

Abstract
Background:
Glucuronidation catalyzed by uridine 5’- diphospho-glucuronosyltransferase (UGT) is a major phase II drug metabolism reaction which facilitates drug elimination. Inhibition of UGT activity can cause drug-drug interaction. Therefore, it is important to determine the inhibitory potentials of drugs on glucuronidation.
Objective:
The objective was to evaluate the inhibitory potentials of mitragynine, 7-hydroxymitragynine, ketamine and buprenorphine, respectively on 4-methylumbelliferone (4-MU) glucuronidation in rat liver microsomes, human liver microsomes and recombinant human UGT1A1 and UGT2B7 isoforms.
Materials and Methods:
The effects of the above four compounds on the formation of 4-MU glucuronide from 4-MU by rat liver microsomes, human liver microsomes, recombinant human UGT1A1 and UGT2B7 isoforms were determined using high-performance liquid chromatography with ultraviolet detection.
Results:
For rat liver microsomes, ketamine strongly inhibited 4-MU glucuronidation with an IC50 value of 6.21 ± 1.51 μM followed by buprenorphine with an IC50 value of 73.22 ± 1.63 μM. For human liver microsomes, buprenorphine strongly inhibited 4-MU glucuronidation with an IC50 value of 6.32 ± 1.39 μM. For human UGT1A1 isoform, 7-hydroxymitragynine strongly inhibited 4-MU glucuronidation with an IC50 value of 7.13 ± 1.16 μM. For human UGT2B7 isoform, buprenorphine strongly inhibited 4-MU glucuronidation followed by 7-hydroxymitragynine and ketamine with respective IC50 values of 5.14 ± 1.30, 26.44 ± 1.31, and 27.28 ± 1.18 μM.
Conclusions:
These data indicate the possibility of drug-drug interaction if 7-hydroxymitragynine, ketamine, and buprenorphine are co-administered with drugs that are UGT2B7 substrates since these three compounds showed significant inhibition on UGT2B7 activity. In addition, if 7-hydroxymitragynine is to be taken with other drugs that are highly metabolized by UGT1A1, there is a possibility of drug-drug interaction to occur.
Keywords: 4-methylumbelliferone, 7-hydroxymitragynine, buprenorphine, glucuronidation, ketamine, mitragynine
INTRODUCTION
Ketum or Mitragyna speciosa Korth from Rubiaceae family is a tropical plant that can be found in the South-East Asia particularly in Thailand and Peninsular of Malaysia.[1] Traditionally, the ketum leaves have been used as a herbal drug to treat cough, diarrhea, hypertension, and to wean off morphine addicts.[2,3] Mitragynine, chemically known as 9-methoxy-corynantheidine, the major alkaloid constituent extracted from ketum leaves and 7-hydroxymitragynine, a minor constituent of ketum had been isolated.[4] Mitragynine had been obtained as a major constituent in ketum leaves as 66% of the total alkaloids in the Thai species and as 12% of the total alkaloids in the Malaysian species.[1] Meanwhile, 7-hydroxymitragynine is present up to 2% of total alkaloids content.[4] 7-hydroxymitragynine demonstrates 13 times higher analgesic potency than morphine and 46 folds higher potency than mitragynine.[5]
Recently, the use of ketum by drug users in Malaysia either as an opium substitute or as a stimulant had escalated.[6] It was also reported that 46% of ketum users are concomitantly taking other drugs[6] including ketamine which is one of the frequently abused drug.[7] In response to the drug abuse problem, buprenorphine, a semi-synthetic opioid was introduced in Malaysia to treat opioid addiction. These situations place individuals being treated with buprenorphine at risk for potentially toxic drug interactions when buprenorphine is ingested concomitantly by ketum users.
Drug-drug interactions may occur through several mechanisms, the main one is through the effects of these drugs on hepatic drug metabolism. Many drug interactions result from the inhibition or induction of enzymes that can have a significant influence on the use and safety of the drugs. Enzyme inhibition refers to the decrease in metabolic enzyme activity because of the presence of an inhibitor. Some drugs may inhibit the enzymes from metabolizing other drugs taken concomitantly causing them to remain active within the body for too long. This can cause high levels of the drug within the body which in turn can lead to adverse reactions and be harmful. Enzyme induction refers to the increase in enzyme activity caused by an inducer. Normally, enzyme induction is related to a reduction in the drug efficacy. However, it may also alter the toxicity of some substances.[8] The effects can take place on phase I metabolism (cytochrome P450 [CYP] enzymes) or on phase II metabolism (uridine 5’-diphospho [UDP]-glucuronosyltransferase [UGT] enzymes).
Glucuronidation is a major phase II metabolism reaction that facilitates detoxification and elimination efficiently for many drugs, numerous dietary, and endogenous compounds such as bilirubin and steroids.[9] Therefore, it is important to determine the inhibitory potentials of drugs on glucuronidation. The aim of this study is to evaluate the inhibitory potentials of the alkaloids from the ketum plant, which are mitragynine and 7-hydroxymitragynine, and the drug compounds which are ketamine and buprenorphine [Figure 1] on 4-methylumbelliferone (4-MU) glucuronidation in rat liver microsomes, human liver microsomes, and recombinant human UGT isoforms. Despite being a minor constituent of ketum, 7-hydroxymitragynine was chosen since it was found to be the most potent agonistic effect compared to the alkaloids present in ketum which consist of mitragynine, speciogynine, speciociliatine, and paynantheine. This suggests that the opioid effect of ketum is due to 7-hydroxymitragynine.[10] In this study, high-performance liquid chromatography (HPLC) will be used to measure the formation rate of 4-MU glucuronide (4-MUG) in the absence and presence of the drugs (potential inhibitors). The concentration of the drugs that reduces UGT enzyme activity by 50% (IC50) will be determined. 4-MU was chosen as the substrate because it is glucuronidated by various UGT isoforms and is considered to be a general substrate for UGTs.[11]
Figure 1.
