| Attention/memory | -.742** | 0.01 | -.664* | 0.03 |
| Gastrointestina | -.636* | 0.04 | -.620* | 0.04 |
| Urinar | -.780** | 0.00 | -.679* | 0.02 |
| Sexual function | -.681* | 0.02 | -0.56 | 0.07 |
| Miscellaneous | -.769** | 0.01 | -0.36 | 0.28 |
| NMSS | -.809** | 0.00 | -.645* | 0.03 |
| Disease duration (years) | -0.21 | 0.54 | -.660* | 0.03 |
| Daily L-dopa dosage (mg) | -.640* | 0.03 | -0.57 | 0.06 |
DISCUSSION
We found several interesting results in this study. First, we found a pronounced reduction of CMRGlc in the frontal lobe and caudate putamen in patients with VP and PD when compared with normal subjects. Second, the VP patients displayed a slight CMRGlc decrease in the caudate putamen and in the frontal lobes in comparison with PD patients. Third, these decreases in CMRGlc in the frontal lobe and caudate putamen were significantly correlated with their H&Y, UPDRS II, UPDRS III, MMSE, cardiovascular, and attention/memory scores in patients with VP and in PD patients. To the best of our knowledge, this report is the first to evaluate the association between motor/non-motor effects and CMRGlc in patients with VP and PD using established and integrated diagnostic scales (NMSS, H&Y, and MMSE).
Several lines of evidence have shown that patients with PD normally display impaired cerebral metabolic rates of glucose [2, 4]. Similar to PD patients displaying clinical syndromes, we propose that patients with VP may have a defect in their cerebral metabolic rates of glucose, which could be correlated with the severity of their motor or non-motor dysfunctions. Patients with VP may progress in terms of the effects of cerebrovascular diseases, while striatal lacunar infarcts and white matter lesions are direct causes of VP [5].
18F-FDG is commonly used to assess levels of resting CMRGlc, and this FDG signal is typically correlated with the clinical severity in PD. In this study, we found a significant decrease in the levels of CMRGlc in the caudate putamen and frontal lobe of VP and PD patients when compared to normal subjects (Table 2). Compared to the PD patients, the VP group showed a slight metabolic reduction in the level of FDG in the caudate putamen and frontal lobe, without any significance (Table 2). Based on our current data, it is not possible to distinguish VP from PD only by the cerebral glucose metabolic profile. This pronounced decrease in CMRGlc in some brain regions in VP patients may be due to the following three major reasons: (1) subcortical lesions resulting from cerebral ischemic damage may induce a more reduced level of CMRGlc; (2) subcortical lesions may result in some constricting vascular lesions in the patients with micro-vascular causes; or (3) decreased rCBF and rCMRO2 in the frontal lobe and the caudate putamen in VP due to the arteriosclerosis or arterioarctia of the small vessels [1, 38-40]. Meanwhile, we cannot exclude other possibilities. For the future study, we will perform other studies to investigate the underlying mechanisms in experimental models. In normal healthy subjects, because glucose is the main substrate for energy metabolism in the brain, this reduction in the cerebral metabolic rates of glucose observed in our study implies a decreased energy metabolism, which may at least partially result from the reduced mitochondrial function and alteration in glucose transport at the Brain-Blood-Barrier (BBB). By combing phosphorus and proton magnetic resonance spectroscopic imaging, Hattingen et al. indicated that, as one of the pathogenetic pathways in PD, impaired mitochondrial dysfunction of mesostriatal neurons was observed in both early and advanced PD. Similarly, another study demonstrated reduced cerebral blood flow and CMRglc in PD patients, implying a decreased intracellular metabolism of glucose and impaired cerebral mitochondrial function [4]. Furthermore, in this study, the significant relative hypometabolism in the frontal cortex may have contributed to the decline in cognition in VP and PD patients in comparison with healthy subjects, which is consistent with previous reports.
