

Summary
Maternal genomic imprints are established during oogenesis. Histone deacetylases (HDACs) 1 and 2 are required for oocyte development in mouse, but their role in genomic imprinting is unknown. We find that Hdac1:Hdac2−/− double mutant growing oocytes exhibit global DNA hypomethylation and fail to establish imprinting marks for Igf2r, Peg3, and Srnpn. Global hypomethylation correlates with increased retrotransposon expression and double-strand DNA breaks. Nuclear-associated DNMT3A2 is reduced in double mutant oocytes and injecting these oocytes with Hdac2 partially restores DNMT3A2 nuclear staining. DNMT3A2 co-immunoprecipitates with HDAC2 in mouse embryonic stem cells. Partial loss of nuclear DNMT3A2 and HDAC2 occurs in Sin3a−/− oocytes, which exhibit decreased DNA methylation of imprinting control regions for Igf2r and Srnpn, but not Peg3. These results suggest seminal roles of HDAC1/2 in establishing maternal genomic imprints and maintaining genomic integrity in oocytes mediated in part through a SIN3A complex that interacts with DNMT3A2.
Graphical Abstract

Introduction
During oogenesis in mouse, formation of a full-grown oocyte capable of undergoing maturation, fertilization, and embryogenesis to term entails epigenetic reprogramming of the genome, global changes in chromatin structure, and silencing of gene expression. DNA methylation, an epigenetic modification, is involved in chromatin remodelling and gene expression during oocyte development. For example, maternal genomic imprints are established in growing oocytes in a locus-dependent manner (Lucifero et al., 2004), and require DNA methyltransferase 3A (DNMT3A) and its cofactor DNMT3L (Bourc'his et al., 2001; Hata et al., 2002). In contrast, although we recently demonstrated seminal roles of histone acetylation during oogenesis (Ma et al., 2012; Ma and Schultz, 2013) histone post-translational modifications (PTMs), an important class of epigenetic modifications, have not been well studied during oocyte development. These PTMs are closely linked to transcriptional regulation and required for many biological processes, e.g., differentiation of pluripotent stem cells into specific tissue lineages (Bhaumik et al., 2007).
Gene expression can be modulated by an interplay between DNA methylation and histone acetylation (Cedar and Bergman, 2009). For example, unmethylated DNA is largely assembled in nucleosomes containing acetylated histones (transcriptionally permissive chromatin), whereas DNA methylated on identical sequences is assembled into nucleosomes containing non-acetylated histone H3 and H4 (transcriptionally repressive chromatin) (Hashimshony et al., 2003). Histone modifications also play a role in establishing the DNA methylation profile during early development. For example, DNMT3L, which lacks methyltransferase activity (Chedin et al., 2002), recruits DNMT3A to DNA by binding to unmethylated H3K4 in nucleosomes, which leads to de novo DNA methylation (Ooi et al., 2007). Furthermore, histone H3K9 trimethylation conferred by Suv39h histone methyltransferase (HMTases) directs DNMT3B-dependent DNA methylation at pericentric heterochromatin repeats in embryonic stem (ES) cells (Lehnertz et al., 2003). Taken together, these results suggest a causal relationship between DNA methylation and histone modifications that is partially mediated through related histone–modifying enzymes (Cedar and Bergman, 2009). Indeed, mouse oocytes lacking KDM1B (lysine demethylase 1B, a histone H3K4 demethylase) show a substantial increase in H3K4 methylation and fail to establish DNA methylation marks at a subset of imprinted genes, suggesting that H3K4 methylation affects DNA methylation imprints during oogenesis (Ciccone et al., 2009).
Deletion of Hdac1 and Hdac2 in mouse oocytes results in global histone hyperacetylation and a precocious decrease in global transcription that is likely a consequence of increased expression of Kdm5b that in turn promotes H3K4 demethylation (Ma et al., 2012). Demethylation of H3K4 in Hdac1/2 double mutant oocytes suggests that DNA methylation is likely perturbed in these oocytes in light of the interactions between these two epigenetic modifications (Ciccone et al., 2009; Ooi et al., 2007). In the present study we assess the effect of deleting Hdac1 and Hdac2 on DNA methylation in mouse oocytes.
Results
Deletion of Hdac1/2 results in global decrease of 5-methylcytosine
The global demethylation of H3K4 in Hdac1/2 double mutant oocytes (Ma et al., 2012) prompted us to investigate whether DNA methylation was also affected. We detected by immunocytochemistry a small but significant decrease (~15%) in 5-methylcytosine (5-mC) staining in Hdac1:2−/−oocytes (Fig. 1A, B). The decrease in 5mC could be due to its conversion to 5-hydroxymethylcytosine (5-hmC) by the Ten-eleven translocation methylcytosine dioxygenase (Tet) family of methylcytosine dioxygenases (Tahiliani et al., 2009). Immunostaining with an antibody that specifically recognizes 5-hmC demonstrated that such was unlikely the case (Fig. 1A, B).
Figure 1. Deletion of both Hdac1 and Hdac2 results in global decrease of 5-mC without affecting 5-hmC in oocytes.

(A) Immunocytochemical detection of 5-mC (green) and HDAC2 (red) in oocytes obtained from wild-type (WT) mice 12-days-of-age and Hdac1:2−/− mice. Shown are representative images and only the nucleus is shown. In this and other figures, the bar corresponds to 10 μm. Quantification of the 5-mC nuclear staining signals shown in lower panel, the nuclear staining intensity of 5-mC in the WT oocytes was set to 1. All data are expressed as mean ± SEM, * P<0.05. (B) Quantification of the 5-mC and 5-hmC nuclear staining signals in panel A. The nuclear staining intensity in the WT oocytes was set to 1 and the data are expressed as mean ± SEM, * P<0.05.
