
Fig. 4.
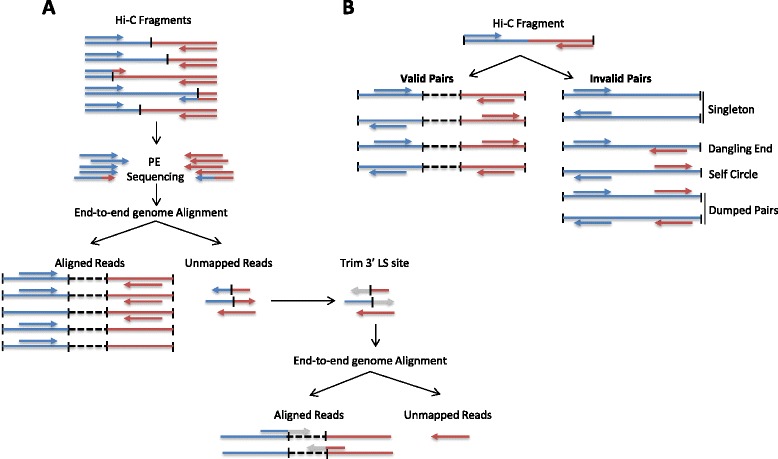
Read pair alignment and filtering. a Read pairs are first independently aligned to the reference genome using an end-to-end algorithm. Then, reads spanning the ligation junction which were not aligned in the first step are trimmed at the ligation site and their 5′ extremity is realigned on the genome. All aligned reads after these two steps are used for further analysis. b According to the Hi-C protocol, digested fragments are ligated together to generate Hi-C products. A valid Hi-C product is expected to involve two different restriction fragments. Read pairs aligned on the same restriction fragment are classified as dangling end or self-circle products, and are not used to generate the contact maps. PE paired end, LS Ligation Site