In the current study, the observation for the reduction of cerebral glucose metabolism in the caudate putamen and frontal lobe of VP and PD patients is similar to previous studies. Carbon and Eidelberg also indicated a cortical hypometabolism in the prefrontal and parieto-occipital regions, which were similar to PD-related metabolic patterns. The reduction in the cerebral glucose metabolism may be closely correlated with the dysfunctions of the cerebral mitochondrial electron transport system (ETS), and the alterations in cerebral metabolic rates of glucose are likely a consequence of the disease process, including the medication intervention. In the current study, we observed associations between the CMRGlc and clinical features. Spearman’s rank correlation coefficients were used to explore the association of CMRGlc with clinical parameters in VP and PD patients. Highly significant correlations were observed between CMRGlc in the caudate putamen/frontal lobes and the MMSE, UPDRS (II)/(III), H&Y and some non-motor clinical manifestations (Table 3). This result suggests that CMRGlc may influence clinical manifestations, and it may be used to predict the longitudinal course or severity of VP and PD patients, especially in activities of daily life and some non-motor dysfunctions.
In this study, we noticed that the UPDRS motor scores and H&Y were significantly inversely correlated with CMRGlc in both the frontal lobes and caudate putamen in both VP and PD patients. Reduced CMRGlc in the caudate putamen contributes to the dopamine deficit in the striatum in VP and PD patients, subsequently leading to motor dysfunction [10]. However, the same phenomenon occurring in the frontal lobes may result from the indirect fact that the frontal lobes modulate motor function through frontostriatal circuitry and the corticostriatal-thalamocortical loop [6]. Hypometabolism of cerebral glucose in the frontal lobes could influence motor dysfunction in VP and PD patients. A global loss of neuronal connections would affect the neuronal integrity in the caudate putamen, which may then affect motor dysfunctions, especially gait disturbance [5]. Based on the evidence above, we propose that the hypometabolism of cerebral glucose can affect motor dysfunction through multiple paths, such as neurotransmitters, neuronal integrity and the corticostriatal-thalamocortical loop.
In addition to motor dysfunction, non-motor dysfunctions were also observed in VP and PD patients, such as cognitive impairment, declined attention/memory activity, and anxiety and stress. Several lines of evidence indicate that local lacunar ischemia in the nigrostriatal brain regions may lead to VP patients with dementia [1]. Microinfarction or demyelination in cortex or white matter lesions can lead to declined cognition. In the present study, significantly reduced hypometabolism in the frontal cortex was found, and profound correlations in FDG were observed between MMSE and glucose metabolism in the frontal lobes, attention/memory and the frontal lobes, and UPDRS III and the frontal lobes in VP and PD patients. This result was consistent with previous reports and would contribute to the decline in cognition in VP and PD. Interestingly, the current result also showed a robust metabolic reduction in the level of FDG in the caudate putamen in VP and PD patients when compared to the normal group (Table 2); while significant correlations of FDG were observed between MMSE and caudate putamen, attention/memory and the caudate putamen, and UPDRS III and the caudate putamen. Consistent with this finding, Polito et al. demonstrated that a lesion in frontostriatal circuits in PD may lead to nonmotor related cognitive impairments, and this disruption could constitute its pathophysiological basis [1]. Similarly, other studies also indicated that striatal dopaminergic denervation was associated with frontostriatal cognitive impairment [2, 15]. Taken together, the dopamine-related impairment of the caudate putamen and reduced cerebral glucose metabolism in the caudate putamen/frontal lobe would be recognized as possible causes and an early pathophysiological basis for impaired cognitive dysfunction in VP and PD.
Our findings provide insight into the metabolic features of VP and PD, and they demonstrate the likelihood that cognitive functional deficits and glucose metabolic alterations could closely connect with one another. Based on our results, we propose that decreased FDG in regional brain areas may be a potential predictor of disease severity and development in VP and PD. It is unknown when the onset of such hypometabolism happens during VP and PD progression. Is it possible that it precedes motor manifestations? If yes, the subtle changes seen during initial motor manifestations of the disease may be used as a reference for future comparisons to predict disease severity and progression. Moreover, if rapid changes were observed initially, such imaging could be quite useful to predict the timeliness of intervention with neuromodulation such as deep brain stimulation, which when instituted early in PD models has demonstrated the potential for neuroprotection. It remains to be examined if such hypometabolism is present in the stage of pre-motor symptoms of PD, as it would act as a powerful non-specific screening test for the disease. These alterations of cerebral glucose metabolism together with others could potentially be useful as a biomarker to predict disease onset and to evaluate disease progression. However, the main limitation of this study is that small number participants were recruited in the current study. Future study needs to include a large number of PD and VP patients. As the scramble to find predictors of VP and PD intensifies, a better understanding of the role of cerebral glucose metabolism may help give us insight into the pathogenesis of VP and PD.
