


Abstract
In humans, DNA double-strand breaks (DSBs) are repaired by two mutually-exclusive mechanisms, homologous recombination or end-joining. Among end-joining mechanisms, the main process is classical non-homologous end-joining (C-NHEJ) which relies on Ku binding to DNA ends and DNA Ligase IV (Lig4)-mediated ligation. Mostly under Ku- or Lig4-defective conditions, an alternative end-joining process (A-EJ) can operate and exhibits a trend toward microhomology usage at the break junction. Homologous recombination relies on an initial MRN-dependent nucleolytic degradation of one strand at DNA ends. This process, named DNA resection generates 3′ single-stranded tails necessary for homologous pairing with the sister chromatid. While it is believed from the current literature that the balance between joining and recombination processes at DSBs ends is mainly dependent on the initiation of resection, it has also been shown that MRN activity can generate short single-stranded DNA oligonucleotides (ssO) that may also be implicated in repair regulation. Here, we evaluate the effect of ssO on end-joining at DSB sites both in vitro and in cells. We report that under both conditions, ssO inhibit C-NHEJ through binding to Ku and favor repair by the Lig4-independent microhomology-mediated A-EJ process.
INTRODUCTION
Single-stranded (ss) DNA has several functions during signaling and processing of DNA damage. For example tracks of ssDNA associated with DNA breakage or replication fork stalling are necessary to activate the ATR protein kinase through the sequential recruitment of RPA, ATRIP and TOPBP1 (1). Also, production of 3′-ssDNA tails at DNA double-strand breaks (DSBs) through the combined 3′- and 5′-exonuclease activities of the CtIP/MRN and Exo1/Blm–Dna2 complexes, respectively, is necessary to initiate repair by homologous recombination (HR) (2). In addition, endonucleolytic processes operating during repair of DNA damage can generate ssDNA oligonucleotides (ssO). During the nucleotide excision repair process in humans, dual incisions remove pyrimidine dimers from the genome as ∼30 nt long, protein-bound, ssO that may serve specialized intracellular functions (3). The Spo11 trans-esterase operating during meiosis as well as DNA topoisomerase II have been shown to attach covalently through a catalytic tyrosine to short oligonucleotides released by endonucleolytic cleavage (4). During DSBs repair also, MRN-associated ssO have been shown to promote activation of the ATM protein kinase (5).
In addition to HR, DSBs can be repaired by direct joining mechanisms. The predominant route is the classical non-homologous end-joining reaction (C-NHEJ) that operates throughout the cell cycle and requires first recognition and tethering of DNA ends followed by processing and final ligation (6). Briefly, C-NHEJ starts with binding of the ring-shaped Ku70/Ku80 heterodimer to each end of the DSB, stabilized by the newly discovered PAXX factor (7,8). Ku binding to a DSB is followed by recruitment of the catalytic subunit of the DNA-dependent protein kinase (DNA-PKcs), forming the DNA-PK (9). After processing of the DNA ends, the XRCC4/Ligase IV (Lig4) complex executes final ligation stimulated by Cernunnos/XLF. Aside from C-NHEJ, a still not fully characterized alternative end-joining process (A-EJ) can operate; it is defined as Ku- or Lig4-independent end-joining and exhibits a trend for microhomology usage at the break junction (for an extensive recent review, (10)).
Little is known about the effect of ssO on the dominant C-NHEJ process. Biochemical experiments with purified DNA-PKcs without Ku under non-physiological low-salt conditions, have shown an interaction with ssDNA in electrophoretic mobility shift assays (EMSA) (11). Under similar conditions, DNA-PKcs was slightly activated with high concentrations of ssO up to 10 nt long while longer ssO impaired its activation by standard double-stranded (ds) DNA oligonucleotides (dsO) (12). It was proposed from these data a model in which optimal DNA-PKcs activation requires unpaired ssDNA at the ends of DSBs (12). Furthermore, it was suggested that strand opening may be promoted by Ku at DNA ends under natural conditions of Ku-dependent DNA-PKcs activation (13,14). Ku exhibits affinity for ssO, albeit in a much lower affinity range than for dsO (15,16).
It is believed from the current literature that the choice between joining and recombination processes at DSBs ends is mainly depending on the initiation of resection that may produce ssDNA ends and/or ssO, possibly implicated in repair regulation (reviewed in (2)). Therefore, we intended to evaluate both in vitro and in cells the effect of ssO on end-joining of DSBs. We report here that under both conditions, ssO inhibit C-NHEJ through binding to Ku and favor repair by a Lig4-independent and microhomology-mediated alternative end-joining process.
MATERIALS AND METHODS
Cell culture
All culture medium were from Life Technologies and were supplemented with 10% fetal calf serum, 2 mM glutamine, 125 U/ml penicillin and 125 μg/ml streptomycin. All cells were grown at 37°C in a humidified atmosphere with 5% CO2. HeLa cells, from the American Type Culture Collection, were grown in Dulbecco's modified Eagle's medium (DMEM). The LIG4-defective (a kind gift from Eric Hendrickson, University of Minnesota Medical School, Minneapolis, USA), PRKDC-defective (PRKDC gene codes for DNA-PKcs; Horizon Discovery Ltd, Cambridge, UK) and parental HCT116 cell lines were maintained in RPMI 1640 medium. RPE-1 hTERT XRCC6+/− and RPE-1 hTERT XRCC6Venus/− were grown in DMEM/Ham's F-12 medium buffered with sodium bicarbonate (XRCC6 gene codes for Ku70 protein). A manuscript detailing the generation of these cells is in preparation (S.B., J.C., S.P.J. and P.C., unpublished data).