Maternally methylated ICRs are hypomethylated in Hdac1:2−/−oocytes
The global decrease in 5-mC in Hdac1:2−/−growing oocytes suggested that DNA methylation of maternal ICRs could be affected because maternal-specific DNA methylation and functional imprints are established during oocyte growth (Bao et al., 2000; Obata and Kono, 2002). The DNA methylation status of imprinted genes in Hdac1:2−/−growing oocytes > 50 μm in diameter (present in secondary follicles) was analyzed by bisufite sequencing. Oocytes of these diameters were used because DNA, which is demethylated during germ cell formation (Kafri et al., 1992), is remethylated only when oocytes have attained a diameter of ~50 μm (Lucifero et al., 2004). The maternally methylated Snrpn, Igf2r, and Peg3 ICRs were hypomethylated in mutant oocytes (Figs. 2A-C, p<0.05, χ2), whereas there were no differences in methylation at H19 ICR between wild-type (WT) and Hdac1:2−/−growing oocytes (Fig. 2D). These results strongly suggest that deletion of Hdac1 and Hdac2 in oocytes disrupts establishment of maternal genomic imprints.
Figure 2. DNA methylation analysis in Hdac1:2−/− growing oocytes.

Bisulfite sequencing analysis of DNA methylation at the ICRs of several maternally [Snrpn (A), Igf2r (B), Peg3 (C)] or paternally [H19 (D)] methylated genes and repetitive elements [Line1 [E), Iap (F)] in WT and Hdac1:2−/− growing oocytes obtained from mice 12 days-of-age. Open circles and filled circles represent unmethylated and methylated CpG sites, respectively, and each row represents data from a single DNA molecule.
During oocyte growth repetitive sequences undergo de novo DNA methylation (Lane et al., 2003). We observed a significant decrease in DNA methylation of long interspersed nuclear elements 1 (Line1)(p<0.05, χ2) whereas there was no significant decrease in DNA methylation of intracisternal A particle (Iap) elements in Hdac1:2−/− growing oocytes (Fig. 2E, F). This latter finding is consistent with Iap maintaining DNA methylation during primordial germ cell reprogramming and therefore does not require de novo DNA methylation during oocyte growth (Kafri et al., 1992; Seisenberger et al., 2012).
Increased retrotransposon expression and DNA DSBs in Hdac1:2−/−oocytes
DNA methylation appears to confer genomic stability and integrity, and DNA hypermethylation at repetitive elements is proposed to protect against expression of transposable elements and endogenous retroviruses (Rakyan et al., 2010; Wilson et al., 2007). The observed decrease in Line1 DNA methylation could therefore facilitate activation of previously silenced transposable elements. Accordingly, we analyzed expression of five retrotransposon families [Line1, Iap, mouse transcript (MT), and short interspersed nucleotide element (SinB1 and SinB2)] in growing Hdac1:2−/−oocytes and found a significant up-regulation of Line1 MT, SinB1 and SinB2 expression (Fig. 3A). Again, no significant change in DNA methylation at Iap elements in Hdac1:2−/−growing oocytes is consistent with unchanged Iap expression in these oocytes (Fig. 3A). These results suggest that HDAC1 and 2 are involved in maintaining transposable elements silencing in oocytes.
Figure 3. Increased expression of repetitive elements and incidence of DNA double-strand breaks (DSBs) in Hdac1:2−/− oocytes.

(A) Relative abundance of repetitive element mRNA in WT and Hdac1:2−/− growing oocytes obtained from mice 12 days-of-age. Data are expressed relative to that in WT oocytes as mean ± SEM. *, p< 0.05. (B) Immunocytochemical detection of DSBs with anti-γH2AX (green). DNA was detected with propidium iodide (red). Shown are representative images and only the nucleus is shown.
Transposable elements integrate into the genome at different sites to produce DNA double-strand breaks (DSBs) (Hedges and Deininger, 2007) and their reactivation generally coincides with elevated levels of DNA damage. Indeed, up-regulation of retrotransposons is associated with increased DSBs in mouse germ cells (Soper et al., 2008; Su et al., 2012). As anticipated there was an increase in nuclear DNA DSBs as detected by γH2AX levels in Hdac1:2−/− growing oocytes (Fig. 3B). Consistent with the increase in DNA damage, gene ontology (GO) analysis of our microarray data from Hdac1:2−/− oocytes showed that up-regulated genes were enriched in apoptosis and DNA damage response related categories ((Ma et al., 2012) and Fig. S1A). Moreover, the mRNA levels of major regulators of DNA damage response were significantly increased (Fig. S1B), suggesting that deletion of Hdac1 and 2 leads to pronounced DNA damage in oocytes, which is probably responsible for the increased incidence of apoptosis observed in Hdac1:2−/−growing oocytes (Ma et al., 2012).
Loss of nuclear DNMT3A in Hdac1/2 mutant oocytes
The maternal-specific de novo DNA methylation that occurs during oocyte growth and coincides with accumulation of transcripts of Dnmt3a, Dnmt3b, and Dnmt3l (Lucifero et al., 2004). Conditional knockout of these Dnmt3 genes in oocytes with Zp3-Cre demonstrates that de novo methylation by DNMT3A in growing oocytes is required to establish maternal imprints, whereas DNMT3B is dispensible for DNA methylation of imprinted genes and repetitive elements (Kaneda et al., 2010). A third member of the DNMT3 family, DNMT3-like (DNMT3L), which has no catalytic activity, functions as a regulator of DNMT3A and DNMT3B (Bourc'his et al., 2001; Hata et al., 2002). Because we observed hypomethylation in maternally methylated ICRs in Hdac1:2−/− oocytes we ascertained whether this defect was due to misregulation of these DNMT3 family methyltransferases.
We first examined expression of DNMT3A, DNMT3B and DNMT3L, as well as DNMT1, by immunoblot analysis (Fig. 4A). No difference in the amount of DNMT3A and DNMT1 was noted, but there was an ~2-fold increase in the amount of DNMT3B and a pronounced decrease in the amount of DNMT3L. The increase in DNMT3B and decrease in DNMT3L protein is consistent with increased Dnmt3b and decreased Dnmt3l transcript abundance, respectively, in Hdac1:2−/− oocytes (Ma et al., 2012). Furthermore, observing impaired DNA methylation in these oocytes despite an increase in the amount of DNMT3B is consistent with DNMT3B not being essential for de novo methylation during oocyte growth. Immunocytochemical detection of DNMT3B, DNMT3L and DNMT1 was consistent with the immunoblotting data (Fig. 4B). In marked contrast, the amount of nuclear-associated DNMT3A protein was significantly reduced (Fig. 4B), suggesting that DNMT3A was now present in the cytoplasm. Dilution of DNMT3A in the cytoplasm would account for the decreased immunocytochemical signal because the nuclear volume is ~3-5% that of total oocyte volume.
