
FIGURE 2.
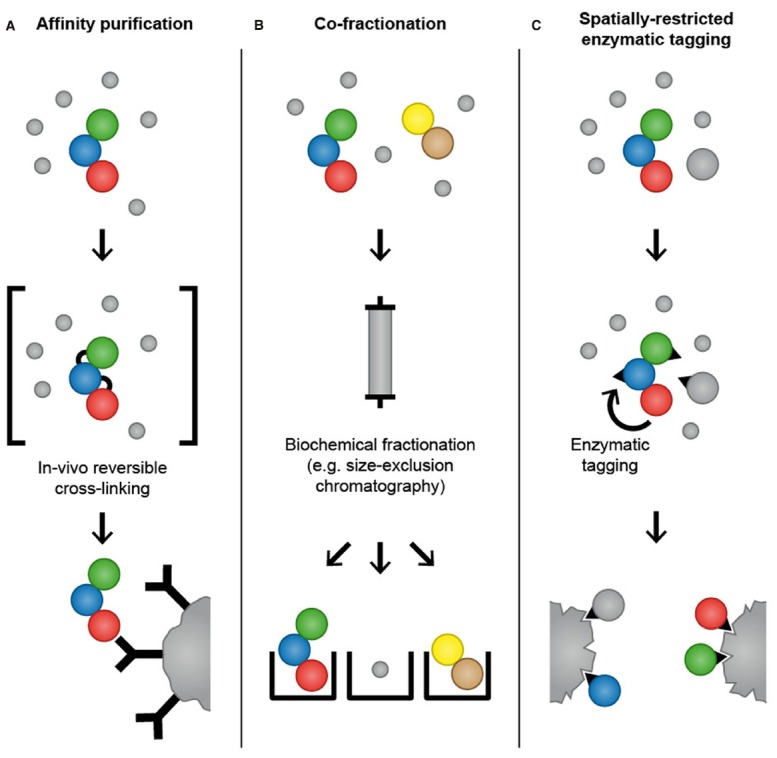
Mass spectrometry-based proteomics methods for analysis of temporal and spatial aspects of protein–protein interactions. In affinity purification approaches, an antibody that specifically binds to endogenously expressed bait protein is used to purify the protein of interest and its interaction partners. Alternatively, a bait protein fused to an epitope tag is ectopically expressed in cells and purified using affinity matrices or tag-specific antibodies. To increase the probability of capturing transient and weak interactions, chemicals that mediate protein–protein crosslinks can be applied to cells before lysis to “freeze” interactions by forming reversible covalent bonds between adjacent amino acids (A). In co-fractionation-based methods, proteins are subjected to extensive fractionation, for instance by high-performance size-exclusion chromatography, and the precise co-elution of two proteins is used as evidence for their interaction (B). In spatially restricted enzymatic tagging BirA* or APEX is fused to a protein of interest and ectopically expressed in cells. Biotinylation of proximal proteins is triggered by the addition of biotin for 24 h (BioID) or biotin-phenol for 1 min (APEX). Cells are lysed under denaturing conditions and biotinylated proteins are recovered using streptavidin followed by LC-MS/MS analysis (C).