Chemicals
Calicheamicin-γ1, a generous gift from P. R. Hamann (Wyeth Research, Pearl River, NY, USA), was dissolved at 4 mM in ethanol and stored at −70°C. NU7441 (Sigma-Aldrich) was dissolved in DMSO (5 mM stock solution) and stored at −20°C. Small aliquots of stock solutions chemicals were used once.
Plasmid substrates, oligonucleotides and DNA fragments
pBluescript-KS-II (−) or pDVG94 plasmids (gift from Dik van Gent, Cancer Genomics Center, Rotterdam, The Netherlands) were digested with FastDigest restriction enzymes HindIII, AfeI/EcoRV (Thermo Scientific) respectively, according to the manufacturer's instructions and linear forms were then purified by Gel and polymerase chain reaction (PCR) Clean-Up System (Promega).
Except other indication, all the oligonucleotides were from Sigma-Aldrich. The ss oligonucleotide used as competitor in NHEJ EMSA were as follows : 6 bases : 5′ GTGTGA; 10 bases : 5′ GTGTGAGTGT; 15 bases : 5′ GTGTGAGTGTGAGTG; 20 bases : 5′ GTGTGAGTGTGAGTGTGAGT; 25 bases : 5′ GTGTGAGTGTGAGTGTGAGTGTGAG; 30 bases : 5′ GTGTGAGTGTGAGTGTGAGTGTGAGTGTGA; 35 bases : 5′ GTGTGAGTGTGAGTGTGAGTGTGAGTGTGAGTGTG. The double-stranded oligonucleotides were obtained by annealing with the complementary oligomers. The DNA probe used in EMSA was 5′ CGAACACCGGTACCCAGCTTTTGTTCCCTTTA, bearing or not a 3′ biotin moiety for the ss probe, or the same oligomer hybridized with its complementary oligomer for the ds probe. The same ss oligomer biotinylated at 3′, 5′ or both ends was used as competitor in the EMSA on radiolabeled DNA probe; the extent of biotinylation was identical for the 3 probes, as measured in a control EMSA in the presence or not of streptavidin (data not shown). The biotinylated 75 bp fragment used in pull-down experiments was amplified by PCR with Phusion Hot Start (Thermo Scientific) from pBluescript-KS-II(−) with 5′-biotinylated reverse primer 5′ GCGTTATCCCCTGATTCTGTGG and forward primer 5′ CGTTCGGCTGCGGCGAGCGGTATCAGC and purified by Gel and PCR Clean-Up System (Promega). The LNA ssO was 5′Cy5-CAG CTC CTC GCC CTT GCT CA in which LNA bases are underlined and all the bonds are phosphorothioate (Exiqon).
End-joining extracts and protein purification
NHEJ-competent cell extracts preparation and Ku protein purification were carried out as previously described (17).
In vitro end-joining assay
Typically, 40 μg cell extracts were incubated with 10 ng linearized plasmid for 1 h at 25°C in 10 μl reaction mixture containing EJ buffer (50 mM Triethanolamine pH 8.0, 0.5 mM magnesium acetate, 1 mM dithiothreitol, 0.1 mg/ml bovine serum albumin (BSA), 60 mM potassium acetate) and 1 mM ATP (Sigma-Aldrich). When indicated, extracts were pre-incubated with ss or ds oligonucleotides, NU7441 or purified Ku protein for 10 min at 4°C as indicated. Samples were then treated with 100 μg/ml RNAse A at 37°C and then deproteinized with proteinase K (Euromedex). Ligation products were separated in 0.7% agarose gels and stained with SYBR-Green (Life Technologies). Fluorescence was detected and analyzed on a Typhoon fluorimager (Molecular Dynamics). Quantitative analysis of the gel was performed with the ImageJ software (version 1.4).
Electrophoretic mobility shift assay (EMSA)
EMSA using a biotinylated DNA probe was performed as follows: purified human Ku protein was incubated for 30 min on ice with 0.01 pmol biotinylated DNA probe and competitor single stranded oligonucleotides when necessary in 10 μl reaction containing 20 mM Tris–HCl, pH 7.5, 2 mM MgCl2, 90 mM potassium acetate, 0.1 mM EDTA, 0.3 mM dithiothreitol, 5% glycerol. The mixture was loaded on a 5% polyacrylamide gel, electrophoresed at 4°C for 1 h at 100 V and then transferred to a Biodyne B nylon membrane (Thermo Scientific). After cross-link under UV light (Spectrolinker XL-1000), signals of probe were detected with the Chemiluminescent Nucleic Acid Detection Module (Thermo Scientific) according to the manufacturer's instructions.
