

Abstract
Overexpression of anti-apoptotic members of the BCL2 family has been found in all types of cancer. A member of the family, BCLxl (B-cell lymphoma extra-large), is known to be associated with the progression of leukemogenesis. In the present study, we focused on understanding the domains of BCLxl responsible for in vivo oncogenic potency. To this end, we utilized engineered BCLxl proteins with alternative transmembrane domains (TM) or chimeric BCLxl proteins containing domains from a less potent BCL2-like protein, BCLb. As expected, mice receiving MYC-only expressing bone marrow develop leukemia by 100 days, whereas co-expression of MYC with wild-type BCLxl led to aggressive myeloid leukemia with an average latency of ~25 days. Interestingly, mice injected with bone marrow co-expressing MYC and BCLxl targeted specifically to either mitochondria or ER also succumbed to leukemia with an average latency of ~25 days. Further, our study was extended to examine the role of the BH4 domain in driving potent leukemogenesis. Mice injected with bone marrow co-expressing MYC and BCLb succumb to leukemia in an average of ~55 days, but interestingly a BCLxl protein containing only the loop region of BCLb drove MYC-induced leukemogenesis with the same latency as wild-type BCLxl. These data suggest that the localization of exogenous BCLxl to either mitochondria or ER is not a steadfast dictator of in vivo oncogenic potency. Further, our findings suggest that the loop domain of BCLb and BCLxl is not responsible for dictating the in vivo leukemogeneic potency. This study provides further mechanistic details into the biochemical functions of BCLxl.
Keywords: apoptosis, leukemia, MYC, BH4, BCL2L1, BCL2L10
Introduction
The B-cell lymphoma 2 (BCL2) family of proteins plays a fundamental role in deciding the fate of cells when challenged with death stimuli by regulating mitochondrial outer membrane permeabilization (MOMP). The family can be divided into pro-apoptotic proteins like BAX, BAK, which potentiate MOMP, cytochrome c release and cell death, and anti-apoptotic proteins like BCL2, BCLxl, BCLw which inhibit MOMP to allow cell survival [1-4]. One remarkable feature of BCL2 family members is that they differentially interact with each other to control apoptosis [1, 3, 5, 6]. Modulating the ability of cells to undergo apoptosis is a key approach in the development of therapies for many diseases, including cancer [7-9]. Therefore, many studies have been previously directed to explore the role of BCL2 family members in driving tumorigenesis and to develop candidate molecules that inhibit anti-apoptotic BCL2-like proteins to be used as therapeutics against hematologic malignancies and solid cancers [3, 8-10]. Previous work from our laboratory directly determined the comparative in vivo oncogenic potency of all six anti-apoptotic proteins; BCL2, BCLxl/BCL2L1, BCLw/BCL2L2, BFL1/BCL2A1, MCL1 and BCLb/BCL2L10 [2]. Of importance, this work also demonstrated that although each anti-apoptotic BCL2-like gene can drive MYC-induced leukemogenesis, there were significant differences in the oncogenic potencies of each family member [2].
Individual anti-apoptotic BCL2 family members are characterized as having four conserved BCL2 homology (BH) domains known as BH1, BH2, BH3 and BH4 and a c-terminal α-helix transmembrane domain (TM) that is imperative for its localization to the mitochondrial outer membranes, endoplasmic reticulum (ER) membranes, and nuclear membranes. The variations in the domains present on each of the members is likely responsible for the structural and functional activity of these proteins. For example, the BH1, BH2 and BH3 domains of all six anti-apoptotic BCL2-like proteins are extremely conserved while, the n-terminal BH4 domain is much less well conserved. The lack of conservation within the BH4 domain would suggest that this domain could be a significant factor in determining how potently each individual BCL2-like gene can drive leukemogenesis [1, 3, 4, 7, 9-12].
There are numerous studies providing evidence that BCL2 family members are localized not only on mitochondrial outer membranes, but also endoplasmic reticulum (ER) or nuclear membranes [3, 6, 13]. Localization at the mitochondrial outer membranes has been shown to be important for inhibiting apoptosis, whereas localization to the ER may be important for regulating ER homeostasis and calcium storage [6, 13, 14]. The in vivo significance of targeting BCLxl to individual sub-cellular compartments has not been examined.