Figure 4. Expression of DNMT1, DNMT3A, DNMT3B and DNMT3L in Hdac1:2−/− oocytes and expressing Hdac2 in Hdac1:2−/− growing oocytes partially restores DNMT3A2 nuclear stainning.

(A) Relative amount of DNMT1, DNMT3A, DNMT3B and DNMT3L determined by immunoblot analysis. Extracts for immunoblot analysis were prepared from oocytes obtained from at least four WT or mutant mice 12 days-of-age and total protein extract equivalent to 150 oocytes was loaded per lane. The experiment was conducted 3 times, and similar results were obtained in each case. ACTB was used as a loading control. (B) Immunocytochemical detection of DNMT31, DNMT3A, DNMT3B and DNMT3L in WT and mutant oocytes obtained from mice 12-days-of-age. Shown are representative images and only the nucleus is shown. (C) Immunocytochemical detection of DNMT3A in oocytes obtained from mice 12-days-of-age and lacking different combinations of Hdac1 and Hdac2. Shown are representative images and only the nucleus is shown. DNA was counterstained with propidium iodide. (D) Quantification of the data shown in C in which the nuclear staining intensity of DNMT3A in WT oocytes was set to 1. All data are expressed as mean ± SEM. *, p < 0.05. (E) Mutant oocytes were injected with either a cRNA (0.4 μg/μl) encoding Hdac2 (Hdac1:2−/−-OE) or MiliQ water (Hdac1:2−/−-C) and incubated in CZB medium; controls were wild-type oocytes (WT-C). Oocytes were removed 30 h after injection for immunoctyochemical detection of DNMT3A. Shown are representative images. DNA was counterstained with propidium iodide. (F) Quantification of the data shown in panel A. Staining intensity of DNMT3A2 in WT oocytes was set to 1 and the data are expressed as mean ± SEM. Signal intensities relative to WT in Hdac1:2−/−-C and Hdac1:2−/−-OE are 45±4%, and 63±6% respectively. *, p< 0.02.
The loss of nuclear DNMT3A could be linked to loss of DNMT3L given that DNMT3L, which binds to H3K4, interacts with DNMT3A. Examination of the nuclear concentration of DNMT3A and DNMT3L in oocytes obtained from mice lacking different combinations of Hdac1 and Hdac2 minimized this possibility. The extent of decreased DNMT3A nuclear staining correlated with the extent of loss of Hdac1 and Hdac2 in oocytes; the nuclear signal intensity of DNMT3A was reduced by ~20% in Hdac2−/− oocytes, ~40% in Hdac1−/+:Hdac2−/− oocytes and ~60% in Hdac1:2−/− oocytes, respectively (Fig. 4 C, D), whereas, no change of nuclear signal intensity of DNMT3L was observed in Hdac2−/− or Hdac1−/+:Hdac2−/− oocytes (Fig. 4 C, D). A decrease in the nuclear signal for DNMT3L was only observed in double mutant oocytes, i.e., the decrease in nuclear DNMT3A preceded the loss of nuclear DNMT3L (Fig. 4C, D). The loss of nuclear DNMT3A in Hdac2−/− oocytes, but not in Hdac1−/−oocytes, is consistent with our previous proposal that HDAC2 is the major HDAC in oocytes (Ma et al., 2012; Ma and Schultz, 2013) and suggests that absence of HDAC2 underlies the decreased nuclear-associated DNMT3A signal. Consistent with this proposal is that a significant decrease in Snrpn DNA methylation was observed in Hdac2−/− the oocytes (Fig. S2). Taken together, a reduction of nuclear-localized DNMT3A could account for the observed defects in maternal genomic imprints.
A DNMT3A isoform termed DNMT3A2, which lacks the amino-terminal 219 amino acid residues of DNMT3A, is present in tissues containing cells that undergo active de novo methylation (Chen et al., 2002). DNMT3A2 protein is also highly expressed in ES cells (Chen et al., 2003) and prospermatogonia of mouse embryos (Sakai et al., 2004), and Dnmt3a2 is highly expressed in mouse ovaries (Chen et al., 2002). The DNMT3A species shown in Figure 4 has a molecular weight of ~100 kDa, which corresponds to that of DNMT3A2 (Fig. S3A).
Immunoblot analysis using an antibody that recognizes both DNMT3A and DNMT3A2 confirmed that the 100-kDa band detected in oocytes is DNMT3A2, as well as the dominant form in preimplantation embryos (Fig. S3A, B). Thus, DNMT3A2 is the major form of DNMT3A expressed in oocytes, a conclusion consistent with PCR results using sense primers specific for Dnmt3a and Dnmt3a2 and a common antisense primer (Fig. S3). The nuclear staining detected with the DNMT3A antibody in oocytes (Fig. 4B, C), therefore, likely represented DNMT3A2 and not DNMT3A.
Expression of HDAC2 in Hdac1:2−/− growing oocytes partially restores DNMT3A2 nuclear staining
Gene deletion experiments indicate that HDAC2 is the major HDAC in oocytes with respect to generating a phenotype (Ma et al., 2012; Ma and Schultz, 2013) and our analysis of mice with compound Hdac1/2 genotypes revealed that deletion of Hdac2, not Hdac1, in growing oocytes is largely responsible for the differences in the extent of loss of nuclear DNMT3A2 (Fig. 4C, D) and likely the defects in maternal genomic imprinting. To test whether HDAC2 was required for nuclear localization of DNMT3A2, we expressed HDAC2 in Hdac1:2−/− oocytes by injecting an Hdac2 cRNA in mutant oocytes. Following culture, the observed DNMT3A2 nuclear staining intensity was restored ~20% compared to controls (Fig. 4E, F), suggesting that HDAC2 is required for nuclear localization of DNMT3A. Attempts to express both HDAC1 and HDAC2 were unsuccessful because, for unknown reasons, little HDAC1 was detected following injecting an Hdac1 cRNA. Attempts to assess whether DNA methylation of ICRs of imprinted genes affected in Hdac1:2−/− oocytes was [partially] restored by bisufite sequencing were unsuccessful, likely due to the Hdac1:2−/− oocytes undergoing apoptosis during culture in vitro (Ma and Schultz, 2013).