For EMSA using a radiolabeled probe, ds DNA probe was 32P-labeled at 5′-ends with T4 polynucleotide kinase in the presence of [γ32P]-ATP (4500 Ci mmol−1, ICN Pharmaceuticals) and unincorporated [γ32P]-ATP was removed twice by gel filtration (ProbeQuant G-50, GE Healthcare Life Sciences). Competitor biotinylated ss-oligomers were preincubated for 5 min at 4°C in the presence or not of streptavidin (Sigma-Aldrich) at a 9/1 streptavidin/oligomer molar ratio. Then 20 ng Ku protein was added for 10 min at 4°C in 10 mM Tris–HCl, pH 7.5, 1 mM MgCl2, 45 mM potassium acetate, 0.1 mM EDTA, 0.25 mM dithiothreitol, 5% glycerol. Then radiolabeled DNA (0.2 pmol) was added for a further incubation 20 min at 20°C in 10 μl final volume of the same buffer. The samples were electrophoresed on a 5% polyacrylamide gel at 4°C, for 1 h at 100V. The gel was dried on Whatman 3MM and DE81 paper and exposed to a storage phosphor screen (Molecular Dynamics) followed by processing with a PhosphorImager (Molecular Dynamics, Storm System TM).
Microhomology detection assay
The microhomology assay was adapted from a published protocol (18). For in vitro experiments, linearized pDVG94 was used as the substrate of end-joining assay as described above. The ligated product was then purified by Gel and PCR Clean-Up System (Promega). For in cellulo experiments, 1 μg of linearized pDVG94 plasmid was transfected for 4 h using Lipofectamine 2000 (Life Technologies) into sub-confluent wild-type and mutant HCT116 cells in 6-well plates and in culture medium without antibiotics. When necessary, single-stranded LNA oligonucleotide (100 pmol) was co-transfected with pDVG94 plasmid. After transfection, cells were washed and incubated in culture medium. Ligated plasmid DNA was recovered from cells 24 h later using a PureYield Plasmid Miniprep System Kit (Promega).
After purification from cell extracts or cells, ligation product was PCR amplified using FM30 (5′-CTCCATTTTAGCT-TCCTTAGCTCCTG) and DAR5 (5′-TGCTTCCGGCTCGTATGTTGGTTGGATT) primers. The PCR product was digested with BstXI (FastDigest, Thermo Scientific). Restriction fragments were separated by electrophoresis along with undigested PCR product in 6% polyacrylamide gel in 90 mM Tris-borate, pH 8.0, 2 mM EDTA (TBE) buffer and stained with SYBR-Green (Life Technologies). The bands representing the undigested (180 bp) or digested (120 + 60 bp) PCR products were detected on a Typhoon fluorimager (Molecular Dynamics)
Antibodies
Rabbit polyclonal anti-XRCC4 was produced in our laboratory against the full-length recombinant human protein and rabbit polyclonal anti-Ligase IV was manufactured by Biotem (Le Rivier d'Apprieu, France) against a peptide. For immunoblotting, were used mouse monoclonal antibodies anti-β-Actin (clone AC-15, Ambion), PARP-1 (clone 4C10–5, BD Pharmingen), Ku80 (clone 111, ThermoScientific), Ligase III (clone 7, BD biosciences), Ligase I (clone 10H5, Genetex), Chk1 (clone G4, Santa-Cruz), Chk2 (clone B4, Santa-Cruz) and anti-DNA-PKcs (clone 18.2, Abcam), rabbit monoclonal antibody anti-γH2AX (Abcam) and rabbit polyclonal antibodies anti-DNA-PKcs PhSer-2056 (Abcam), KAP-1 (Abcam), KAP-1 PhSer-824 (Bethyl Laboratories), H2AX (Abcam), Chk1 PhSer-345 and Chk2 PhThr-68 (Cell Signaling). Peroxidase-conjugated goat anti-mouse or anti-rabbit secondary antibodies were from Jackson Immunoresearch Laboratories. For flow cytometry, mouse monoclonal anti-γH2AX primary antibody (clone JBW301, EMD Millipore) and AlexaFluor488-conjugated goat anti-mouse secondary antibody (Life technologies) were used.
Protein extraction after DNA damaging treatment
Cells were rinsed with phosphate buffered saline (PBS), lysed in for 20 min at 4°C in buffer (50 mM Hepes-KOH pH 7.5, 450 mM NaCl, 1 mM EDTA, 1% Triton, 1 mM DTT, 1× Protease-phosphotase inhibitors cocktail (Pierce)) and then sonicated. Protein concentration was measured by the Bradford assay (Biorad).
Western blotting
Protein samples were mixed with loading sample buffer to 1× final concentration (50 mM Tris–HCl pH 6.8, 10% glycerol, 1% sodium dodecyl sulphate, 300 mM 2-mercaptoethanol, 0.01% bromophenol blue), boiled, separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis on 4–15% precast gels (Biorad) and blotted onto Immobilon-P polyvinylidene difluoride membranes (Millipore). Membranes were blocked for 60 min with 5% dry milk in PBS, 0.1% Tween-20 (Sigma-Aldrich), incubated for 1 h with primary antibody diluted in PBS containing 0.02% Tween-20 and 1% BSA (fraction V, Sigma-Aldrich) and washed three times with PBS-T; membranes were incubated for 1 h with secondary antibodies in PBS-T. Membranes were washed five times with PBS-T and immuno-blots were visualized by enhanced chemiluminescence (ImmunofaxA, Yelen). For blots with antibodies against non- and phosphorylated forms of the same protein, two loadings of the same protein samples were run, transferred and blotted in parallel.