Overexpression of BCLxl has been found in many human cancers. Increased level of BCLxl results in decreased overall patient survival and is a marker for tumor progression [2, 15-18]. In addition, the increased level of BCLxl protein causes tumor cells to gain resistance to chemotherapeutic drugs and inhibit tumor necrosis factor (TNF) related or p53 mediated apoptosis [15, 19]. Studies suggest that inhibition of anti-apoptotic BCL2-like proteins with BH3-mimetics is a potential therapeutic approach. For example, inhibiting BCL2, BCLxl and BCLw with ABT-737 (or ABT-263) sensitizes many different cancer cell lines to apoptosis [20-22]. These findings support the importance of exploring the detailed biochemical mechanisms by which anti-apoptotic BCL2-like family members drive leukemogenisis.
To date, most studies examining the anti-apoptotic ability of mutants of BCL2-like members have been exclusively performed in cell culture with defined apoptotic stimuli. The contribution of the different domains in vivo in the regulation of apoptosis is largely unknown. Therefore, in the present study we wanted to explore the contribution of the loop domain of BCLb and subcellular localization of BCLxl on in vivo oncogenic potency. Using our previously published MYC-induced leukemogenesis assay [23], we demonstrate that while the BH4 domain of BCLxl is critical for determining full oncogenic potency, the intracellular localization of BCLxl does not impact oncogenic potency. Taken together these studies suggest that understanding the biochemical details of how individual BCL2-like family members regulate apoptosis and tumorigenesis will provide insight into mechanisms that regulate apoptosis.
Results and Discussion
Targeting BCLxl only to mitochondria or ER does not alter in vivo oncogenic potency
Previous studies have shown that targeting exogenous BCLxl to the mitochondria or the ER does not alter the apoptotic response to toxic stimuli of mouse embryonic fibroblasts (MEF) cells expressing endogenous BCLxl [6]. In the same study, researchers targeted BCLxl to the mitochondria or ER in MEFs deficient for endogenous BCLxl. These studies demonstrated that endogenous BCLxl was required for the ability of ER targeted BCLxlcb5 to block apoptosis, whereas endogenous BCLxl was dispensable for mitochondrially targeted BCLxlActA to inhibit cell death [6, 24]. Herein, we wanted to determine the oncogenic potency of these BCLxl mutants in an in vivo MYC-induced model of leukemogenesis. We subcloned the described BCLxl constructs (BCLxlWT, BCLxldTM, BCLxlActA, and BCLxlcb5) into a retroviral plasmid expressing the tetracycline transactivator (tTA) (Figure 1a and 1b). To confirm construct overexpression, HEK293T cells were transfected and western blot analysis was performed (Figure 1c). Retroviruses expressing tTA and BCLxlWT, BCLxldTM, BCLxlcb5, BCLxlActA or GFP were used to infect bone marrow harvested from mice harboring a tetracycline transactivator (tTA)-dependent MYC allele (TET-O-MYC). The MIT-BCLxl retrovirus allows for constitutive co-expression of MYC and the individual BCLxl constructs. After infection of the bone marrow and transplantation into syngeneic recipients, mice were monitored for disease progression and then euthanized for further analysis and their survival noted on a Kaplan-Meier curve (Figure 1d). Previous studies have shown that BCLxlWT and MYC co-expression causes lethal myeloid leukemia in less than 30 days [2]. Mice expressing only MYC usually develop myeloid leukemia with a long latency, but in some instances mice will develop CD4+ T-cell leukemia, judged by a large abberant population of T-cells in the bone marrow, as we have shown previously [2, 23, 25] In this study, mice that received cells co-expressing MYC and BCLxl constructs accelerated leukemogenesis compared to mice that received cells co-expressing MYC and GFP (Figure 1d). All BCLxl constructs accelerated disease in the mice, when compared to GFP, suggesting that oncogenic potency is not affected by BCL localization, in vivo. These data are consistent with previous in vitro biochemical studies using the same constructs [6]. Interestingly, BCLxl confined to the cytosol only (BCLxldTM), accelerated leukemogenesis compared to GFP, but subjects showed a longer survival (~60 days) compared to the mitochondrial or ER targeted constructs. To demonstrate that all BCLxl constructs were expressed at similar levels in the leukemias, spleen samples were examined by western blot analysis (Figure 1e). All leukemic spleens showed a comparable increase in the expression of BCLxl compared to non-leukemic spleens or a spleen from a leukemic spleen expressing MYC and GFP. The leukemia that developed in all mice was consistent with our previous observations. The mice developed an acute myeloid leukeima (AML) defined by an increase in cells with the myeloid cell markers GR-1 and CD11b in the spleen and bone marrow (Figure 2a), an enlarged spleen and leukemic infiltration of the blood, liver, and spleen also confirm the cancerous disease (Figure 2b).