In conclusion, to the best of our knowledge, this is the first report to use CMRGlc to clinically compare normal subjects, VP, and PD. This cross-sectional study demonstrated that motor and non-motor dysfunctions in patients with VP and PD are heralded by cerebral glucose hypometabolism in the frontal lobes and caudate putamen. Our surprising findings bring to light the fact that the observed changes in CMRGlc in the frontal lobes and the caudate putamen may provide a common foundation underlying mechanisms of glucose metabolism in patients with VP and PD, while the low levels of glucose metabolism in frontal lobe and caudate putamen can also be detected in early stages of disease. Our findings indicate that low levels of CMRGlc in the frontal lobes and the caudate putamen are good predictors for the severity and progression of VP and PD and may precede the clinical manifestation of these diseases.
Acknowledgments
YX, XW, XL, JL and QW conceived and designed the experiments. YX, XW, XL, CL, and JL performed the experiments. XL, XW, CD, YX, XM, DX, and QW analyzed the data. QW, CZ, TB, HZ, and YX contributed reagents/materials/analysis tools. QW, YX, MC, and AJF wrote the paper. The authors are grateful to Dr. Fengwei Zeng for the preparation of the FDG imaging for assessment on some patients.
This work was supported by the 973 Project (2011CB510000), National Natural Science Foundation of China (Grant NO: 81271427, 81471291), Scientific Research Foundation of Guangzhou (Grant NO: 2014J4100210), and Guangdong Department of Science & Technology Translational Medicine Center Grant (2011A080300002) to Q. W. YX was supported by NIH (HD-034852 and AT-004422) and the Vivian L. Smith Neurologic Foundation.
Financial Disclosures
The authors have reported no actual or potential conflicts of interest.
Contributor Information
Ying Xia, Email: Ying.Xia@UTH.TMC.edu.
Qing Wang, Email: denniswq@yahoo.com.
References
- [1].Aalto S,Bruck A,Laine M,Nagren K,Rinne JO (2005). Frontal and temporal dopamine release during working memory and attention tasks in healthy humans: a positron emission tomography study using the high-affinity dopamine D2 receptor ligand [11C]FLB 457. J Neurosci, 25:2471-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Baba T,Takeda A,Kikuchi A,Nishio Y,Hosokai Y,Hirayama K, et al. (2011). Association of olfactory dysfunction and brain. Metabolism in Parkinson's disease. Mov Disord, 26: 621-8. [DOI] [PubMed] [Google Scholar]
- [3].Backes H,Walberer M,Endepols H,Neumaier B,Graf R,Wienhard K, et al. (2011). Whiskers Area as Extracerebral Reference Tissue for Quantification of Rat Brain Metabolism Using 18F-FDG PET: Application to Focal Cerebral Ischemia. J Nucl Med, 52:1252-60. [DOI] [PubMed] [Google Scholar]
- [4].Benamer HT,Grosset DG (2009). Vascular parkinsonism: a clinical review. Eur Neurol, 61:11-5. [DOI] [PubMed] [Google Scholar]
- [5].Bohnen NI,Koeppe RA,Minoshima S,Giordani B,Albin RL,Frey KA, et al. (2011). Cerebral glucose metabolic features of Parkinson disease and incident dementia: longitudinal study. J Nucl Med, 52:848-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Borghammer P,Chakravarty M,Jonsdottir K Y,Sato N,Matsuda H,Ito K, et al. (2010). Cortical hypometabolism and hypoperfusion in Parkinson's disease is extensive: probably even at early disease stages. Brain Struct Funct, 214:303-17. [DOI] [PubMed] [Google Scholar]
- [7].Borghammer P,Cumming P,Østergaard K,Gjedde A,Rodell A,Bailey CJ, et al. (2012). Cerebral oxygen metabolism in patients with early Parkinson's disease. J Neurol Sci, 313:123-8. [DOI] [PubMed] [Google Scholar]
- [8].Braidy N,Gai W,Xu YH,Sachdev P,Guillemin GJ,Jiang XM, et al. (2013). Uptake and mitochondrial dysfunction of alpha-synuclein in human astrocytes, cortical neurons and fibroblasts. Transl Neurodegener, 2: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Brooks DJ,Pavese N (2011). Imaging biomarkers in Parkinson's disease. Prog Neurobiol, 95:614-28. [DOI] [PubMed] [Google Scholar]