In vitro pull-down experiment
About 1 pmol of 75 bp biotinylated DNA fragment was immobilized on 10 μl streptavidin magnetic beads (Dynabeads M280 streptavidin, Life Technologies) as recommended by the manufacturer, leading to ∼80% binding efficiency. For ATP-depletion, 60 μg NHEJ-competent extract were incubated with 10 mM glucose and 1 U hexokinase (Calbiochem) 5 min at 20°C, then 100 pmol of ss or dsO were added when necessary and incubation went on for 10 min at 4°C. Extract was then mixed with 10 μl of mocked or DNA-treated beads washed in PBS 0.5× and then incubated in a final 10 μl of 50 mM Triethanolamine pH 8.0, 0.5 mM magnesium acetate, 1 mM DTT, 0.1 mg/ml BSA, 40 mM potassium chloride at 10°C for 30 min under gentle agitation. The beads were washed rapidly twice with two-fold diluted PBS buffer and then processed for western blotting.
In cellulo ssO tranfection and pull-down experiment
Sub-confluent HeLa cells in 6-well plates were transfected for 4 h with or without 100 pmol cy5-LNA ssO using lipofectamine 2000 (Life Technologies) according to the manufacturer's instruction. For the pull-down experiment, cells were washed in PBS, incubated in culture medium for 2 h and then transfected with or without 4 pmol of the 75 bp biotinylated DNA fragment using jetPEI (Polyplus tranfection). After 1 h, cells were collected by scrapping in a buffer containing 100 mM Tris–Cl, pH 8, 0.2% Triton X-100, 2 mM MgCl2, 1 mM EDTA and centrifuged at 600 g in a microcentrifuge. The supernatant was further centrifuged at 20 000 g. Protein concentration was measured by the Bradford assay (Biorad). Streptavidin beads (Dynabeads M280 streptavidin, Life Technologies) were prepared by incubation with control extracts for 1 h at 4°C under gentle agitation followed by three washes in lysis buffer. Then Biotinylated DNA from a volume of supernatant equivalent to 100 μg protein was trapped on wet streptavidin beads from a 50 μl prepared beads suspension under incubation conditions similar as above. After beads removal, proteins were retrieved from the supernatant by precipitation overnight at −20°C in three volumes of acetone:trichloraoactic acid (3:1 vol:vol) and centrifugation at 20 000 g in a microcentrifuge.
Live-cell microscopy and micro-irradiation
Cells were grown and observed in 35-mm glass-bottom culture dishes (MatTek). Experiments were carried out with a ZEISS LSM 710 confocal laser scanning microscope equipped with a coherent chameleon Vision II tunable laser (690–1080 nm) and a 40X/1.3 oil immersion objective. Venus fluorescence was excited using a 488 nm Ar-laser line and Cy-5 with a 561 nm Ne-laser line. The microscope was equipped with a heated environmental chamber set at 37°C in 5% CO2 atmosphere.
Confocal image series were recorded with a frame size of 512 × 512 pixels. Nuclei micro-irradiation was carried out at 800 nm at 30% of max power (mean max power was 3300 mW) in spots of 2.5 μm diameter at 3× zoom during 7.75 s as previously described (19). Before and after micro-irradiation, confocal image series of one mid z-section were recorded at 7.75 s time interval (typically two pre-irradiation and 30–40 post-irradiation frames). For evaluation of the recruitment kinetics, fluorescence intensities of the irradiated region were corrected for total nuclear loss of fluorescence over the time course and normalized to the pre-irradiation value. Data from micro-irradiation of individual cells obtained in several independent experiments performed on different days were averaged, analyzed and displayed using PRISM software.
Cell cycle and DNA repair analysis by flow cytometry
Confluent RPE-1 hTERT XRCC6+/− hemizygous for Ku70 were transfected for 2 h with cy5-LNA ssO using lipofectamine 2000 (Life Technologies) as described before. After transfection, medium was refreshed with complete RPE-1 medium with or without 3 μM DNA-PK inhibitor NU7441. One hour later, cells were treated with 80 pM Cali for 30 min in medium with or without inhibitor. After two washes with medium, a 14 h post-incubation was performed with or without inhibitor. Cells were harvested by trypsination, washed with PBS 1% BSA and fixed 15 min with 2% paraformaldehyde. After a 30 min permeabilization at room temperature (RT) with PBS 0.2% Triton X-100, cells were incubated at RT with mouse monoclonal anti-γH2AX antibody at 1:1000 dilution in PBS 1% BSA. AlexaFluor-488 secondary antibody was incubated at RT for 30 min at 1:200 dilution in PBS 1% BSA. Finally, DNA was stained by incubating cells 30 min at RT in PBS containing 2 μg/ml DAPI (4′,6-Diamidino-2-Phenylindole) and 0.25 μg/ml RNase A. A LSR II cytometer (BD Biosciences) was used to monitor γH2AX and cy-5 fluorescence intensity in the different phases of the cell cycle.