Figure 1. Targeting BCLxl to mitochondria or ER does not alter in vivo oncogenic potency.

a) Schematic of BCLxl constructs (BCLxlwt; BCLxldTM; BCLxlActA and BCLxlcb5) used in the study. b) Schematic of MIT-RX retroviral plasmid used in the studies. c) Western blot of lysates expressing BCLxl constructs. d) Kaplan-Meier survival curve of mice receiving Tet-O-MYC bone marrow infected with the indicated retrovirus [23]. Mantel-Cox Log-rank test was used to determine statistical significance, compared to BCLxlwt survival (BCLxldTM: p=<0.0001; BCLxlActA: p=0.063; BCLxlcb5: p=0.324; GFP: p=<0.0001).e) Western blot of splenocytes from leukemic mice.
Figure 2. Characterization of leukemias that arise in mice receiving Tet-O-MYC bone marrow infected with MIT-BCLxl retroviruses.

a) Flow cytometry of bone marrow (BM), thymus and splenocytes from a non-manipulated 12 week old FVB/n mouse or a representative MIT-BCLxl leukemic mouse. Percentages indicate the percent of GR1/CD11b and CD4/CD8 cells in each sample, as determined by FACS analysis. b) Histological analysis from a non-manipulated 12 week old FVB/n mouse or a representative MIT-BCLxl leukemic mouse.
The result that oncogenic potency is not modified when BCLxl is targeted to different organelles may be somewhat expected based on our previous observations in MEF cells [6]. However, it is interesting to note that there is a correlation between apoptotic sensitivity in vitro and oncogenic potency in vivo. This correlation could be further investigated by implementing the exogenous BCLxl constructs into BCLxlWT deficient hematopoietic progenitor cells.
Oncogenic role of the loop domain of BCLb
If BCLxl localization to ER or mitochondria does not alter oncogenic potency, then what does? It has been shown previously that a BH4 domain peptide is sufficient for anti-apoptotic activity in mouse models [26] but the role of the loop domain of BCL2-like proteins in oncogenic transformation is unknown. We therefore wanted to determine the role that the BCLb loop domain plays in dictating in vivo oncogenic potency of BCLxl. We developed a novel chimeric gene that has the loop domain from the less oncogenically potent BCLb engineered onto BCLxl. BCLb has been shown in previous studies, that although its expression is up regulated in some leukemic tumors, it is a less potent oncogene than BCLxl, in the same TET-O-MYC mouse model [2]. The n-terminal loop domain (loop) from BCLb was engineered to replace the corresponding domains of BCLxl (Figure 3a). To investigate if the stability of the BCLxl/b construct is altered in vitro, we treated cells expressing either wild-type BCLxl or the BCLxl/b chimera with MG132 (a proteasome inhibitor) or cycloheximide (CH; a protein biosynthesis inhibitor) (Figure 3b and 3c). No obvious differences were observed after either treatment, suggesting that the loop domain of BCLb is not responsible for dictating the half-life of the protein. Next, we determined the oncogenic potency of chimera in vivo, using the same TET-O-MYC mouse model of leukemia described above. The oncogenic potency of the BCLb loop domain construct was equivalent to wild type BCLxl (20-30 day survival) (Figure 3d). This data suggests that loop domain of BCLb does not change the survival span of mice in comparison to wild type BCLxl.
Figure 3. Oncogenic potency of loop domain of BCLb.

