
Fig. 11.
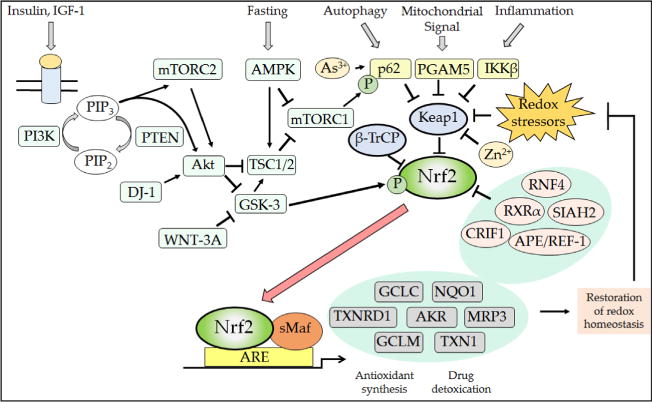
Posttranslational mechanisms that regulate Nrf2. The Nrf2 transcription factor is repressed under normal homeostatic conditions by the dual actions of β-TrCP and Keap1, thereby ensuring that the expression of antioxidant proteins and detoxication systems is restricted when the cell is not exposed to stress. However, numerous signaling processes allow derepression of Nrf2 by β-TrCP and Keap1, and this ensures the induction of ARE-driven genes to enable the elimination of electrophiles and oxidative stressors as well as metabolic adaptation to nutrients and growth cues. Thus, from the top left, the binding of insulin and growth factors to their cognate receptors will activate PI3K, which in turn increases Akt and mTORC2 activity while decreasing that of mTORC1 (not shown, but see Fig. 12). Increases in the PI3K–Akt–mTORC2 signaling system will inhibit GSK-3 activity, causing a decrease in the rate of β-TrCP-mediated Nrf2 turnover through reduced formation of the DSGIS-containing phosphodegron. In addition to insulin and growth factor signaling, the activity of mTORC1 is also decreased under conditions of low nutrient availability by the activation of AMPK. As shown top center, the activity of mTORC1 increases competitive inhibition of Keap1 by p62/SQSTM1 because it phosphorylates the STGE motif in the autophagy cargo receptor, which increases the affinity of p62/SQSTM1 for Keap1 and stimulates elimination of Keap1 by autophagy. Thus, during fasting, phosphorylation by mTORC1 of the STGE motif in p62/SQSTM1 is diminished and Keap1 is able to repress Nrf2 more effectively. As shown top right, signaling from autophagy, mitochondria, and inflammation pathways will diminish Keap1-mediated degradation of Nrf2 via competitive binding by p62/SQSTM1, PGAM5, and IKKβ to Keap1. Electrophiles and ROS also antagonize Keap1 substrate adaptor activity by modifying its sensor Cys-151, Cys-273, Cys-288, Cys-226/Cys-613, and Cys-434 residues. When small-molecule inducers inactivate Keap1, the conformation and/or orientation of the Keap1 dimer relative to other proteins within the Cul3–RBX1 complex is altered, producing a stalling of ligase activity that traps bound Nrf2 within CRLKeap1. As a consequence, newly translated Nrf2 evades degradation by CRLKeap1. This free fraction of Nrf2 accumulates and translocates to the nucleus where it heterodimerizes with sMaf proteins before binding to the ARE sequences in the promoters of target genes, causing induction of cytoprotective proteins. The increased expression of ARE-driven genes results in the synthesis and recycling of antioxidants, detoxification of harmful agents, and excretion of drugs. The physiological consequence of the induction of ARE-driven genes is the restoration of cellular redox homeostasis, through a negative feedback loop that ultimately allows Keap1 repression of Nrf2 to be reinstated through a variety of mechanisms that may include reactivation of oxidized Keap1 by TXNRD1, the induction of p62/SQSTM1 leading to increased degradation of electrophile-modified Keap1 by autophagy, and increased synthesis of Keap1.