- [10].Brownell AL,Canales K,Chen YI,Jenkins BG,Owen C,Livni E, et al. (2003). Mapping of brain function after MPTP-induced neurotoxicity in a primate Parkinson's disease model. Neuroimage, 20:1064-75. [DOI] [PubMed] [Google Scholar]
- [11].Buratti L,Viticchi G,Falsetti L,Cagnetti C,Luzzi S,Bartolini M, et al. (2014).Vascular impairment in Alzheimer's disease: the role of obstructive sleep apnea. J Alzheimers Dis, 38(2):445-53. [DOI] [PubMed] [Google Scholar]
- [12].Burghaus L,Eggers C,Timmermann L,Fink GR,Diederich NJ (2012). Hallucinations in neurodegenerative diseases. CNS Neurosci Ther, 18: 149-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Carbon M,Eidelberg D (2002). Modulation of regional brain function by deep brain stimulation: studies with positron emission tomography. Curr Opin Neurol, 15: 451-5. [DOI] [PubMed] [Google Scholar]
- [14].Chen Z,Zhong C (2013). Decoding Alzheimer's disease from perturbed cerebral glucose metabolism: implications for diagnostic and therapeutic strategies. Prog Neurobiol, 108: 21-43. [DOI] [PubMed] [Google Scholar]
- [15].Cropley V L,Fujita M,Bara-Jimenez W,Brown A K,Zhang X Y,Sangare J, et al. (2008). Pre- and post-synaptic dopamine imaging and its relation with frontostriatal cognitive function in Parkinson disease: PET studies with [11C]NNC 112 and [18F]FDOPA. Psychiatry Res, 163:171-82. [DOI] [PubMed] [Google Scholar]
- [16].De Reuck J,Siau B,Decoo D,Santens P,Crevits L,Strijckmans K, et al. (2001). Parkinsonism in patients with vascular dementia: clinical, computed- and positron emission-tomographic findings. Cerebrovasc Dis, 11: 51-8. [DOI] [PubMed] [Google Scholar]
- [17].De Reuck JL (2012). The significance of small cerebral bleeds in neurodegenerative dementia syndromes. Aging Dis, 3:307-12. [PMC free article] [PubMed] [Google Scholar]
- [18].Doorn KJ,Lucassen PJ,Boddeke HW,Prins M,Berendse HW,Drukarch B, et al. (2012). Emerging roles of microglial activation and non-motor symptoms in Parkinson's disease. Prog Neurobiol, 98:222-38. [DOI] [PubMed] [Google Scholar]
- [19].Duarte JM,Schuck PF,Wenk GL,Ferreira GC (2013). Metabolic disturbances in diseases with neurological involvement. Aging Dis, 5:238-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Eidelberg D,Moeller JR,Dhawan V,Spetsieris P,Takikawa S,Ishikawa T, et al. (1994). The metabolic topography of parkinsonism. J Cereb Blood Flow Metab, 14:783-801. [DOI] [PubMed] [Google Scholar]
- [21].Eidelberg D,Takikawa S,Moeller JR,Dhawan V,Redington K,Chaly T, et al. (1993). Striatal hypometabolism distinguishes striatonigral degeneration from Parkinson's disease. Ann Neurol, 33:518-27. [DOI] [PubMed] [Google Scholar]
- [22].Fahn S,Elton R, Members of the UPDRS development committee (1987). The unified Parkison disease rating scale. Macmillan Health Care Information. Recent developments in Parkinson's disease, 153-163 and 293-304 [Google Scholar]
- [23].Farkas E,Luiten P G (2001). Cerebral microvascular pathology in aging and Alzheimer's disease. Prog Neurobiol, 64: 575-611. [DOI] [PubMed] [Google Scholar]
- [24].Folstein MF,Folstein SE,McHugh PR (1975). "Mini-mental state". A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res, 12: 189-98. [DOI] [PubMed] [Google Scholar]
- [25].González-Redondo R,Toledo J,Clavero P,Lamet I,García-García D,García-Eulate R, et al. (2012). The impact of silent vascular brain burden in cognitive impairment in Parkinson's disease. Eur J Neurol, 19: 1100-7. [DOI] [PubMed] [Google Scholar]
- [26].Grammas P,Martinez J,Sanchez A,Yin X,Riley J,Gay D, et al. (2014). A new paradigm for the treatment of Alzheimer's disease: targeting vascular activation. J Alzheimers Dis, 40:619-30. [DOI] [PubMed] [Google Scholar]