a) Schematic of BCLb cl#1 - wild-type BCLb; BCLxl cl#2- wildtype BCLxl; BCLxl/b (BH4/loop) and cl#7- BCLxl engineered to harbor the loop domain of BCLb. constructs. b) Western blot of lysates expressing indicated BCLxl constructs followed by treatment with MG132 at 25 μM for the indicated times. c) Western blot of indicated BCLxl constructs after treatment with Cycloheximide (CH) at 20 μM for the indicated times Whereas (V) is empty vector. d) Kaplan-Meier survival curve of mice receiving Tet-O-MYC bone marrow infected with the indicated MIT-RX containing retroviruses. Mantel-Cox Log rank test was used to determine statistical significance, compared to BCLxlwt (#1: p=<0.0001; #7: p=0.568).
To support the notion that the observed results are due to intrinsic differences in biochemical properties of the tested BCLxl constructs and not simply due to alterations in the number of cells infected, a simple viral dilution experiment was carried out. Different ratios of non-potent MIT-GFP plasmids to potent MIT-BCLxl plasmids (5:1, 20:1, 80:1) were transfected into HEK293T cells and confirmed by western blot (Figure 4a). These viruses were then used to infect TET-O-MYC mouse bone marrow and monitored for the expected leukemic symptoms, as noted previously. It is clear that simply decreasing the amount of potent BCLxl plasmid up to 20 times less does not significantly alter in vivo disease onset (Figure 4b). In fact, a mouse that was transplanted with 20:1 GFP to BCLxl virus infected cells expressed similar amounts of BCLxl in leukemic splenocytes, indicating the strong selective pressure for cells expressing BCLxl even when there is 20 times less BCLxl virus present (Figure 4c). It is also interesting that at lower dilutions although all leukemias are clearly driven by infection with the tTA and BCLxl expressing virus some leukemias also express GFP. This represents the co-infection of the cells that become the leukemia with both viruses. However at the larger dilution, the leukemia is either driven by tTA and BCLxl or only by the tTA and GFP expressing virus. This experiment reinforces the idea that it may be intrinsic biochemical properties of individual BCL2-like proteins, such as expression levels, can dictate oncogenic potential.
Figure 4. Determining the correlation between amount of BCLxl and oncogenic potency.
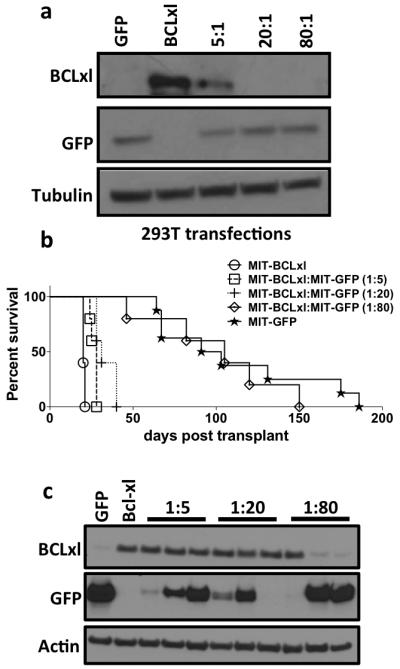
a) Western blot of HEK293T cells transfected with the indicated ratios of MIT-GFP and MIT-BCLxl. b) Kaplan-Meier survival curve of mice receiving Tet-O-MYC bone marrow infected with equal amounts of the indicated retroviral supernatant containing decreasing amounts of MIT-BCLxl virus. Five mice received bone marrow infected with each viral supernatant. c) Western blot of splenocytes from leukemic mice were performed as indicated above.
The results herein demonstrate the importance of moving experiments done in cell culture into in vivo assays. Critically, it is always of significance to determine if the ability of BCL2-like proteins to inhibit apoptosis induced by selected stimuli translates into the ability to more potently cause disease in a model system. Since leukemias generated in this model are readily transplantable, future experiments can be directed at understanding if decreased initiation of leukemogenesis, by certain BCLxl constructs, also means that these leukemias will respond better to cytotoxic therapeutics. These studies will increase our general knowledge of the biochemical mechanisms required for BCL2-like proteins to drive tumorigenesis and therapeutic responses.