RESULTS
ssDNA oligomers switch end-joining in vitro from C-NHEJ to A-EJ
C-NHEJ can operate in vitro upon incubation of protein cell extracts with linear plasmid DNA (reviewed in (20)). Under these conditions, end-joining activity from extracts of HCT116 colon cancer cells produces predominantly linear multimers and lesser amounts of circular species (Figure 1A). This DNA end-joining activity is mediated by C-NHEJ since the appearance of these ligation products is blocked by the specific DNA-PK inhibitor NU7441 (Figure 1A, without ssO) and abolished in extracts devoid of the core C-NHEJ components Lig4 or DNA-PKcs (Figure 1B and C, without ssO). First, we wondered whether the presence of ssDNA might affect C-NHEJ activity under our experimental conditions. When increasing concentrations of a 34 base single-stranded DNA oligonucleotide (ssO) were added to the reactions containing HCT116 cell extracts, we observed a dose-dependent stimulation of plasmid end-joining up to 2.9-fold (Figure 1A). However, in the presence of ssO, the end-joining activity was not mediated anymore by C-NHEJ since it became resistant to DNA-PK inhibition (Figure 1A). Identical results were obtained with HeLa extracts (Supplementary Figure S1A).
Figure 1.
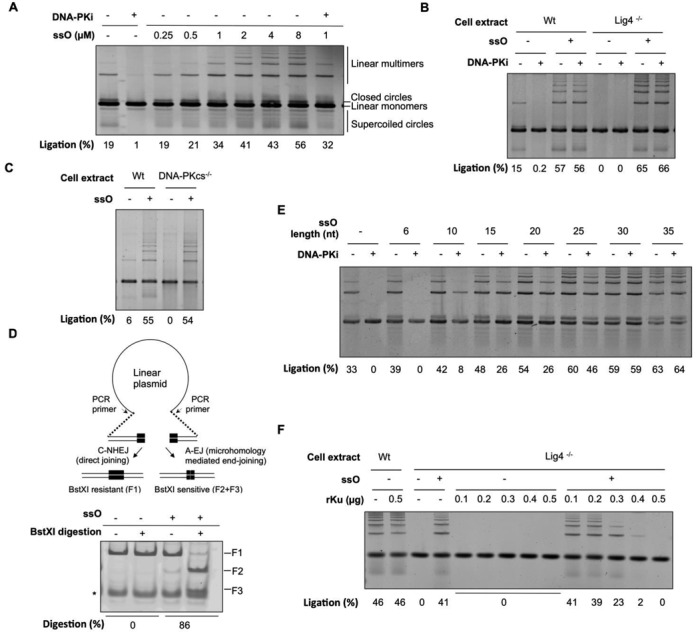
Impact of ssDNA oligonucleotides on DNA end-joining activity in cell extracts. (A) Analysis of end-joining activity under standard reaction conditions using HCT116 extracts, in the presence or not of the specific DNA-PK inhibitor NU7441 (DNA-PKi) and ssO at the indicated concentration. Plasmid ds ends and ssO amount in reactions were 0.01 pmol and from 2.5 to 80 pmol, respectively. DNA ligation products were separated by agarose gel electrophoresis followed by DNA staining. (B) End-joining assay with extracts from wild-type or Lig4−/- HCT116 cells, in the presence or not of the specific DNA-PK inhibitor NU7441 (DNA-PKi) and ssO as indicated. Plasmid ds ends and ssO amount in reactions were 0.01 pmol and 40 pmol, respectively. (C) End-joining with extracts from wild-type or DNA-PKcs−/− HCT116 cells, in the presence or not of ssO as indicated, under conditions as in (B). (D) Upper panel: scheme of the assay used for ligation products characterization. After rejoining of the pDVG94 linearized plasmid, the F1 fragment obtained from PCR-amplification across the junction is incubated with BstXI restriction enzyme. Generation of F2 and F3 sub-fragments relies on microhomology-mediated end-joining which creates a BstXI restriction site. Lower panel: junction characterization after end-joining of linearized pDVG94 plasmid with HeLa cell extracts under conditions as in (B). After ligation, plasmids were purified, the junction was PCR-amplified and digested by BstXI. Digestion products were separated by acrylamide gel electrophoresis followed by DNA staining. Asterisk: non-specific PCR product. (E) End-joining with HCT116 cell extracts, in the presence or not of the specific DNA-PK inhibitor NU7441 (DNA-PKi) and ssO of various lengths as indicated, under conditions as in (B). (F) End-joining assay with extracts from wild-type or Lig4−/− HCT116 cells, in the presence or not of DNA-PK specific inhibitor NU7441 (DNA-PKi), ssO under conditions as in (B) and various amount of purified human recombinant Ku heterodimer (rKu), as indicated.
To further explore the involvement of a C-NHEJ independent ligation mechanism in the presence of ssO, we used extracts from HCT116 deficient for DNA Ligase IV (Lig4−/−) (21), the ligase mediating C-NHEJ. Strikingly, while extracts of Lig4−/− cells exhibited virtually no ligation activity under standard conditions, they produced up to >65% of ligated species in the presence of ssO (Figure 1B). Again, the end-joining activity promoted by ssO in Lig4-deficient extracts was insensitive to DNA-PK inhibition and was dependent on the concentration of ssO (Figure 1B and Supplementary Figure S1B). Notably, the effect of ssO was not mimicked by a same size dsDNA which, in contrast, was completely inactive in promoting end-joining in Lig4-defective extracts at concentrations identical to those used for ssO (Supplementary Figure S1C).