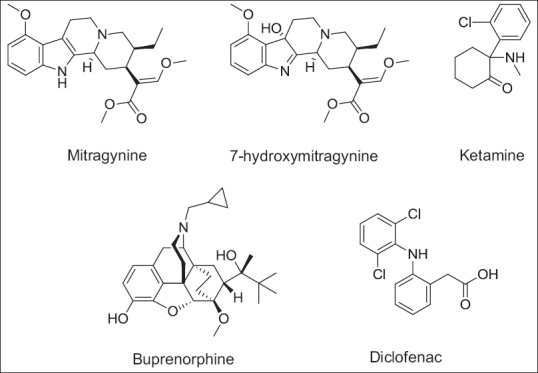
Chemical structures of potential inhibitory compounds investigated
MATERIALS AND METHODS
Chemicals and reagents
4-methylumbelliferone, 4-MU β-D-glucuronide (4-MUG), UDP-glucuronic acid (UDPGA, trisodium salt) and diclofenac (sodium salt) were purchased from Sigma-Aldrich (St. Louis, MO, United States). Ketamine hydrochloride and buprenorphine hydrochloride were purchased from Toronto Research Chemicals Inc. (North York, ON, Canada). Mitragynine (94.3% purity) was purchased from Chromadex (Irvine, CA, United States). 7-hydroxymitragynine (99% purity) was a kind gift from Assoc. Prof. Dr. Mohd Nizam Mordi (Centre for Drug Research, Universiti Sains Malaysia [USM]). Solvents and other reagents were of analytical reagent grade.
Enzyme sources
Male Sprague-Dawley rats weighing 150–200 g were obtained from the Animal House Unit of USM. Animals were housed in the animal room under controlled environment (temperature 25°C ± 2°C and 12 h light and dark cycle) and had free access to food and water ad libitum. Animals were acclimatized for 1-week before sacrificed. Animals were maintained and handled following the recommendations of the USM Ethical Committee which approved the design of the animal experiments with reference to the number USM/Animal Ethics Approval/2011/(72)(340).
Rat liver microsomes were prepared by differential centrifugation, as described by Azizi et al.[12] Briefly, rat livers were removed from the sacrificed animals, washed with ice-cold distilled water followed by ice-cold 67 mM potassium phosphate buffer (pH 7.4) containing 1.15% (w/v) KCl and then homogenized in three volumes of the same solution. The liver homogenates were centrifuged at 12,500 × g for 20 min at 4°C. The supernatants were further centrifuged at 100,000 × g for 60 min at 4°C, resulting in the sedimentation of the microsomal pellet, which was resuspended in 300 μL of 67 mM potassium phosphate buffer with 1.15% (w/v) KCl and 20% (v/v) glycerol. The rat liver microsomes were kept frozen at −80°C. Protein concentration of the rat liver microsomes was estimated by the method of Lowry et al.[13] using serum albumin as standard with slight modification according to Pomory.[14]
Pooled human liver microsomes were purchased from Sigma-Aldrich (St. Louis, MO, United States) and microsomes from baculovirus-infected insect cells expressing recombinant human UGT1A1 and UGT2B7 were obtained from BD Biosciences (Woburn, MA, United States).
4-Methylumbelliferone glucuronidation assay in rat liver microsomes and human liver microsomes
4-methylumbelliferone glucuronidation was evaluated in microsomal incubation using 4-MU as the probe substrate. Substrate concentration corresponds to the Km value.[15] Microsomal protein concentration, incubation time and percentage of detergent used (Triton X-100) for rat liver microsomes and human liver microsomes were optimized through range-finding studies (data not shown). The enzyme activity assay mixture (250 μL) contained Tris-HCl (100 mM) (pH 7.4), MgCl2 (5 mM), microsomal protein (0.25 mg/mL for rat liver microsomes or 0.1 mg/mL for human liver microsomes), Triton X-100 (0.01% for rat liver microsomes assay or 0.005% for human liver microsomes assay), UDPGA (3 mM), and 4-MU (substrate) (100 μM). After preincubation at 37°C for 5 min in a shaking water bath, reactions were initiated by the addition of UDPGA and were further incubated at 37°C for 30 min. Reactions were then terminated by the addition of 20% (w/v) trichloroacetic acid (50 μL). Samples were centrifuged (14,000 rpm for 5 min) and the supernatant fraction (20 μL) was injected into the HPLC column for analysis.
4-Methylumbelliferone glucuronidation assays in recombinant human uridine 5’- diphospho-glucuronosyltransferase isoforms (UGT1A1 and UGT2B7)
4-methylumbelliferone glucuronidation was measured using a previously published procedure[11] with slight modifications. The amount of protein, the incubation time and the concentration of 4-MU used in the measurement of 4-MU glucuronidation were described in the method by Uchaipichat et al.[11] Briefly, incubations (total volume 250 μL) contained phosphate buffer (100 mM) (pH 7.4), MgCl2 (5 mM), cell lysate (83.3 μg/mL for UGT1A1 or 62.5 μg/mL for UGT2B7), UDPGA (5 mM), and 4-MU (substrate) (100 μM for UGT1A1 assay or 350 μM for UGT2B7 assay). After preincubation at 37°C for 5 min in a shaking water bath, reactions were initiated by the addition of UDPGA and were further incubated at 37°C for 120 min. Reactions were then terminated by addition of 24% (v/v) perchloric acid (10 μL). Samples were centrifuged (6,200 rpm for 10 min) and the supernatant fraction (20 μL) was injected into the HPLC column for analysis.