HDAC2 and DNMT3A2 reside in a complex in mouse ES cells
The results described above suggest that nuclear localization of DNMT3A2 is a consequence of an interaction, direct or indirect, with HDAC2, and therefore independent of HDAC2 activity. Consistent with this proposal is that no nuclear loss of DNMT3A was observed when wild-type oocytes were treated with the HDAC inhibitor trichostatin A (TSA), which results in histone hyperacetylation (Fig. S4); TSA treatment would mimic loss of HDAC activity in Hdac1:2−/− oocytes.
To ascertain whether HDAC2 and DNMT3A2 reside in the same complex, we first conducted co-IP experiments using mouse ES cells because the amount of readily obtainable oocyte material is limiting; DNMT3A2 is the predominant isoform in ES cells (Chen et al., 2003). The ES cell lyste was immunoprecipitated with an anti-HDAC2 antibody and immunoblotting analysis of the immunoprecipitate with an anti-DMNT3A antibody detected DNMT3A2, whereas virtually no DNMT3A2 was detected in the control (Fig. S5A). Thus, HDAC2 and DNMT3A presumably reside in the same protein complex. When similar co-IP experiments were conducted with 1,800 full-grown oocytes, which corresponds to ~36-40 μg of protein, DNMT3A2 was not detected (Fig. S5B). This failure, however, likely reflected an insufficient amount of starting material because DNMT3A was not co-immunoprecipitated when this amount of ES cell protein extract was used (Fig. S5C). Taken together, the results suggest that HDAC2 and DNMT3A2 reside is a complex and that loss of HDAC2 in turn results in partial failure of DNMT3A2 to remain localized in the nucleus.
A SIN3A-DNMT3A2 complex mediates DNA methylation of imprinted loci
SIN3A is a major HDAC1 and 2-containing complex (Kadamb et al., 2013). To ascertain whether a SIN3A complex interacts with DNMT3A, likely via HDAC2 present in the complex, we analyzed oocytes in which Sin3a was conditionally deleted (Fig. 6A). Similar to double mutant Hdac oocytes, there was no change in the amount of DNMT3A2 or HDAC2 (or HDAC1) as detected by immunoblotting (Fig. 5A), but there was a decrease not only in nuclear DNMT3A2 but also nuclear HDAC2 (Fig. 5 B, C); there was no apparent decrease in the staining intensity of nuclear HDAC1. Analysis of DNA methylation in Sin3a−/− oocytes revealed no change in H19 DNA methylation as anticipated (Fig. 5G), but also no significant change in DNA methylation of Peg3, Line1, and Iap (Fig. 5F, H, I). There was, however, a significant decrease in DNA methylation of Snrpn (p < 0.01, χ2) (Fig. 5D) and Igf2r (p < 0.001, χ2) (Fig. 5E). Note that the decrease in DNA methylation for Snrpn and Igf2r was not as pronounced as that observed in Hdac1:2−/− oocytes. Results of these experiments suggest that a SIN3A complex containing HDAC2 is responsible for interacting with DNMT3A2 that mediates DNA methylation of a subset of genes.
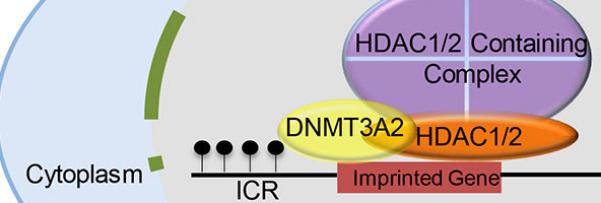
Figure 5. Expression of HDAC1, HDAC2 and DNMT3A2 and DNA methylation in Sin3a−/− Oocytes.

(A) Relative amount of SIN3A, HDAC1, HDAC2 and DNMT3A2 was determined by immunoblot analysis by using total protein extracts from WT and Hdac1:2−/− growing oocytes obtained from mice 12 days-of-age. Equal numbers (200) of oocytes were loaded per lane and and beta-actin (ACTB) was used as a loading control. The experiment was conducted 2 times, and similar results were obtained in each case. (B) Immunocytochemical detection of SIN3A, HDAC1, HDAC2 and DNMT3A2 in WT and Sin3a−/− oocytes obtained from mice 12-days-of-age. Shown are representative images and only the nucleus is shown. (C) Quantification of the data of HDAC2 and DNMT3A2 imunostaining shown in panel B. Nuclear staining intensity of HDAC2 and DNMT3A2 in WT oocytes was set to 1, and the data are expressed as mean ± SEM. Bisulfite sequencing analysis of DNA methylation at the ICRs of maternally methylated genes, Snrpn (D), Igf2r (E), Peg3 (F) or H19 (G) that is a paternally methylated gene, and repetitive elements, LINE1 (H) and IAP (I) in WT and Sin3a−/− growing oocytes obtained from mice 12 days-of-age. Filled circles represent methylated sites and open circles represent unmethylated sites. Each row represents data from a single DNA molecule. The numbers below each set of DNA strands indicate the percent of methylated CpG sites.
Discussion
We report that loss of Hdac1 and Hdac2 is associated not only with impaired acquisition of DNA methylation imprinting marks during oocyte growth but also with a mild reduction in global DNA methylation and increased expression of repetitive elements and compromised genomic integrity; loss of DNA methylation in repetitive elements likely accounts for their increased expression. The observed impairment of DNA methylation could arise from several sources in these mutant oocytes, e.g., changes in histone modification that modulate DNMT3A2 binding to nucleosomes; changes in gene expression that compromise the DNA methylation machinery; decreased transcription, a process recently implicated in DNA methylation in oocytes (Tomizawa et al., 2012); and failure of DNMT3A2 to localize to the nucleus as an outcome of its inability to interact with HDAC1/2 in an HDAC-containing complex.