Methods
Plasmids
All constructs (BCLxlwt - wildtype BCLxl; BCLxldTM - BCLxl lacking the c-terminal transmembrane domain; BCLxlActA - BCLxl engineered to have the mitochondrial targeted transmembrane domain from Listeria ActA protein; BCLxlcb5 - BCLxl engineered to have the ER targeted transmembrane domain from cytochrome b5) were produced in Li laboratory, Departments of Medicine, Pharmacology, and Toxicology, University of Louisville, Louisville and epitope tagged BCLb cl#1 - wild-type BCLb; BCLxl cl#2 - wildtype BCLxl; BCLxl/b (BH4/loop); cl#7- BCLxl engineered to harbor the loop domain of BCLb were produced in our laboratory and sub-cloned into MIT-RX; a murine retroviral vector, as described in our earlier study [27]. Where, LTR - long terminal repeats; MCS- multiple cloning sites; IRES- internal ribosomal entry sequence; tTA- tetracycline transactivator. Details of BCLxl constructs have also been previously characterized [6]. Clones were then transformed into 5-α competent strain of E. coli cells (NEB, Ipswich, MA, USA, #C2987H) and DNA was isolated using eZNA kit as per the manufacturer’s protocol (Omega, Norcross, GA, USA, #D6924).
Cell culture and transfection
Human embryonic kidney 293T cells (HEK293T) was procured from American Type Culture Collection (ATCC, Rockville, MD, USA) and cultured in DMEM medium (Hyclone, Logan, UT, USA, #SH30243) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, USA, #SH30070) and 1% antibiotic/antimycotic (Hyclone, Logan, UT, USA, #SV30010). DNA transfections were done with BCLxl constructs using polyethylenimine (Polysciences, Warrington, PA, USA, #23966-2) in HEK293T cells and viruses were collected as described [23].
Bone marrow transplantation, Western Blots and flow cytometer
In vivo experiments were done as described previously [2, 23, 27]. Briefly, bone marrow was flushed from untreated donor mice that carried the Tet-O-MYC transgene. Red blood cells were lysed and viral infections were then carried out in the presence of polybrene (Millipore, Danvers, MA, USA, #TR-1003-G) using retroviral BCLxl construct. After infection, cells were immediately transplanted into the tail vein of lethally irradiated FVB/n recipients acquired from Taconic. Spleens from leukemic mice were snap frozen in liquid nitrogen and pulverized with mortar and pestle. Cells were lysed with 1% CHAPS lysis buffer and protein was estimated as described in our previous study [28]. Western blots were performed in Bolt Bis-Tris gels (Life Technologies, Grand island, NY, USA, #BG4120BOX) as per manufacturers protocol using anti-BCLxl (Cell Signaling Technology, Danvers, MA, # 2764) or anti-Actin (Sigma, St. Louis, MO, # A2228). Spleen, bone marrow and thymus were analyzed by flow cytometer. In brief, single cells were isolated from indicated organ; red blood cells were lysed and blocked for 10 mins at room temperature with Fc Block (BD Biosciences, Miami, FL, USA, #553142). Afterward, FACS analysis were performed by standard techniques using anti-PE Ly-6G & Ly-6C (BD, San Jose, CA, # 553128) and anti-PE-Cy7 CD11b (BD, # 552850) on Becton Dickinson FACScan with FlowJo.
Histology and Biochemical Analysis
For histological staining peripheral blood from the indicated mice was smeared on a glass slide and stained with Hema 3 (Fisher, Hampton, NH, # 22-122-911) as indicated by manufacturer’s protocol. Liver and Spleen were fixed in neutral buffered formalin and embedded in paraffin. Sections were stained with Hematoxylin and eosin (H&E) using standard techniques and photographed at 10X or 20X. For biochemical analysis MIT-BCLxl (cl#1, cl#2 & cl#7) or empty vector (V) were transfected into HEK293T cells and 36 hrs after transfection, the cells were treated with MG132 at 25 μM for the 4, 8 & 16 hrs followed by western blot with the anti-epitope tagged antibodies (FLAG and Tubulin, Sigma, # F1804). The same constructs were also transfected into HEK293T cells and 36 hrs later cells were treated with Cycloheximide (CH) at 20 μM for the 4 and 16 hrs followed by western blot with FLAG and anti-tTA (Clontech, Mountain View, CA, # 631108). To determine the correlation between amount of BCLxl and oncogenic potency, HEK293T cells were transfected with the ratios of the MIT-GFP and MIT-BCLxl (3 μg of MIT-GFP; BCLxl, 3 μg of MIT-BCLxl; 1:5, 2.4 μg of MIT-GFP and 0.6 μg of MIT-BCLxl; 1:20, 2.85 μg of MIT-GFP and 0.15 μg of MIT-BCLxl; 1:80, 2.9625 μg of MIT-GFP and 0.0375 μg of MIT-BCLxl), virus were produced, infected in bone marrow and transplanted as described above. Western blot of splenocytes from leukemic mice were also performed using anti-BCLxl, GFP and actin.
