
Fig. 3.
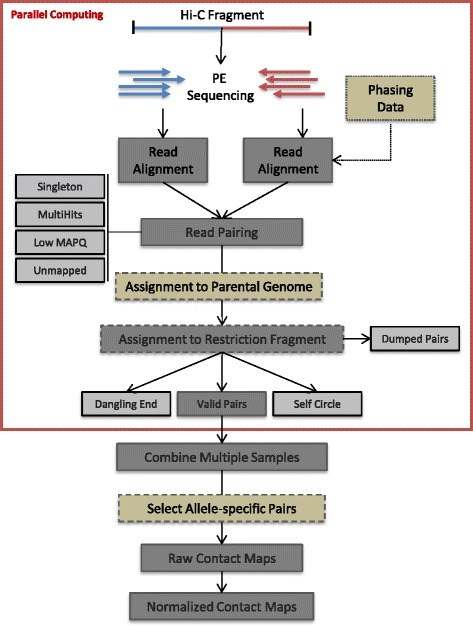
HiC-Pro workflow. Reads are first aligned on the reference genome. Only uniquely aligned reads are kept and assigned to a restriction fragment. Interactions are then classified and invalid pairs are discarded. If phased genotyping data and N-masked genome are provided, HiC-Pro will align the reads and assign them to a parental genome. For the Hi-C protocol based on restriction enzyme digestion, the read pairs will then be assigned to a restriction fragment and invalid ligation products will be filtered out. These first steps can be performed in parallel for each read chunk. Data from multiple chunks are then merged and binned to generate a single genome-wide interaction map. For allele-specific analysis, only pairs with at least one allele-specific read are used to build the contact maps. The normalization is finally applied to remove Hi-C systematic bias on the genome-wide contact map. MAPQ Mapping Quality , PE paired end