4-Methylumbelliferone glucuronidation inhibition assays
For 4-MU glucuronidation inhibition assays performed in rat liver microsomes, human liver microsomes and recombinant human UGT1A1 and UGT2B7 isoforms, compounds that were investigated were diclofenac (positive inhibitor), mitragynine, 7-hydroxymitragynine, ketamine and buprenorphine. Diclofenac, ketamine and buprenorphine were fully dissolved in water. Mitragynine was prepared in methanol whereas 7-hydroxymitragynine was prepared in dimethyl sulfoxide (DMSO). Inhibitor was added to the incubation assay to reach the final desired concentrations. Final concentrations used for diclofenac, ketamine and buprenorphine in the inhibition screening experiments were 0.01, 0.1, 1, 10, 100, and 1000 μM, respectively. However, final concentrations used for mitragynine and 7-hydroxymitragynine were up to only 100 μM due to limited solubility of both of the compounds in methanol and DMSO, respectively. The final concentration of both solvents in the incubation mixture was 0.5% (v/v).
Apparatus and high-performance liquid chromatography conditions
High-performance liquid chromatography determination of 4-MUG was performed using a method modified from Lewis et al.[16] The peak areas of 4-MUG were analyzed using reversed-phase HPLC with ultraviolet (UV) detection. The HPLC system used was an Agilent 1200 series instrument consisting of G1322A degasser, G1311A quaternary pump, G1329B autosampler, G1315B diode array detector, and G1316A thermostatted column compartments. Chromatographic separation was achieved using a Gemini NX C18 column (4.6 mm × 150 mm, 5 μm particle sizes). The column was kept at 37°C and the column eluant was monitored by UV absorbance at a wavelength of 316 nm. The mobile phase was delivered at a flow rate of 1 mL/min using gradient elution consisting of solvent A (10% [v/v] acetonitrile in an aqueous solution of 10 mM triethylamine adjusted to pH 2.5 with 60% [v/v] perchloric acid) and solvent B (acetonitrile) in the (v/v) ratios of 95% A: 5% B from 0 to 7 min, 70% A: 30% B from 7.1 to 12 min, and 95% A: 5% B from 12.1 to 15 min. Concentrations of 4-MUG in incubation samples were quantified by comparison of peak areas to those of standard curves prepared over concentrations ranging from 0.5 to 100 μM.
Statistical analysis
Results are represented as mean ± standard error of mean (SEM) for three independent measurements (n = 3). IC50 values were calculated graphically on the basis of plots created using the GraphPad Prism 6 (Version 6.01, GraphPad Software, Inc., USA) under dose-response inhibition (log [inhibitor] vs. normalized response – -variable slope) model. One-way ANOVA followed by Dunnett test were performed using the same software to determine statistical significance between a range of 0–1000 μM for diclofenac, ketamine and buprenorphine and 0–100 μM for mitragynine and 7-hydroxymitragynine.
RESULTS
Inhibition of 4-methylumbelliferone glucuronidation by mitragynine, 7-hydroxymitragynine, ketamine, buprenorphine and diclofenac in rat liver microsomes, human liver microsomes and recombinant human uridine 5’- diphospho-glucuronosyltransferase isoforms (UGT1A1 and UGT2B7)
Figures 2-5 show the effects of five compounds, mitragynine, 7-hydroxymitragynine, ketamine, buprenorphine and diclofenac on the 4-MUG formation by rat liver microsomes, human liver microsomes, human UGT1A1 and human UGT2B7 isoforms, respectively.
Figure 2.
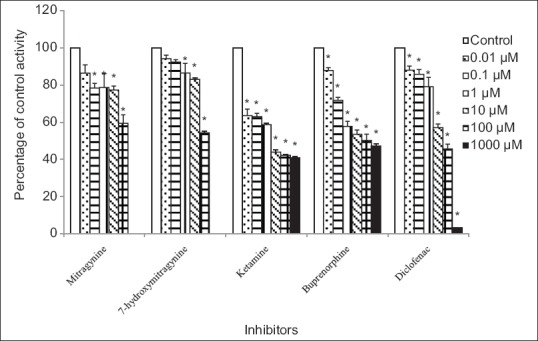
Inhibitory effects of mitragynine, 7-hydroxymitragynine, ketamine, buprenorphine, and diclofenac on 4-methylumbelliferone glucuronide formation by rat liver microsomes. Panels to the right show inhibitor concentrations. Each bar represents the mean percentage activity relative to control ± standard error of mean for three independent measurements (n = 3). Statistical analysis was conducted using one-way ANOVA and Dunnett test. *P < 0.05 versus control (no inhibitor)
Figure 5.