- [27].Guan J,Pavlovic D,Dalkie N,Waldvogel HJ,O'Carroll SJ,Green CR, et al. (2013). Vascular degeneration in Parkinson's disease. Brain Pathol, 23:154-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hattingen E,Magerkurth J,Pilatus U,Mozer A,Seifried C,Steinmetz H, et al. (2009). Phosphorus and proton magnetic resonance spectroscopy demonstrates mitochondrial dysfunction in early and advanced Parkinson's disease. Brain, 132(Pt 12):3285-97. [DOI] [PubMed] [Google Scholar]
- [29].Heiss WD,Graf R,Löttgen J,Ohta K,Fujita T,Wagner R, et al. (1997). Repeat positron emission tomographic studies in transient middle cerebral artery occlusion in cats: residual perfusion and efficacy of postischemic reperfusion. J Cereb Blood Flow Metab, 17:388-400. [DOI] [PubMed] [Google Scholar]
- [30].Heller J,Dogan I,Schulz JB,Reetz K (2013). Evidence for gender differences in cognition, emotion and quality of life in Parkinson's disease? Aging Dis, 5:63-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hirano S,Eckert T,Flanagan T,Eidelberg D (2009). Metabolic networks for assessment of therapy and diagnosis in Parkinson's disease. Mov Disord, 24 Suppl 2: S725-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hoehn MM,Yahr MD (1967). Parkinsonism: onset, progression and mortality. Neurology, 17: 427-442. [DOI] [PubMed] [Google Scholar]
- [33].Ihara M,Tomimoto H,Ishizu K,Yoshida H,Sawamoto N,Hashikawa K, et al. (2007). Association of vascular parkinsonism with impaired neuronal integrity in the striatum. J Neural Transm, 114:577-84. [DOI] [PubMed] [Google Scholar]
- [34].Jellinger KA (2013). Pathology and pathogenesis of vascular cognitive impairment-a critical update. Front Aging Neurosci, 5:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Jesse S,Steinacker P,Lehnert S,Gillardon F,Hengerer B,Otto M (2009). Neurochemical approaches in the laboratory diagnosis of Parkinson and Parkinson dementia syndromes: a review. CNS Neurosci Ther, 15: 157-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kadir A,Nordberg A (2010). Target-specific PET probes for neurodegenerative disorders related to dementia. J Nucl Med, 51:1418-30. [DOI] [PubMed] [Google Scholar]
- [37].Kalbe E,Voges J,Weber T,Haarer M,Baudrexel S,Klein JC, et al. (2009). Frontal FDG-PET activity correlates with cognitive outcome after STN-DBS in Parkinson disease. Neurology, 72 42-49. [DOI] [PubMed] [Google Scholar]
- [38].Kalra S,Grosset DG,Benamer HT (2010). Differentiating vascular parkinsonism from idiopathic Parkinson's disease: a systematic review. Mov Disord, 25:149-56. [DOI] [PubMed] [Google Scholar]
- [39].Kono S,Ouchi Y,Terada T,Ida H,Suzuki M,Miyajima H (2010). Functional brain imaging in glucocerebrosidase mutation carriers with and without parkinsonism. Mov Disord, 25:1823-9. [DOI] [PubMed] [Google Scholar]
- [40].Lee PH,Yong SW,An YS (2008). Changes in cerebral glucose metabolism in patients with Parkinson disease with dementia after cholinesterase inhibitor therapy. J Nucl Med, 49:2006-11. [DOI] [PubMed] [Google Scholar]
- [41].Lewis SJ,Dove A,Robbins TW,Barker RA,Owen AM (2003). Cognitive impairments in early Parkinson's disease are accompanied by reductions in activity in frontostriatal neural circuitry. J Neurosci, 23:6351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Li H,Zhang M,Chen L,Zhang J,Pei Z,Hu A,Wang Q (2010). Nonmotor symptoms are independently associated with impaired health-related quality of life in Chinese patients with Parkinson's disease. Mov Disord, 25:2740-6. [DOI] [PubMed] [Google Scholar]
- [43].Lyoo CH,Jeong Y,Ryu YH,Rinne JO,Lee MS (2010). Cerebral glucose metabolism of Parkinson's disease patients with mild cognitive impairment. Eur Neurol, 64:65-73. [DOI] [PubMed] [Google Scholar]