Histones are hyperacetylated in oocytes deficient in HDAC1 and HDAC2 and given that acetylation and methylation on the same site are mutually exclusive histone acetylation can result in decreased histone methylation (Ma et al., 2012). Unmethylated histone H3K4 is required for binding not only of DNMT3L (Ooi et al., 2007) but also for DNMT3A (Hashimoto et al., 2010; Zhang et al., 2010) and presumably DNMT3A2. The increase in hyperacetylated H3K4 in Hdac1:2−/− oocytes could inhibit direct binding of DNMT3L, which would then recruit DNMT3A2 to nucleosomes, as well as DNMT3A2 and result in the observed reduced nuclear concentration of DNMT3A2. The loss of nuclear DNMT3A2 prior to that of DNMT3L in the allelic series of Hdac1 and Hdac2 mutants (Fig. 4) suggests that loss of DNMT3A2 is not coupled with decreased ability of DNMT3L to bind to acetylated H3K4-containing nucleosomes. In addition, loss of KDM1B, a histone H3K4 demethylase, has no effect on the amount and localization of DNMT3A2 and DNMT3L in growing oocytes (Ciccone et al., 2009).
DNMT3A (and presumably DNMT3A2) also directly interacts with histone H3K36me3 (Dhayalan et al., 2010), which is enriched in the body of transcribed genes (Rando, 2007), and could account for the widespread DNA methylation found in oocytes that extends far beyond imprinting control regions (Kobayashi et al., 2012; Smallwood et al., 2011). A decrease in H3K36me3 in mutant oocytes unlikely leads to a loss of nuclear DNMT3A2 because there is no apparent change in H3K36me3 in mutant oocytes (Ma et al., 2012). Taken together, these results suggest that changes in histone modification in mutant oocytes are not the primary cause for DNA hypomethylation.
Changes in expression of genes critical for DNA methylation mutant oocytes could lead to impaired DNA methylation during oocyte growth. We observed pronounced hypomethylation in the Srnpn ICR, which requires ZFP57 for proper DNA methylation (Li et al., 2008). Zfp57 expression, however, is not altered in Hdac1:2−/− mutants (Ma et al., 2012) and therefore it is unlikely that its misexpression is the molecular basis for hypomethylation of Srnpn. The decreased amount of DNMT3L in mutant oocytes, on the other hand, could lead to decreased DNMT3A2 activity that affects DNA methylation of both imprinted genes and non-imprinted chromosome regions. Although we cannot totally rule out this possibility, two lines of evidence suggest that such is not the case. First, we find no change in Dnmt3l transcript abundance in Hdac2−/− oocytes (Ma et al., 2012) but still observe a marked decrease in DNA methylation of the Snrpn ICR. Second, DNA methylation during oocyte growth requires DNMT3L. In oocytes, CpG islands associated with ICRs of maternal imprinted genes are methylated—CpG islands in somatic cells are typically not methylated—but the majority of DNA methylation occurs on CpG islands not associated with ICRs but rather within the gene body (Kobayashi et al., 2012; Smallwood et al., 2011). In addition, non-CpG methylation is common in oocytes (Shirane et al., 2013). It is unlikely that the decrease in DNMT3L protein in Hdac1:2−/− oocytes is the primary source of impaired DNA methylation because little DNA methylation of ICRs and intragenic regions, as well as non-CpGs, is observed in Dnmt3l−/− oocytes (Kobayashi et al., 2012; Smallwood et al., 2011) whereas only a modest decrease in global DNA methylation is observed in Hdac1:2−/− oocytes. Furthermore, only a subset of the genes we assayed for DNA methylation is hypomethylated in Sin3a−/− oocytes that exhibit no apparent change in nuclear concentration of DNMT3L.
Transcription is required to establish DNA methylation of certain ICRs in oocytes (Tomizawa et al., 2012). Transcription may also be linked to DNA methylation through remodeling histone modifications (e.g., the ability of DNMT3A to interact with histone H3K36me3, which marks actively transcribed genes) or creating chromatin domains permissive for de novo methylation (Tomizawa et al., 2012). Global transcription is reduced by ~40% (Ma et al., 2012) but this decrease is unlikely the major source of impaired DNA methylation in Hdac1:2−/− oocytes that only display a 15% decrease in global DNA methylation. In addition, although there is no decrease in transcription in Hdac2−/− oocytes (Ma and Schultz, 2013) we noted hypomethylation of Snrpn in these oocytes. Thus, a decrease in transcription-driven DNA methylation is unlikely the basis for impaired DNA methylation in Hdac1:2−/− oocytes.
The most likely cause for impaired DNA methylation in Hdac1:2−/− oocytes is the inability of DNMT3A2 to interact with HDAC2, which leads to less nuclear DNMT3A2. It is unlikely that HDAC activity is required because inhibiting HDAC activity with TSA does not lead to loss on nuclear DNMT3A2. In somatic cells, DNMT3A interacts with HDAC1 and HDAC2 (Ling et al., 2004) and presumably DNMT3A2 interacts with HDAC1/2 because the ATRX-homology domain required for this interaction (Fuks et al., 2001) is present in DNMT3A2. Although we were unable to demonstrate an interaction between DNMT3A2 and HDAC1/2 in oocytes due to limiting amounts of obtainable biological material, we find that DNMT3A2, the major DNMT3A species in oocytes, interacts with HDAC2 in ES cells in which DNMT3A2 is the major isoform. Also consistent with an interaction between DNMT3A2 and HDAC2 is that exogenous expression of HDAC2 in Hdac1:2−/− oocytes results in an increase in nuclear concentration of DNMT3A2. Finally, the decrease in nuclear DNMT3A2 and HDAC2 in Sin3a−/− oocytes and loss of DNA methylation in a subset of imprinted genes suggests that impaired DNA methylation observed in Hdac1:2−/− oocytes is due, at least in part, to DNMT3A2 interacting with a SIN3A complex.
In summary, we find a mechanistic linkage between HDAC1/2 in HDAC-containing complexes of which SIN3A is one and DNA methylation mediated by DNMT3A2 during oocyte growth. This DNA methylation is essential for establishing maternal imprinting DNA marks in ICRs as well as repressing expression of repetitive elements, whose expression can compromise genomic integrity. These data, in combination with our previous results (Ma et al., 2012; Ma and Schultz, 2013), suggest two distinct epigenetic activities of HDAC1/2 in oocytes. HDAC1/2 regulates chromosomal structure and gene expression by deacetylating histone or non-histone proteins through their catalytic activity and as structural components of HDAC-containing complexes HDAC1/2 are involved in de novo DNA methylation by recruiting DNMT3A2.