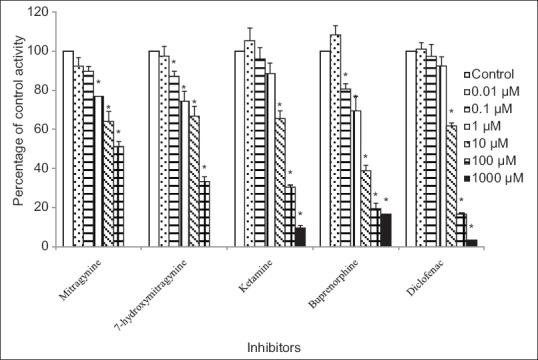
Inhibitory effects of mitragynine, 7-hydroxymitragynine, ketamine, buprenorphine, and diclofenac on 4-methylumbelliferone glucuronide formation by recombinant human UGT2B7 isoform. Panels to the right show inhibitor concentrations. Each bar represents the mean percentage activity relative to control ± standard error of mean for three independent measurements (n = 3). Statistical analysis was conducted using one-way ANOVA and Dunnett test. *P < 0.05 versus control (no inhibitor)
Using rat liver microsomes, diclofenac which was used as the positive inhibitor gave an IC50 value of 21.14 ± 1.32 μM [mean ± SEM, Table 1]. Among the other four compounds, ketamine and buprenorphine had the highest inhibition on 4-MU glucuronidation in rat liver microsomes. Buprenorphine showed inhibition with IC50 value of 73.22 ± 1.63 μM [mean ± SEM, Table 1], meanwhile, ketamine gave an IC50 value of 6.21 ± 1.51 μM [mean ± SEM, Table 1]. However, the IC50 values for the mitragynine and 7-hydroxymitragynine could not be determined accurately since their inhibitions on 4-MU glucuronidation were <50%. 4-MU glucuronidation was reduced by 7-hydroxymitragynine ranged from 6% ± 2% to 46% ± 1% and mitragynine ranged from 14% ± 4% to 40% ± 5% at concentrations of 0.01–100 μM [Figure 2].
Table 1.
Inhibitory effects of mitragynine, 7-hydroxymitragynine, ketamine, buprenorphine and diclofenac on 4-MUG formation by rat liver microsomes, human liver microsomes and recombinant human UGT1A1 and UGT2B7 isoforms

Using human liver microsomes, diclofenac showed inhibition with IC50 value of 86.44 ± 1.28 μM [mean ± SEM, Table 1]. Buprenorphine showed inhibition with IC50 value of 6.32 ± 1.39 μM [mean ± SEM, Table 1]. The percentage inhibition of UGT activity is varied from 16% ± 3% to 33% ± 4% for mitragynine, 8% ± 3% to 32% ± 1% for ketamine and 5% ± 1% to 17% ± 1% for 7-hydroxymitragynine at concentrations that ranged from 0.01 to 1000 μM for ketamine whereas 0.01 to 100 μM for mitragynine and 7-hydroxymitragynine [Figure 3]. The IC50 values for these three compounds toward 4-MU glucuronidation in human liver microsomes were all greater than the highest concentrations used (IC50 > 1000 μM for ketamine whereas IC50 > 100 μM for mitragynine and 7-hydroxymitragynine) since inhibition at more than 50% did not occur at the highest concentration [Table 1].
Figure 3.
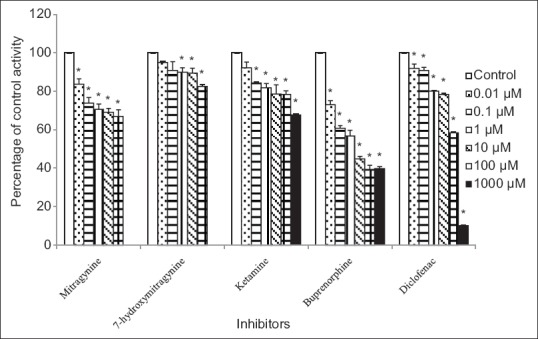
Inhibitory effects of mitragynine, 7-hydroxymitragynine, ketamine, buprenorphine, and diclofenac on 4-methylumbelliferone glucuronide formation by human liver microsomes. Panels to the right show inhibitor concentrations. Each bar represents the mean percentage activity relative to control ± standard error of mean for three independent measurements (n = 3). Statistical analysis was conducted using one-way ANOVA and Dunnett test. *P < 0.05 versus control (no inhibitor)
For the human UGT1A1 isoform, diclofenac gave an IC50 value of 2.34 ± 1.28 μM [mean ± SEM, Table 1]. Meanwhile, 7-hydroxymitragynine gave an IC50 value of 7.13 ± 1.16 μM [mean ± SEM, Table 1]. The percentage inhibition of UGT activity varied from 12% ± 4% to 46% ± 5% for buprenorphine and 9% ± 1% to 25% ± 1% for ketamine at concentrations that ranged from 0.01 to 1000 μM and 8% ± 6% to 35% ± 4% for mitragynine at concentrations that ranged from 0.01 to 100 μM [Figure 4]. For these three compounds, accurate IC50 values could not be determined because the maximum inhibition (>50%) did not occur at the highest concentration used. Therefore, the IC50 values for these three compounds toward 4-MU glucuronidation were all greater than the highest concentrations used (IC50 > 1000 μM for buprenorphine and ketamine whereas IC50 > 100 μM for mitragynine) [Table 1].
Figure 4.