The DNA-PKcs- and Lig4-independent properties of the end-joining activity promoted by ssO were reminiscent of the A-EJ process, which operates mostly in the absence of C-NHEJ factors (10). Given that this process also exhibits a trend toward microhomology usage at the break-junctions, we further explored the end-joining activity promoted by ssO by using a blunt-ended plasmid containing microhomologies in the form of 6 bp terminal direct repeats (18) (Figure 1D). A-EJ converts the two repeats to one, simultaneously generating a BstXI restriction enzyme recognition site easily detectable in the PCR fragment amplified from the plasmid junction (18,21) (Figure 1D). As we had observed previously (Figure 1A and Supplementary Figure S1A), ligation of this plasmid by extracts from HeLa or HCT116-wt cells became resistant to DNA-PK inhibitor in the presence of ssO (Supplementary Figure S1D). As shown in Figure 1D, while ligation products were exclusively BstXI-resistant under standard conditions using HeLa cell extracts, nearly 90% of these products became BstXI-sensitive upon addition of ssO in the ligation reaction. A similar switch toward BstXI-resistant ligation products was observed in HCT116 Lig4-proficient and -deficient extracts in presence of ssO (Supplementary Figure S1E). We concluded that ssO not only abolish C-NHEJ in extracts from wild-type cells but also promote a strong A-EJ activity in extracts proficient or deficient for Lig4.
As A-EJ has been shown to operate preferentially under conditions of deficiency of C-NHEJ factors (reviewed in (10)), we speculated that ssO somehow inactivate one of the C-NHEJ factors. However, since we found that ssO stimulated ligation in Lig4-deficient extracts, this indicated that the ssO target must be distinct from Lig4. Some reports have concluded that ssO could interact with DNA-PKcs under specific conditions (11,12). Therefore, we tested the impact of ssO on extracts from HCT116-DNA-PKcs−/− cells (22). As shown in Figure 1C, while extracts from DNA-PKcs−/− cells exhibited essentially no ligation activity under standard conditions, they produced ∼55% of ligated species in the presence of ssO, indicating that, as for Lig4, DNA-PKcs was not the target of ssO. We have previously observed that Ku depletion from cell extracts decreased the sensitivity of the remaining end-joining activity to DNA-PK inhibition or antibody-mediated XRCC4 inactivation, both hallmarks of A-EJ (23). In light of this, and since Ku has been reported to have a low affinity for ssDNA (15), we hypothesized that ssO could target Ku in cell extracts. In this regard, we noted that the ssO effect on end-joining in vitro with HCT116 extracts containing or lacking Lig4 was almost undetectable for oligomers shorter than 10 bases and optimal for oligomers longer than 25 bases (Figure 1E and Supplementary Figure S1F). These length requirements were reminiscent of Ku binding properties to ds DNA ends (24). We reasoned that if ssO inhibited C-NHEJ through titrating out Ku in cell extracts, increasing the Ku concentration might overcome this inhibitory effect. Indeed, as shown in Figure 1F, while addition of purified recombinant human Ku to extracts of wild-type or Lig4−/− cells did not affect their ligation activity in the absence of ssO, it impaired the promotion of A-EJ by ssO in a dose-dependent manner. Collectively, these data suggested that in cell extracts, ssDNA oligomers divert Ku from dsDNA ends, thereby preventing C-NHEJ while promoting A-EJ at DSB sites.
ssDNA oligomers bind Ku and prevent C-NHEJ machinery assembly on ds DNA ends
When an EMSA was used to assess direct binding of purified Ku to 32 bp dsDNA and 32 nt ssDNA, detectable binding of Ku to ssDNA was observed but with a much lower affinity than for dsDNA (Figure 2A), in agreement with published data (15). Despite this low affinity, ssO showed a dose-dependent competition activity for the binding of Ku to the dsDNA probe, leading to an increasing inhibition of Ku binding over a range of 30- to 1000-fold molar ratio of ss versus ds oligomers (Figure 2B). From these values and using a published calculation ((25), Lin and Riggs equation herein), we estimated a Kd of Ku for ssO at least 10-fold higher than for ds oligomers. Under these conditions, we found a length-dependence for this competition effect which was optimal for ssO longer than 25 bases (Supplementary Figure S2), in close agreement with the length requirement observed for A-EJ promotion by ssO in cell extracts (Figure 1E). To analyze whether Ku binding on ssDNA was DNA-end orientated, EMSA was used to monitor the competition for Ku binding between a radiolabeled dsDNA probe and ssO blocked at one or both ends by a biotin–streptavidin complex. The three biotinylated ssO displaced Ku from the dsDNA probe as efficiently as the non-biotinylated control ssO in the absence of streptavidin (Figure 2C). However, blocking the 5′-end by streptavidin partially released competition by ssO while blocking the 3′-end had no detectable effect (Figure 2C). These data indicated that optimal Ku binding to ssDNA requires a free 5′-end and a 25-nt length.
Figure 2.