Experimental Procedures
Generation of mouse lines
Details for generating mutant mouse lines are described previously (Ma et al., 2012). Hdac1 or Hdac2 mutants in which the gene is deleted only oocytes are referred to as Hdac1−/− or Hdac2−/−, respectively; Hdac1-Hdac2 mutants (double mutant) are referred to as Hdac1:2−/−; Hdac1 heterozygotes-Hdac2 null oocytes are referred to as Hdac1−/+/Hdac2−/− and Hdac1 null-Hdac2 heterozygote oocytes are referred to as Hdac1−/−/Hdac2−/+ (Ma and Schultz, 2013). Female mice carrying Sin3a floxed alleles were crossed with Gdf9-Cre males to generate oocyte-conditioanl mutant Sin3a mice (Sin3a−/−); genotyping was performed as previously described (Dannenberg et al., 2005; Lan et al., 2004). Mice were maintained in accordance with institutional guidelines established by the University of Pennsylvania IACUC committee, and all experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Oocyte collection and culture
Growing oocytes were isolated from ovaries mutant and wild-type of mice 12 days-of-age and cultured as previously described (Ma et al., 2012). Full-grown oocytes were collected as previously described (Ma et al., 2012).
Microarray analysis
Preparation of oocyte RNA for microarray analysis was performed as previously described (Ma et al., 2012).
Oocyte microinjection
Growing oocytes were injected with 10 pl of Hdac2 cRNA as previously described (Ma and Schultz, 2013). Following microinjection, the oocytes were cultured in CZB medium (Chatot et al., 1989) for 30 h.
DNA methylation analysis
Oocytes were collected from mice 12 days-of-age and only oocytes whose diameters were > 50 μm were used for DNA methylation analysis. Genomic DNA was isolated from 60 growing oocytes with a QIAamp DNA micro kit (Qiagen) and subjected to bisulfite conversion with an EpiTect Bisulfate kit (Qiagen) according to the manufacturer's instructions with a minor modification as previously described (Ma et al., 2010). Target sequences of the bisulfite-converted DNA were amplified by PCR, and 24 clones for each sample were sequenced. The allele-specific DNA methylation patterns were examined for ICRs of H19 (Tremblay et al., 1997), Peg3 (Market-Velker et al., 2010), Igf2r (Sato et al., 2003), Line1 and IAPs (Lane et al., 2003) and Snrpn (Mann et al., 2004). The sequencing data was analyzed using the online tool BDPC (http://biochem.jacobs-university.de/BDPC/index.php) (Rohde et al., 2008). Strands from a PCR that contained an identical pattern of methylated cytosines and that could not be distinguished from other strands by polymorphisms were only counted once.
RNA extraction and RT-PCR
Total RNA was isolated from meiotically incompetent oocytes using the Absolutely RNA Microprep Kit (Stratagene, La Jolla, CA) according to the manufacturer's instructions. Details for RT-PCR are described in Supplemental Experimental Procedures.
Antibodies
The antibodies used in this study are described in Supplemental Experimental Procedures.
Immunostaining of oocytes and quantification of fluorescence intensity
Immunostaining of oocytes and quantification of fluorescence intensity was conducted as previously described (Ma et al., 2012); at least 20 oocytes per group were analyzed and the experiment conducted three times, except for Figure 5 in which 16 oocytes were analyzed and Figure 6 in which the experiment was conducted once.
Co-immunoprecipitation
Lysates were prepared from 106 ES cells and 1800 mouse full-grown oocytes as previously described (Ding et al., 2011). Immunoprecipitation was performed using a polyclonal anti-HDAC2 antibody (2545; Cell Signaling; IP: 1:50) in which a rabbit IgG (Santa Cruz, Dallas, Texas) served as the control. After incubating the sample overnight at 4°C with gentle rocking, previously washed protein A agarose beads (50% slurry, Millipore, Billerica, MA) were added to each sample, which was incubated at 4°C with gentle rocking for 3 h. The agarose beads were then collected by centrifugation at 12,000×g for 30 sec at 4° C, and the supernatants kept for a later immunoblot analysis. The protein A beads were washed with 500 μl of lysis buffer four times at 4° C, then 20 μl of 2x SDS sample buffer added and the samples heated at 95–100°C for 5 min. Immunoblot analysis was carried out as described above. The mouse ES cell lysate served as non-IP control. The experiment was conducted twice and similar results were obtained.
Immunoblot analysis
Protein samples from oocytes/ovary were solubilized in Laemmli sample buffer (Laemmli, 1970), resolved by SDS-PAGE (15% gel), and transferred to a PVDF membrane. Immunoblotting was then performed as previously described (Ma et al., 2012) except the membrane was incubated with a secondary antibody conjugated with horseradish peroxidase for 1 h and washed five times with TBST.
Statistics
Experiments were performed at least three times and the values are presented as mean ± SEM. All proportional data were subjected to an arcsine transformation before statistical analysis. Statistics were calculated with Microsoft Excel software. Differences in DNA methylation were determined by χ2. A P value of < 0.05 was considered to be statistically significant.
Supplementary Material
Acknowledgements
This research was supported by a grant from NIH to RMS (HD022681) and MSB and RMS (HD06817). EDW was supported by a Lalor Foundation Fellowship and an NIH Training Grant (5T32ES019851), and JRW was supported by an NIH Training Grant (5T32GM007229). The authors thank Eric N. Olson and Rusty L. Montgomery for providing mice containing floxed alleles of Hdac1 and Hdac2.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
PM designed and conducted experiments, interpreted data, and wrote manuscript; EDW conducted experiments; JRW conducted experiments; MSB designed experiments, interpreted data, and wrote manuscript; RMS designed experiments, interpreted data, and wrote manuscript.