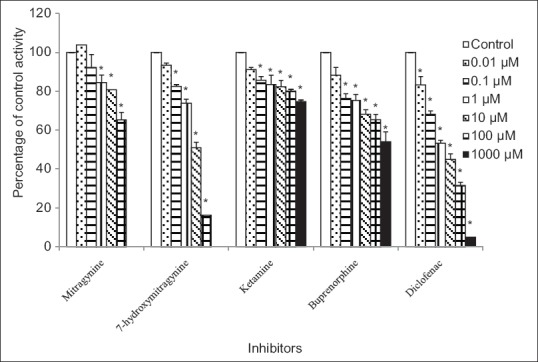
Inhibitory effects of mitragynine, 7-hydroxymitragynine, ketamine, buprenorphine, and diclofenac on 4-methylumbelliferone glucuronide formation by recombinant human UGT1A1 isoform. Panels to the right show inhibitor concentrations. Each bar represents the mean percentage activity relative to control ± standard error of mean for three independent measurements (n = 3). Statistical analysis was conducted using one-way ANOVA and Dunnett test. *P < 0.05 versus control (no inhibitor)
For the human UGT2B7 isoform, diclofenac gave an IC50 value of 16.97 ± 1.11 μM [mean ± SEM, Table 1]. Among the other four compounds, buprenorphine had the highest inhibition on 4-MUG formation by human UGT2B7 isoform followed by 7-hydroxymitragynine and ketamine. The IC50 values for buprenorphine, 7-hydroxymitragynine, and ketamine were 5.14 ± 1.30, 26.44 ± 1.31, and 27.28 ± 1.18 μM, respectively [mean ± SEM, Table 1]. However, the IC50 value for mitragynine could not be determined accurately since its inhibition on 4-MU glucuronidation was <50%. The percentage inhibition by mitragynine against UGT2B7 reducing 4-MU glucuronidation was from 8% ± 4% to 49% ± 3% at concentrations that ranged from 0.01 to 100 μM [Figure 5].
DISCUSSION
Various drugs have been identified as inhibitors using in vitro assays of UGT-mediated glucuronidation reactions in drug metabolism. In the present study, for all the inhibition assays, diclofenac which is a nonsteroidal anti-inflammatory drug (NSAID) was chosen as the positive inhibitor. Diclofenac, a NSAID is a comparatively potent UGT inhibitor for all UGT isoforms.[11] Therefore, the ability of the glucuronidation inhibition assay to detect the effect of test compounds on UGT was demonstrated by performing the assay in the presence of a known UGT inhibitor which is diclofenac.
Under the inhibition assay employed in this study, the result demonstrated that diclofenac, ketamine and buprenorphine had the highest inhibition on 4-MU glucuronidation in rat liver microsomes with the IC50 values of 21.14 ± 1.32, 6.21 ± 1.51, and 73.22 ± 1.63 μM, respectively. There is a differential degree of inhibition among the compounds. Hence, the IC50 value of diclofenac as a potent UGTs inhibitor was determined in order to compare the IC50 ‘s of buprenorphine and ketamine with its value. In this study, buprenorphine appears to be about three orders of magnitude less potent than diclofenac. Meanwhile, ketamine appears to be about three orders of magnitude more potent than diclofenac. In contrast, 7-hydroxymitragynine and mitragynine showed <50% inhibition at the highest concentration used. Therefore no IC50 values could be determined. However, 7-hydroxymitragynine and mitragynine gave the percentages inhibition of 46% and 40%, respectively at the highest concentration used. The inhibitory potential shown in rat liver microsomes was in order: Ketamine > buprenorphine > 7-hydroxymitragynine > mitragynine according to the IC50 values and percentage inhibition obtained.
The results obtained using rat liver microsomes showed a good inhibition on 4-MUG formation. In order to compare the inhibitory effects of these five compounds using human models, human liver microsomes were used. The results demonstrated that diclofenac and buprenorphine had the highest inhibition on 4-MU glucuronidation in human liver microsomes with the IC50 values of 86.44 ± 1.28 and 6.32 ± 1.39 μM, respectively. There is a differential degree of inhibition among these two compounds. Hence, the IC50 value of diclofenac as a potent UGTs inhibitor was determined in order to compare its IC50 to that of buprenorphine. The results illustrate that buprenorphine appears to be about 13 orders of magnitude more potent than diclofenac. However, ketamine, mitragynine, and 7-hydroxymitragynine did not exhibit strong inhibitory effects on 4-MUG formation by human liver microsomes. The inhibitory potential shown in human liver microsomes could be ranked as: Buprenorphine > mitragynine > ketamine > 7-hydroxymitragynine with the respective percentage inhibition of 60% ± 1%, 33% ± 4%, 32% ± 1%, and 17% ± 1% at the highest concentration used.
As a comparison, the inhibitory effects of the inhibitors observed for rat liver microsomes and human liver microsomes were different. This is explained by the fact that UGT isoforms are present at different levels in rat liver microsomes and human liver microsomes, thus, contributing to different glucuronidation capacities between the two species.[17]
Human liver microsomes used in the current study as the enzymes source contained a mixture of UGT isoforms. In order to investigate which specific UGT isoform was being inhibited by those four compounds in human liver microsomes, two recombinant human UGT isoforms, human UGT1A1, and human UGT2B7 were selected for further study. Both of these UGT isoforms are expressed in human liver. Human UGT1A1 was chosen since this UGT isoform is primarily responsible for the glucuronidation of bilirubin and many important clinical drugs such as irinotecan. In addition, of the top 200 drugs prescribed in the United States in 2002, 15% of the drugs undergo glucuronidation by UGT1A1 as their clearance mechanism, for example, levothyroxine, acetaminophen, and raloxifene.[18] Meanwhile, human UGT2B7 was chosen since this UGT isoform glucuronidates endogenous compound such as bile acid and retinoids and several xenobiotics including morphine, zidovudine, and NSAIDs. In fact, UGT2B7 is involved in the metabolism of 35% drugs responsible for glucuronidation of the top 200 prescribed drugs in the United States 2002.[18]
Using human UGT1A1 isoform as the source of enzyme, the results showed that 7-hydroxymitragynine had the highest inhibition on 4-MU glucuronidation. However, buprenorphine, ketamine, and mitragynine did not exhibit potent inhibitory effects on human UGT1A1 activity. From a previous report, it has been demonstrated that ketamine showed a weak inhibitory effect on 4-MU glucuronidation in human UGT1A1 isoform.[19] It is also reported that buprenorphine had no significant inhibitory effect on bilirubin glucuronidation catalyzed by UGT1A1 except at highly extraordinary concentrations.[20] 7-hydroxymitragynine which is the alkaloid that is present in a lesser amount in the ketum extract compared to mitragynine inhibited human UGT1A1 with an IC50 value of 7.13 ± 1.16 μM. There is no report on the effect of this compound on any UGT isoform activity. However, Azizi et al.[12] had reported that the ketum plant extracts had no significant inhibition on 4-nitrophenol UGT activity in rat liver microsomes but 4-nitrophenol UGT is mostly catalyzed by UGT1A6. 7-hydroxymitragynine which has a hydroxyl group at the C7 position of mitragynine [Figure 1] is susceptible to metabolism catalyzed by UGT (s). However, this requires further study.