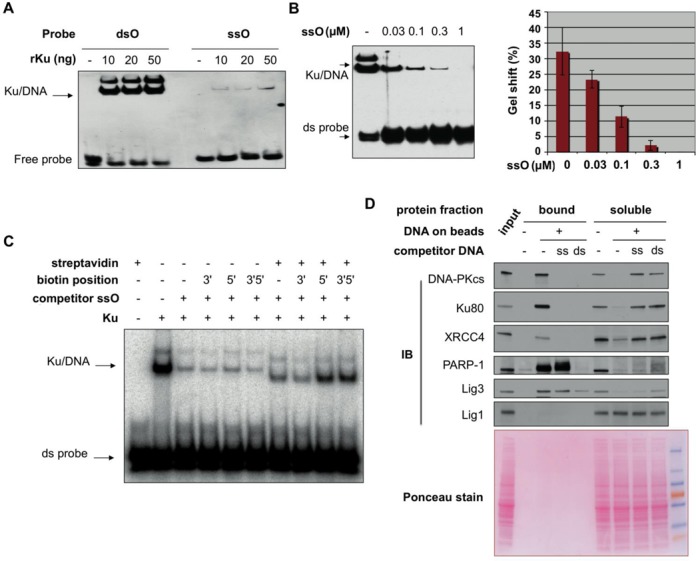
Properties of ssDNA oligonucleotides to bind Ku and affect DNA repair proteins assembly at DNA ends. (A) Electrophoretic mobility shift assay of ds or ss biotinylated DNA probe incubated with various amount of purified human recombinant Ku protein. After acrylamide gel electrophoresis, transfer on membrane and UV-cross-linking, the position of free and retarded probe was revealed by chemiluminescence. (B) Electrophoretic mobility shift assay of ds biotinylated DNA probe incubated with purified human recombinant Ku protein in the presence of various amount of competitor ssO as indicated. ds probe and ssO amount in reactions were 0.01 pmol and from 0.3 to 10 pmol, respectively. Quantification of retarded probe in competition experiments as in (B) (mean ± SD, n = 3). (C) Electrophoretic mobility shift assay of radiolabeled ds DNA probe incubated with purified human recombinant Ku protein in the presence of various competitor ssO bearing a biotin moiety at the 3′-, 5′- or 3′- and 5′- end, blocked or not by streptavidin, as indicated. ds probe and ssO amount in reactions were 0.2 and 10 pmol, respectively. After acrylamide gel electrophoresis, the gel was dried and auto-radiographed. (D) Pull-down experiment on streptavidin magnetic beads bearing or not ds DNA probe, incubated with HCT116 cell extracts mixed or not with competitor ss- or ds-DNA, as indicated. ds probe and ssO amount in reactions were 0.8 pmol and 100 pmol, respectively. A fraction of the initial amount of protein used during the pull-down experiment was loaded as an input control. Proteins bound to the beads or remaining in the soluble fraction were denatured, separated on SDS-PAGE gel and electrotransferred on a membrane that was immunoblotted (IB) with the indicated antibodies.
Assembly of the C-NHEJ protein machinery at DNA ends can be assessed in vitro by pull-down on a dsDNA probe, biotinylated at one end and attached to streptavidin-coated magnetic beads (26). Since Ku is necessary for the recruitment of both DNA-PKcs and C-NHEJ ligation complex to DSBs (9,26), we checked the effect of ssO on C-NHEJ proteins assembly. After incubation of cell extracts with a 75 bp biotinylated DNA fragment, bound proteins were separated from soluble proteins over a magnet and analyzed by western-blotting. Since ATP promotes the dissociation of DNA-PKcs from DNA (27), reactions were performed in extracts depleted of ATP. Thus, we found that both DNA-PK (including Ku and DNA-PKcs components) and ligation (XRCC4 component) complexes of the C-NHEJ machinery were recovered specifically on the dsDNA probe and were essentially titrated out from the soluble fraction under standard pull-down conditions (Figure 2D). DNA-PK and XRCC4 binding to the probe was competed out with excess of dsO as expected. Strikingly, ssO also competed for DNA-PK and XRCC4 binding to the ds DNA probe (Figure 2D). Pioneering experiments have supported the involvement of PARP-1 and DNA Ligase III (Lig3) in an alternative end-joining activity distinct from Ku- and Lig4-dependent DSBs ligation (28,29), findings that were later confirmed in several cellular models (reviewed in (10)). Although several data have reported a competition between Ku and PARP-1 for binding to ds DNA ends (23,30), both proteins were recovered on the ds DNA probe under our conditions, likely due to a non-limiting amount of free ds DNA ends in the assay (Figure 2D). Strikingly and in contrast to their effects on DNA-PK and XRCC4, ssO did not compete for the binding of PARP-1 and Lig3 to the dsDNA probe (Figure 2D). Nevertheless, PARP-1 and Lig3 were largely released from the ds DNA beads in the presence of soluble dsDNA oligomers, while in all conditions used, Lig1 was not recovered on the dsDNA beads (Figure 2D; note that PARP-1 automodification resulting in a smear in the supernatant impaired detection in the last lane). In agreement, end-joining activity promoted by ssO in HCT116 Lig4-/- extracts was partly sensitive to SCR-7 which efficiently inhibits Lig4 and also Lig3 to a lesser extent, but not Lig1 (31) (data not shown). Moreover, ablation of Lig3 expression through shRNA in HeLa negatively impacted on ssO-mediated stimulation of A-EJ in the corresponding cell extracts (Supplementary Figure S3). Taken together, these data suggested that Ku binding to ssO in vitro can prevent assembly of DNA-PK and XRCC4-dependent ligation complexes at ds DNA ends while allowing PARP-1 and Lig3 recruitment that favors A-EJ activity at these ends.