References
- Bao S, Obata Y, Carroll J, Domeki I, Kono T. Epigenetic modifications necessary for normal development are established during oocyte growth in mice. Biol Reprod. 2000;62:616–621. doi: 10.1095/biolreprod62.3.616. [DOI] [PubMed] [Google Scholar]
- Bhaumik SR, Smith E, Shilatifard A. Covalent modifications of histones during development and disease pathogenesis. Nat Struct Mol Biol. 2007;14:1008–1016. doi: 10.1038/nsmb1337. [DOI] [PubMed] [Google Scholar]
- Bourc'his D, Xu GL, Lin CS, Bollman B, Bestor TH. Dnmt3L and the establishment of maternal genomic imprints. Science. 2001;294:2536–2539. doi: 10.1126/science.1065848. [DOI] [PubMed] [Google Scholar]
- Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- Chatot CL, Ziomek CA, Bavister BD, Lewis JL, Torres I. An improved culture medium supports development of random-bred 1-cell mouse embryos in vitro. J Reprod Fertil. 1989;86:679–688. doi: 10.1530/jrf.0.0860679. [DOI] [PubMed] [Google Scholar]
- Chedin F, Lieber MR, Hsieh CL. The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. Proc Natl Acad Sci U S A. 2002;99:16916–16921. doi: 10.1073/pnas.262443999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Ueda Y, Dodge JE, Wang Z, Li E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol Cell Biol. 2003;23:5594–5605. doi: 10.1128/MCB.23.16.5594-5605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Ueda Y, Xie S, Li E. A novel Dnmt3a isoform produced from an alternative promoter localizes to euchromatin and its expression correlates with active de novo methylation. J Biol Chem. 2002;277:38746–38754. doi: 10.1074/jbc.M205312200. [DOI] [PubMed] [Google Scholar]
- Ciccone DN, Su H, Hevi S, Gay F, Lei H, Bajko J, Xu G, Li E, Chen T. KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature. 2009;461:415–418. doi: 10.1038/nature08315. [DOI] [PubMed] [Google Scholar]
- Dannenberg JH, David G, Zhong S, van der Torre J, Wong WH, Depinho RA. mSin3A corepressor regulates diverse transcriptional networks governing normal and neoplastic growth and survival. Genes Dev. 2005;19:1581–1595. doi: 10.1101/gad.1286905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhayalan A, Rajavelu A, Rathert P, Tamas R, Jurkowska RZ, Ragozin S, Jeltsch A. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J Biol Chem. 2010;285:26114–26120. doi: 10.1074/jbc.M109.089433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Swain JE, Smith GD. Aurora kinase-A regulates microtubule organizing center (MTOC) localization, chromosome dynamics, and histone-H3 phosphorylation in mouse oocytes. Mol Reprod Dev. 2011;78:80–90. doi: 10.1002/mrd.21272. [DOI] [PubMed] [Google Scholar]
- Fuks F, Burgers WA, Godin N, Kasai M, Kouzarides T. Dnmt3a binds deacetylases and is recruited by a sequence-specific repressor to silence transcription. EMBO J. 2001;20:2536–2544. doi: 10.1093/emboj/20.10.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto H, Vertino PM, Cheng X. Molecular coupling of DNA methylation and histone methylation. Epigenomics. 2010;2:657–669. doi: 10.2217/epi.10.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimshony T, Zhang J, Keshet I, Bustin M, Cedar H. The role of DNA methylation in setting up chromatin structure during development. Nat Genet. 2003;34:187–192. doi: 10.1038/ng1158. [DOI] [PubMed] [Google Scholar]
- Hata K, Okano M, Lei H, Li E. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development. 2002;129:1983–1993. doi: 10.1242/dev.129.8.1983. [DOI] [PubMed] [Google Scholar]
- Hedges DJ, Deininger PL. Inviting instability: Transposable elements, double-strand breaks, and the maintenance of genome integrity. Mutat Res. 2007;616:46–59. doi: 10.1016/j.mrfmmm.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadamb R, Mittal S, Bansal N, Batra H, Saluja D. Sin3: insight into its transcription regulatory functions. Eur J Cell Biol. 2013;92:237–246. doi: 10.1016/j.ejcb.2013.09.001. [DOI] [PubMed] [Google Scholar]
- Kafri T, Ariel M, Brandeis M, Shemer R, Urven L, McCarrey J, Cedar H, Razin A. Developmental pattern of gene-specific DNA methylation in the mouse embryo and germ line. Genes Dev. 1992;6:705–714. doi: 10.1101/gad.6.5.705. [DOI] [PubMed] [Google Scholar]
- Kaneda M, Hirasawa R, Chiba H, Okano M, Li E, Sasaki H. Genetic evidence for Dnmt3a-dependent imprinting during oocyte growth obtained by conditional knockout with Zp3-Cre and complete exclusion of Dnmt3b by chimera formation. Genes Cells. 2010;15:169–179. doi: 10.1111/j.1365-2443.2009.01374.x. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Sakurai T, Imai M, Takahashi N, Fukuda A, Yayoi O, Sato S, Nakabayashi K, Hata K, Sotomaru Y, et al. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet. 2012;8:e1002440. doi: 10.1371/journal.pgen.1002440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lan ZJ, Xu X, Cooney AJ. Differential oocyte-specific expression of Cre recombinase activity in GDF-9-iCre, Zp3cre, and Msx2Cre transgenic mice. Biol Reprod. 2004;71:1469–1474. doi: 10.1095/biolreprod.104.031757. [DOI] [PubMed] [Google Scholar]
- Lane N, Dean W, Erhardt S, Hajkova P, Surani A, Walter J, Reik W. Resistance of IAPs to methylation reprogramming may provide a mechanism for epigenetic inheritance in the mouse. Genesis. 2003;35:88–93. doi: 10.1002/gene.10168. [DOI] [PubMed] [Google Scholar]
- Lehnertz B, Ueda Y, Derijck AA, Braunschweig U, Perez-Burgos L, Kubicek S, Chen T, Li E, Jenuwein T, Peters AH. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr Biol. 2003;13:1192–1200. doi: 10.1016/s0960-9822(03)00432-9. [DOI] [PubMed] [Google Scholar]