Using human UGT2B7 isoform, the results demonstrated that buprenorphine, ketamine, and 7-hydroxymitragynine exhibited strong inhibition against human UGT2B7 whereas mitragynine exhibited only minor inhibitory effects. Among these three compounds, buprenorphine showed the highest inhibitory effect on 4-MUG formation by human UGT2B7 isoform followed by 7-hydroxymitragynine and ketamine.
In agreement with its effect on human liver microsomes, buprenorphine exhibited a strong inhibitory effect on the human UGT2B7 isoform. Buprenorphine inhibits 4-MUG formation by human UGT2B7 isoform with an IC50 value of 5.14 ± 1.30 μM. Buprenorphine is a potent opioid analgesic used in the treatment of moderate to severe pain and has also been used for the treatment of drug addiction. The glucuronidation of buprenorphine is primarily carried out by human UGT1A3 followed by human UGT2B7 and human UGT1A1 isoforms.[21] Based on the literature, the Km value for buprenorphine glucuronidation by human UGT2B7 isoform is 20.9 μM,[21] which is much lower than the Km value for 4-MU glucuronidation by human UGT2B7 isoform that is 335 μM.[11] Therefore, the affinity of buprenorphine toward human UGT2B7 is much higher than the affinity of 4-MU toward human UGT2B7, which might have resulted in inhibition of 4-MU glucuronidation by human UGT2B7.
7-hydroxymitragynine also had the highest inhibition on 4-MUG formation by human UGT2B7 isoform with an IC50 value of 26.44 ± 1.31 μM. The inhibition of this compound was also observed in human UGT1A1 isoform in this study but the IC50 value obtained was 7.13 ± 1.16 μM which was lower than found in human UGT2B7 isoform.
Similar to buprenorphine and 7-hydroxymitragynine, ketamine also inhibit 4-MUG formation by human UGT2B7 isoform in this study. Ketamine had been used clinically as an anesthetic and prescription agent. However, its abuse among drug addicts had recently escalated. In this study, ketamine inhibited the formation of 4-MUG with an IC50 value of 27.28 ± 1.18 μM. The result observed in this study concurred with a previous study for ketamine in which potent inhibition was observed with human UGT2B7 and human UGT2B15 isoforms with estimated IC50 values of 55 and 95 μM, respectively.[19] Previous data had shown that ketamine inhibited the formation of morphine-3-glucuronide in a non-competitive manner. It was interpretated that ketamine might have occupied a site on the UGT (s), which had caused a reduction in the formation of morphine-3-glucuronide and accounted for the interaction observed between these two compounds in the isolated perfused rat liver model.[22]
According to Krippendorff et al.,[23] the potential of CYP450 inhibition of compounds can be classified into low, medium or high potential inhibitors in drug-drug interactions based on their IC50 values. Compounds exhibiting IC50 values > 10 μM (IC50 > 10 μM) are low potential inhibitors. Meanwhile, compounds exhibiting IC50 values from 1 to 10 μM (1 μM < IC50 < 10 μM) are moderate potential inhibitors. However, the compounds exhibiting IC50 values < 1 μM (IC50 < 1 μM) are high potential inhibitors. Since the interpretation of UGT drug-drug interactions determined with UGT inhibitors is an ongoing and still unresolved challenge,[24] therefore, this classification may also be applicable and sufficient for ranking the compounds as potential inhibitors in the early drug discovery. Further evidence to support this statement can also be obtained from Oechsler and Skopp[25] and Venkatakrishnan et al.[26]
Another way to assess the possible clinical inhibitory potentials against drugs catalyzed mainly by human UGT1A1 and human UGT2B7 is by comparing the IC50 values of buprenorphine, ketamine, and 7-hydroxymitragynine with their therapeutic plasma concentrations on an unbound concentration basis if possible.
It has been reported that the mean plasma concentrations of buprenorphine were 37.52 ng/ml (0.08 μM) and 6.3 ng/ml (0.012 μM) after a single intravenous dose (1.2 mg) and a single sublingual dose (16 mg), respectively.[27,28] The IC50 value of buprenorphine obtained in this study was 5.14 μM which is much higher than the therapeutic unbound plasma concentration. Therefore, it is highly unlikely that buprenorphine would inhibit human UGT2B7 in clinical use.
Meanwhile, Idvall et al.[29] reported that a mean plasma concentration of ketamine was 9.3 μM during steady-state anesthesia after induction with 2 mg/kg and a maintenance dose of approximately 40 μg/kg/min. After a low-dose (0.125–0.250 mg/kg intravenous) administration, ketamine concentration of > 0.42 μM (100 μg/l) was found.[30] The IC50 value of ketamine obtained in this study was 27.28 μM which is higher than the therapeutic unbound plasma concentration in both studies. Therefore, we postulate that ketamine would also not inhibit human UGT2B7 in clinical setting.