- [44].Müller T (2012). Drug therapy in patients with Parkinson's disease. Transl Neurodegener, 1:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].McInnes J (2013). Insights on altered mitochondrial function and dynamics in the pathogenesis of neurodegeneration. Transl Neurodegener, 2:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Muresanu DF,Popa-Wagner A,Stan A,Buga AM,Popescu BO (2014). The vascular component of Alzheimer's Disease. Curr Neurovasc Res, 11:168-76. [DOI] [PubMed] [Google Scholar]
- [47].Murray DK,Sacheli MA,Eng JJ,Stoessl AJ (2014). The effects of exercise on cognition in Parkinson's disease: a systematic review. Transl Neurodegener, 3:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ohlin KE,Sebastianutto I,Adkins CE,Lundblad C,Lockman PR,Cenci MA (2012). Impact of L-DOPA treatment on regional cerebral blood flow and metabolism in the basal ganglia in a rat model of Parkinson's disease. Neuroimage, 61:228-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Owen AM (2004). Cognitive dysfunction in Parkinson's disease: the role of frontostriatal circuitry. Neuroscientist, 10:525-37. [DOI] [PubMed] [Google Scholar]
- [50].Piert M,Koeppe R A,Giordani B,Minoshima S,Kuhl D E (1996). Determination of regional rate constants from dynamic FDG-PET studies in Parkinson's disease. J Nucl Med, 37:1115-22. [PubMed] [Google Scholar]
- [51].Polito C,Berti V,Ramat S,Vanzi E,De Cristofaro MT,Pellicano G, et al. (2012). Interaction of caudate dopamine depletion and brain metabolic changes with cognitive dysfunction in early Parkinson's disease. Neurobiol Aging, 33: 206, e29-39. [DOI] [PubMed] [Google Scholar]
- [52].Powers WJ,Videen TO,Markham J,Black KJ,Golchin N,Perlmutter JS (2008). Cerebral mitochondrial metabolism in early Parkinson's disease. J Cereb Blood Flow Metab, 28:1754-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Puchades M,Sogn CJ,Maehlen J,Bergersen LH,Gundersen V (2013). Unaltered lactate and glucose transporter levels in the MPTP mouse model of Parkinson's disease. J Parkinsons Dis, 3:371-85. [DOI] [PubMed] [Google Scholar]
- [54].Rapoport SI,Nelson PT (2011). Biomarkers and evolution in Alzheimer disease. Prog Neurobiol, 95:510-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Saracchi E,Fermi S,Brighina L (2013). Emerging candidate biomarkers for Parkinson's disease: a review. Aging Dis, 5:27-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Shim YS,Roe CM,Buckles VD,Morris JC (2013). Clinicopathologic study of Alzheimer's disease: Alzheimer mimics. J Alzheimers Dis, 35:799-811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Spieles-Engemann AL,Behbehani MM,Collier TJ,Wohlgenant SL,Steece-Collier K,Paumier K, et al. (2010). Stimulation of the rat subthalamic nucleus is neuroprotective following significant nigral dopamine neuron loss. Neurobiol Dis, 39:105-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Targarona EM,Balague C,Pernas JC,Martinez C,Berindoague R,Gich I, et al. (2008). Can we predict immediate outcome after laparoscopic rectal surgery? Multivariate analysis of clinical, anatomic, and pathologic features after 3-dimensional reconstruction of the pelvic anatomy. Ann Surg, 247:642-9. [DOI] [PubMed] [Google Scholar]
- [59].Xu Y,Yan J,Zhou P,Li J,Gao H,Xia Y, et al. (2012). Neurotransmitter receptors and cognitive dysfunction in Alzheimer's disease and Parkinson's disease. Prog Neurobiol, 97:1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zhang L,Yan J,Xu Y,Long L,Zhu C,Chen X, et al. (2011). The combination of homocysteine and C-reactive protein predicts the outcomes of Chinese patients with Parkinson's disease and vascular parkinsonism. PLoS One, 6, e19333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Zijlmans JC,Daniel SE,Hughes AJ,Révész T,Lees AJ (2004). Clinicopathological investigation of vascular parkinsonism, including clinical criteria for diagnosis. Mov Disord, 19:630-40. [DOI] [PubMed] [Google Scholar]