- Li X, Ito M, Zhou F, Youngson N, Zuo X, Leder P, Ferguson-Smith AC. A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev Cell. 2008;15:547–557. doi: 10.1016/j.devcel.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling Y, Sankpal UT, Robertson AK, McNally JG, Karpova T, Robertson KD. Modification of de novo DNA methyltransferase 3a (Dnmt3a) by SUMO-1 modulates its interaction with histone deacetylases (HDACs) and its capacity to repress transcription. Nucleic Acids Res. 2004;32:598–610. doi: 10.1093/nar/gkh195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucifero D, Mann MR, Bartolomei MS, Trasler JM. Gene-specific timing and epigenetic memory in oocyte imprinting. Hum Mol Genet. 2004;13:839–849. doi: 10.1093/hmg/ddh104. [DOI] [PubMed] [Google Scholar]
- Ma P, Lin S, Bartolomei MS, Schultz RM. Metastasis tumor antigen 2 (MTA2) is involved in proper imprinted expression of H19 and Peg3 during mouse preimplantation development. Biol Reprod. 2010;83:1027–1035. doi: 10.1095/biolreprod.110.086397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma P, Pan H, Montgomery RL, Olson EN, Schultz RM. Compensatory functions of histone deacetylase 1 (HDAC1) and HDAC2 regulate transcription and apoptosis during mouse oocyte development. Proc Natl Acad Sci U S A. 2012;109:E481–489. doi: 10.1073/pnas.1118403109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma P, Schultz RM. Histone deacetylase 2 (HDAC2) regulates chromosome segregation and kinetochore function via H4K16 deacetylation during oocyte maturation in mouse. PLoS Genet. 2013;9:e1003377. doi: 10.1371/journal.pgen.1003377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann MR, Lee SS, Doherty AS, Verona RI, Nolen LD, Schultz RM, Bartolomei MS. Selective loss of imprinting in the placenta following preimplantation development in culture. Development. 2004;131:3727–3735. doi: 10.1242/dev.01241. [DOI] [PubMed] [Google Scholar]
- Market-Velker BA, Zhang L, Magri LS, Bonvissuto AC, Mann MR. Dual effects of superovulation: loss of maternal and paternal imprinted methylation in a dose-dependent manner. Hum Mol Genet. 2010;19:36–51. doi: 10.1093/hmg/ddp465. [DOI] [PubMed] [Google Scholar]
- Obata Y, Kono T. Maternal primary imprinting is established at a specific time for each gene throughout oocyte growth. J Biol Chem. 2002;277:5285–5289. doi: 10.1074/jbc.M108586200. [DOI] [PubMed] [Google Scholar]
- Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z, Erdjument-Bromage H, Tempst P, Lin SP, Allis CD, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, Whittaker P, McCann OT, Finer S, Valdes AM, et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010;20:434–439. doi: 10.1101/gr.103101.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando OJ. Global patterns of histone modifications. Curr Opin Genet Dev. 2007;17:94–99. doi: 10.1016/j.gde.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Rohde C, Zhang Y, Jurkowski TP, Stamerjohanns H, Reinhardt R, Jeltsch A. Bisulfite sequencing Data Presentation and Compilation (BDPC) web server--a useful tool for DNA methylation analysis. Nucleic Acids Res. 2008;36:e34. doi: 10.1093/nar/gkn083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai Y, Suetake I, Shinozaki F, Yamashina S, Tajima S. Co-expression of de novo DNA methyltransferases Dnmt3a2 and Dnmt3L in gonocytes of mouse embryos. Gene Expr Patterns. 2004;5:231–237. doi: 10.1016/j.modgep.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Sato S, Yoshimizu T, Sato E, Matsui Y. Erasure of methylation imprinting of Igf2r during mouse primordial germ-cell development. Mol Reprod Dev. 2003;65:41–50. doi: 10.1002/mrd.10264. [DOI] [PubMed] [Google Scholar]
- Seisenberger S, Andrews S, Krueger F, Arand J, Walter J, Santos F, Popp C, Thienpont B, Dean W, Reik W. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol Cell. 2012;48:849–862. doi: 10.1016/j.molcel.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirane K, Toh H, Kobayashi H, Miura F, Chiba H, Ito T, Kono T, Sasaki H. Mouse oocyte methylomes at base resolution reveal genome-wide accumulation of non-CpG methylation and role of DNA methyltransferases. PLoS Genet. 2013;9:e1003439. doi: 10.1371/journal.pgen.1003439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood SA, Tomizawa S, Krueger F, Ruf N, Carli N, Segonds-Pichon A, Sato S, Hata K, Andrews SR, Kelsey G. Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat Genet. 2011;43:811–814. doi: 10.1038/ng.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soper SF, van der Heijden GW, Hardiman TC, Goodheart M, Martin SL, de Boer P, Bortvin A. Mouse maelstrom, a component of nuage, is essential for spermatogenesis and transposon repression in meiosis. Dev Cell. 2008;15:285–297. doi: 10.1016/j.devcel.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su YQ, Sugiura K, Sun F, Pendola JK, Cox GA, Handel MA, Schimenti JC, Eppig JJ. MARF1 regulates essential oogenic processes in mice. Science. 2012;335:1496–1499. doi: 10.1126/science.1214680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomizawa S, Nowacka-Woszuk J, Kelsey G. DNA methylation establishment during oocyte growth: mechanisms and significance. Int J Dev Biol. 2012;56:867–875. doi: 10.1387/ijdb.120152gk. [DOI] [PubMed] [Google Scholar]
- Tremblay KD, Duran KL, Bartolomei MS. A 5' 2-kilobase-pair region of the imprinted mouse H19 gene exhibits exclusive paternal methylation throughout development. Mol Cell Biol. 1997;17:4322–4329. doi: 10.1128/mcb.17.8.4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AS, Power BE, Molloy PL. DNA hypomethylation and human diseases. Biochim Biophys Acta. 2007;1775:138–162. doi: 10.1016/j.bbcan.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Jurkowska R, Soeroes S, Rajavelu A, Dhayalan A, Bock I, Rathert P, Brandt O, Reinhardt R, Fischle W, et al. Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res. 2010;38:4246–4253. doi: 10.1093/nar/gkq147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.