Since there are no published reports on the therapeutic plasma concentration of 7-hydroxymitragynine and percent plasma unbound in human, the possible clinical inhibitory potentials of 7-hydroxymitragynine against drugs catalyzed mainly by human UGT1A1 and human UGT2B7 cannot be predicted at present.
CONCLUSIONS
To summarize, for rat liver microsomes, among the four compounds investigated, the highest inhibition was observed for ketamine followed by buprenorphine. For human liver microsomes, only buprenorphine gave strong inhibitory effect on 4-MU glucuronidation. Using human UGT1A1 isoform, only 7-hydroxymitragynine had the highest inhibitory potential, whereas using human UGT2B7 isoform, the highest inhibition was observed for buprenorphine followed by 7-hydroxymitragynine and ketamine. This in vitro study indicates that the interaction of these four compounds with rat liver microsomes, human liver microsomes and recombinant human UGT1A1 and UGT2B7 isoforms is complex and differs among the source of enzymes. However, these data indicate the possibility of drug-drug interaction if 7-hydroxymitragynine, ketamine and buprenorphine are co-administered with drugs that are UGT2B7 substrates since these three compounds showed significant inhibition on human UGT2B7 activity. In addition, there is also possibility of drug interaction if 7-hydroxymitragynine is to be taken with other drugs that are highly metabolized by UGT1A1.
Footnotes
Source of Support: Short term research grant from Universiti Sains Malaysia, Penang, Malaysia (Grant No: 304/CDADAH/6311105).
Conflicts of Interest: None declared.
REFERENCES
- 1.Takayama H. Chemistry and pharmacology of analgesic indole alkaloids from the rubiaceous plant, Mitragyna speciosa. Chem Pharm Bull (Tokyo) 2004;52:916–28. doi: 10.1248/cpb.52.916. [DOI] [PubMed] [Google Scholar]
- 2.Suwanlert S. A study of kratom eaters in Thailand. Bull Narc. 1975;27:21–7. [PubMed] [Google Scholar]
- 3.Jansen KL, Prast CJ. Ethnopharmacology of kratom and the Mitragyna alkaloids. J Ethnopharmacol. 1988;23:115–9. doi: 10.1016/0378-8741(88)90121-3. [DOI] [PubMed] [Google Scholar]
- 4.Ponglux D, Wongseripipatana S, Takayama H, Kikuchi M, Kurihara M, Kitajima M, et al. A new indole alkaloid, 7 alpha-Hydroxy-7H-mitragynine, from Mitragyna speciosa in Thailand. Planta Med. 1994;60:580–1. doi: 10.1055/s-2006-959578. [DOI] [PubMed] [Google Scholar]
- 5.Matsumoto K, Horie S, Ishikawa H, Takayama H, Aimi N, Ponglux D, et al. Antinociceptive effect of 7-hydroxymitragynine in mice: Discovery of an orally active opioid analgesic from the Thai medicinal herb Mitragyna speciosa. Life Sci. 2004;74:2143–55. doi: 10.1016/j.lfs.2003.09.054. [DOI] [PubMed] [Google Scholar]
- 6.Vicknasingam B, Narayanan S, Beng GT, Mansor SM. The informal use of ketum (Mitragyna speciosa) for opioid withdrawal in the northern states of peninsular Malaysia and implications for drug substitution therapy. Int J Drug Policy. 2010;21:283–8. doi: 10.1016/j.drugpo.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 7.Policy, Planning and Research Division; National Anti-drugs Agency. Laporan Dadah Bulan Disember 2013. NADA (Malaysia) 2013:1–39. [Google Scholar]
- 8.Leucuta SE, Vlase L. Pharmacokinetics and metabolic drug interactions. Curr Clin Pharmacol. 2006;1:5–20. doi: 10.2174/157488406775268183. [DOI] [PubMed] [Google Scholar]
- 9.Radominska-Pandya A, Bratton S, Little JM. A historical overview of the heterologous expression of mammalian UDP-glucuronosyltransferase isoforms over the past twenty years. Curr Drug Metab. 2005;6:141–60. doi: 10.2174/1389200053586127. [DOI] [PubMed] [Google Scholar]
- 10.Horie S, Koyama F, Takayama H, Ishikawa H, Aimi N, Ponglux D, et al. Indole alkaloids of a Thai medicinal herb, Mitragyna speciosa, that has opioid agonistic effect in guinea-pig ileum. Planta Med. 2005;71:231–6. doi: 10.1055/s-2005-837822. [DOI] [PubMed] [Google Scholar]
- 11.Uchaipichat V, Mackenzie PI, Guo XH, Gardner-Stephen D, Galetin A, Houston JB, et al. Human udp-glucuronosyltransferases: Isoform selectivity and kinetics of 4-methylumbelliferone and 1-naphthol glucuronidation, effects of organic solvents, and inhibition by diclofenac and probenecid. Drug Metab Dispos. 2004;32:413–23. doi: 10.1124/dmd.32.4.413. [DOI] [PubMed] [Google Scholar]
- 12.Azizi J, Ismail S, Mansor SM. Mitragyna speciosa korth leaves extracts induced the CYP450 catalyzed aminopyrine-N-demethylase (APND) and UDP-glucuronosyl transferase (UGT) activities in male Sprague-Dawley rat livers. Drug Metabol Drug Interact. 2013;28:95–105. doi: 10.1515/dmdi-2012-0039. [DOI] [PubMed] [Google Scholar]