Acknowledgements
We would like to thank members of the Beverly laboratory for technical support, especially Lavona Casson. Portions of this work were performed in the laboratory of Harold Varmus at MSKCC, we extend our gratitude for his generosity and support. This work was supported by start-up funds from James Graham Brown Cancer Center to LJB, The Kosair Pediatric Cancer Research Program, Molecular Targets COBRE 8P20GM103482-10 from NIH, a Wendy Will Case Cancer Fund grant to LJB, R25-CA-134283 from the National Cancer Institute to AS.
Footnotes
Conflict of Interest
The authors declare no conflict of interest
References
- 1.Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 2.Beverly LJ, Varmus HE. MYC-induced myeloid leukemogenesis is accelerated by all six members of the antiapoptotic BCL family. Oncogene. 2009;28:1274–1279. doi: 10.1038/onc.2008.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kvansakul M, Hinds MG. Structural biology of the Bcl-2 family and its mimicry by viral proteins. Cell Death Dis. 2013;4:e909. doi: 10.1038/cddis.2013.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shamas-Din A, Kale J, Leber B, Andrews DW. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb Perspect Biol. 2013;5:a008714. doi: 10.1101/cshperspect.a008714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reed JC, Jurgensmeier JM, Matsuyama S. Bcl-2 family proteins and mitochondria. Biochim Biophys Acta. 1998;1366:127–137. doi: 10.1016/s0005-2728(98)00108-x. [DOI] [PubMed] [Google Scholar]
- 6.Eno CO, Eckenrode EF, Olberding KE, Zhao G, White C, et al. Distinct roles of mitochondria- and ER-localized Bcl-xL in apoptosis resistance and Ca2+ homeostasis. Mol Biol Cell. 2012;23:2605–2618. doi: 10.1091/mbc.E12-02-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fischer U, Schulze-Osthoff K. New approaches and therapeutics targeting apoptosis in disease. Pharmacol Rev. 2005;57:187–215. doi: 10.1124/pr.57.2.6. [DOI] [PubMed] [Google Scholar]
- 8.Gerl R, Vaux DL. Apoptosis in the development and treatment of cancer. Carcinogenesis. 2005;26:263–270. doi: 10.1093/carcin/bgh283. [DOI] [PubMed] [Google Scholar]
- 9.Yip KW, Reed JC. Bcl-2 family proteins and cancer. Oncogene. 2008;27:6398–6406. doi: 10.1038/onc.2008.307. [DOI] [PubMed] [Google Scholar]
- 10.Elkholi R, Floros KV, Chipuk JE. The Role of BH3-Only Proteins in Tumor Cell Development, Signaling, and Treatment. Genes Cancer. 2011;2:523–537. doi: 10.1177/1947601911417177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Strasser A, O'Connor L, Dixit VM. Apoptosis signaling. Annu Rev Biochem. 2000;69:217–245. doi: 10.1146/annurev.biochem.69.1.217. [DOI] [PubMed] [Google Scholar]
- 12.Frenzel A, Grespi F, Chmelewskij W, Villunger A. Bcl2 family proteins in carcinogenesis and the treatment of cancer. Apoptosis. 2009;14:584–596. doi: 10.1007/s10495-008-0300-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, et al. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300:135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- 14.Thomenius MJ, Distelhorst CW. Bcl-2 on the endoplasmic reticulum: protecting the mitochondria from a distance. J Cell Sci. 2003;116:4493–4499. doi: 10.1242/jcs.00829. [DOI] [PubMed] [Google Scholar]
- 15.Schott AF, Apel IJ, Nunez G, Clarke MF. Bcl-XL protects cancer cells from p53-mediated apoptosis. Oncogene. 1995;11:1389–1394. [PubMed] [Google Scholar]
- 16.Olopade OI, Adeyanju MO, Safa AR, Hagos F, Mick R, et al. Overexpression of BCL-x protein in primary breast cancer is associated with high tumor grade and nodal metastases. Cancer J Sci Am. 1997;3:230–237. [PubMed] [Google Scholar]