- 13.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–75. [PubMed] [Google Scholar]
- 14.Pomory CM. Color development time of the Lowry protein assay. Anal Biochem. 2008;378:216–7. doi: 10.1016/j.ab.2008.04.015. [DOI] [PubMed] [Google Scholar]
- 15.Hanioka N, Jinno H, Tanaka-Kagawa T, Nishimura T, Ando M. Determination of UDP-glucuronosyltransferase UGT1A6 activity in human and rat liver microsomes by HPLC with UV detection. J Pharm Biomed Anal. 2001;25:65–75. doi: 10.1016/s0731-7085(00)00491-x. [DOI] [PubMed] [Google Scholar]
- 16.Lewis BC, Mackenzie PI, Elliot DJ, Burchell B, Bhasker CR, Miners JO. Amino terminal domains of human UDP-glucuronosyltransferases (UGT) 2B7 and 2B15 associated with substrate selectivity and autoactivation. Biochem Pharmacol. 2007;73:1463–73. doi: 10.1016/j.bcp.2006.12.021. [DOI] [PubMed] [Google Scholar]
- 17.Coughlin JL, Thomas PE, Buckley B. Inhibition of genistein glucuronidation by bisphenol A in human and rat liver microsomes. Drug Metab Dispos. 2012;40:481–5. doi: 10.1124/dmd.111.042366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams JA, Hyland R, Jones BC, Smith DA, Hurst S, Goosen TC, et al. Drug-drug interactions for UDP-glucuronosyltransferase substrates: A pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab Dispos. 2004;32:1201–8. doi: 10.1124/dmd.104.000794. [DOI] [PubMed] [Google Scholar]
- 19.Uchaipichat V, Raungrut P, Chau N, Janchawee B, Evans AM, Miners JO. Effects of ketamine on human UDP-glucuronosyltransferases in vitro predict potential drug-drug interactions arising from ketamine inhibition of codeine and morphine glucuronidation. Drug Metab Dispos. 2011;39:1324–8. doi: 10.1124/dmd.111.039727. [DOI] [PubMed] [Google Scholar]
- 20.Rios GR, Tephly TR. Inhibition and active sites of UDP-glucuronosyltransferases 2B7 and 1A1. Drug Metab Dispos. 2002;30:1364–7. doi: 10.1124/dmd.30.12.1364. [DOI] [PubMed] [Google Scholar]
- 21.Rouguieg K, Picard N, Sauvage FL, Gaulier JM, Marquet P. Contribution of the different UDP-glucuronosyltransferase (UGT) isoforms to buprenorphine and norbuprenorphine metabolism and relationship with the main UGT polymorphisms in a bank of human liver microsomes. Drug Metab Dispos. 2010;38:40–5. doi: 10.1124/dmd.109.029546. [DOI] [PubMed] [Google Scholar]
- 22.Qi X, Evans AM, Wang J, Miners JO, Upton RN, Milne RW. Inhibition of morphine metabolism by ketamine. Drug Metab Dispos. 2010;38:728–31. doi: 10.1124/dmd.109.030957. [DOI] [PubMed] [Google Scholar]
- 23.Krippendorff BF, Lienau P, Reichel A, Huisinga W. Optimizing classification of drug-drug interaction potential for CYP450 isoenzyme inhibition assays in early drug discovery. J Biomol Screen. 2007;12:92–9. doi: 10.1177/1087057106295897. [DOI] [PubMed] [Google Scholar]
- 24.Kiang TK, Ensom MH, Chang TK. UDP-glucuronosyltransferases and clinical drug-drug interactions. Pharmacol Ther. 2005;106:97–132. doi: 10.1016/j.pharmthera.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 25.Oechsler S, Skopp G. An in vitro approach to estimate putative inhibition of buprenorphine and norbuprenorphine glucuronidation. Int J Legal Med. 2010;124:187–94. doi: 10.1007/s00414-010-0418-8. [DOI] [PubMed] [Google Scholar]
- 26.Venkatakrishnan K, von Moltke LL, Obach RS, Greenblatt DJ. Drug metabolism and drug interactions: Application and clinical value of in vitro models. Curr Drug Metab. 2003;4:423–59. doi: 10.2174/1389200033489361. [DOI] [PubMed] [Google Scholar]
- 27.Kuhlman JJ, Lalani S, Magluilo J, Levine B, Darwin WD, Johnson RE, et al. Human pharmacokinetics of intravenous, sublingual and buccal buprenorphine. J Anal Toxicol. 1996;20:369–78. doi: 10.1093/jat/20.6.369. [DOI] [PubMed] [Google Scholar]
- 28.Greenwald MK, Johanson CE, Moody DE, Woods JH, Kilbourn MR, Koeppe RA, et al. Effects of buprenorphine maintenance dose on mu-opioid receptor availability, plasma concentrations, and antagonist blockade in heroin-dependent volunteers. Neuropsychopharmacology. 2003;28:2000–9. doi: 10.1038/sj.npp.1300251. [DOI] [PubMed] [Google Scholar]
- 29.Idvall J, Ahlgren I, Aronsen KR, Stenberg P. Ketamine infusions: Pharmacokinetics and clinical effects. Br J Anaesth. 1979;51:1167–73. doi: 10.1093/bja/51.12.1167. [DOI] [PubMed] [Google Scholar]
- 30.Clements JA, Nimmo WS. Pharmacokinetics and analgesic effect of ketamine in man. Br J Anaesth. 1981;53:27–30. doi: 10.1093/bja/53.1.27. [DOI] [PubMed] [Google Scholar]