- 17.Zhuang L, Lee CS, Scolyer RA, McCarthy SW, Zhang XD, et al. Mcl-1, Bcl-XL and Stat3 expression are associated with progression of melanoma whereas Bcl-2, AP-2 and MITF levels decrease during progression of melanoma. Mod Pathol. 2007;20:416–426. doi: 10.1038/modpathol.3800750. [DOI] [PubMed] [Google Scholar]
- 18.Cao X, Yap JL, Newell-Rogers MK, Peddaboina C, Jiang W, et al. The novel BH3 alpha-helix mimetic JY-1-106 induces apoptosis in a subset of cancer cells (lung cancer, colon cancer and mesothelioma) by disrupting Bcl-xL and Mcl-1 protein-protein interactions with Bak. Mol Cancer. 2013;12:42. doi: 10.1186/1476-4598-12-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walczak H, Bouchon A, Stahl H, Krammer PH. Tumor necrosis factor-related apoptosis-inducing ligand retains its apoptosis-inducing capacity on Bcl-2- or Bcl-xL-overexpressing chemotherapy-resistant tumor cells. Cancer Res. 2000;60:3051–3057. [PubMed] [Google Scholar]
- 20.Chauhan D, Velankar M, Brahmandam M, Hideshima T, Podar K, et al. A novel Bcl-2/Bcl-X(L)/Bcl-w inhibitor ABT-737 as therapy in multiple myeloma. Oncogene. 2007;26:2374–2380. doi: 10.1038/sj.onc.1210028. [DOI] [PubMed] [Google Scholar]
- 21.Lin X, Morgan-Lappe S, Huang X, Li L, Zakula DM, et al. 'Seed' analysis of off-target siRNAs reveals an essential role of Mcl-1 in resistance to the small-molecule Bcl-2/Bcl-XL inhibitor ABT-737. Oncogene. 2007;26:3972–3979. doi: 10.1038/sj.onc.1210166. [DOI] [PubMed] [Google Scholar]
- 22.Casson L, Howell L, Mathews LA, Ferrer M, Southall N, et al. Inhibition of ceramide metabolism sensitizes human leukemia cells to inhibition of BCL2-like proteins. PLoS One. 2013;8:e54525. doi: 10.1371/journal.pone.0054525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beverly LJ. Oncogenic driver supersedes BM preparation as the critical determinant of leukemic outcome: a one day protocol for BM harvest, infection and transplantation. Bone Marrow Transplant. 2013;48:1003–1005. doi: 10.1038/bmt.2012.273. [DOI] [PubMed] [Google Scholar]
- 24.Zhu W, Cowie A, Wasfy GW, Penn LZ, Leber B, et al. Bcl-2 mutants with restricted subcellular location reveal spatially distinct pathways for apoptosis in different cell types. Embo j. 1996;15:4130–4141. [PMC free article] [PubMed] [Google Scholar]
- 25.Saurabh K, Scherzer MT, Shah PP, Mims AS, Lockwood WW, et al. The PIM family of oncoproteins: small kinases with huge implications in myeloid leukemogenesis and as therapeutic targets. Oncotarget. 2014 doi: 10.18632/oncotarget.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sugioka R, Shimizu S, Funatsu T, Tamagawa H, Sawa Y, et al. BH4-domain peptide from Bcl-xL exerts anti-apoptotic activity in vivo. Oncogene. 2003;22:8432–8440. doi: 10.1038/sj.onc.1207180. [DOI] [PubMed] [Google Scholar]
- 27.Beverly LJ, Lockwood WW, Shah PP, Erdjument-Bromage H, Varmus H. Ubiquitination, localization, and stability of an anti-apoptotic BCL2-like protein, BCL2L10/BCLb, are regulated by Ubiquilin1. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E119–126. doi: 10.1073/pnas.1119167109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shah PP, Lockwood WW, Saurabh K, Kurlawala Z, Shannon SP, et al. Ubiquilin1 represses migration and epithelial-to-mesenchymal transition of human non-small cell lung cancer cells. Oncogene. 2014 doi: 10.1038/onc.2014.97. [DOI] [PMC free article] [PubMed] [Google Scholar]