ssDNA oligomers impair C-NHEJ and promote alternative error-prone end-joining in cells
In order to assess ssO interference with C-NHEJ in cells, we used a modified oligomer containing both Locked Nucleic Acids (LNA) bases and phosphorothioate linkages that have been shown to increase stability and resistance to nucleases (32). In addition, a cyanine-5 fluorochrome (Cy5) was present at the 5′-end of the oligomer. This stabilized LNA ssO was as efficient as a control unmodified ss oligomer in trapping Ku in EMSA competition assays and in switching end-joining activity to A-EJ in ligation assays with cell extracts (data not shown). The LNA ssO was transfected into cells by lipofection and as shown in Figure 3A, it accumulated mainly in the cell nucleus after 4 hours, as reported after electrotransfection with the same LNA ssO (33).
Figure 3.

Effect of ssDNA oligonucleotides on the recruitment and activation of DNA repair proteins at DSBs in cells. (A) Intracellular localization of LNA ssO 4 h after lipofection in HCT116 cells. a and c panels correspond to mock-transfected cells and b and d panels to transfected cells. Red images correspond to Cy5 fluorescence from LNA ssO and gray images to light transmission acquisition. Cells were visualized with an Olympus IX73 inverted fluorescence microscope equipped with a DP26 digital camera and a 20× objective lens. (B) Characterization of the damage response induced by cell transfection with oligonucleotides. The LNA ssO alone or annealed with the complementary strand (dsO) (150 pmol each per well) was transfected for 2 h into HCT116 cells in 6-well plates. After cell lysis, proteins were separated on SDS-PAGE gel and electro-transferred on a membrane that was blotted with the indicated antibodies. (C) Pull-down experiment. HCT116 cells were transfected with a biotinylated ds DNA ± LNA ssO as indicated. ds probe and ssO amount per well were 4 and 100 pmol, respectively. After cell lysis, the extracts were incubated with streptavidin magnetic beads. Proteins bound to the beads or remaining in the soluble fraction were denatured and separated on SDS-PAGE gel; then electro-transfer on membrane and immuno-blotting (IB) with the indicated antibodies were performed. (D) Effect of oligonucleotides on the cellular response to DSBs. The LNA ssO (150 pmol per well) was transfected for 2 h into HCT116 cells in 6-well plates. After washing, cells were incubated for 30 min in culture medium containing 80 pM Calicheamicin-γ1. After cell lysis, proteins were separated on SDS-PAGE gel and electro-transferred on a membrane that was blotted with the indicated antibodies.
We first investigated whether ssO activate the DNA-damage response (DDR) in cells. Hence, we compared established DDR-induced protein phosphorylation sites after transfection of HCT116 cells with the LNA ssO or its dsO counterpart. While dsO triggered a strong phosphorylation of DNA-PKcs, KAP1, Chk1, Chk2 and H2AX as reported previously (34), phosphorylation of these proteins was undetectable after transfection of HCT116 cells with an equivalent amount of ssO (Figure 3B), again supporting that ssO and dsO promote very different effects. In line with this, no cell cycle effect was detected after ssO tranfection in cells (Supplementary Figure S4).
Then, we proceeded to test the ability of LNA ssO to interfere with the interaction between Ku and DSBs in cells. A 75-bp biotinylated DNA fragment was transfected into cells, then bound proteins were isolated by their retrieval on streptavidin-coated magnetic beads. As shown in Figure 3C, components of both DNA-PK and C-NHEJ ligation complexes were specifically recovered on the beads at the expense of their soluble fraction in the presence of the dsDNA probe. In cells co-transfected with the modified ssO, however, the amounts of C-NHEJ components that co-purified with the dsDNA probe were substantially reduced, with much now remaining in the soluble fraction (Figure 3C). These data suggested that DSBs recognition by the C-NHEJ machinery in cells was impaired in the presence of ss DNA oligomers.
DSBs repair by C-NHEJ relies on several key steps, including DNA-PKcs autophosphorylation on Ser-2056. DNA-PKcs autophosphorylation at Ser-2056 needs DNA-PKcs recruitment and synapsis of adjacent DNA ends (32), both depending on proper Ku binding at the break extremities (22). We checked the effect of ssO on DNA-PKcs autophosphorylation at Ser-2056 after treatment of HCT116 cells with Calicheamicin-γ1 which yields a high ratio of DSBs to DNA single-strand breaks (33). In parallel, we assessed phosphorylation of KAP1 at Ser-824 and H2AX at Ser-139 which are mainly ATM-targeted in response to DSBs (34). From published data, we calculated that calicheamicin treatment induced ∼300 DSB per nucleus (35). Under our conditions and based on a minimum of 1% transfection efficiency of ssO, we estimated a ssO versus DSBs ratio of ∼1000 per nucleus, in the range reached in vitro. As shown in Figure 3D, ATM-dependent phosphorylation of KAP1 and H2AX were preserved following Calicheamicin treatment in the presence of ssO. By contrast, DNA-PKcs autophosphorylation was significantly impaired, suggesting that ssO compromised DNA-PK activation and C-NHEJ at DSBs in cells.
Next, the impact of ssO on Ku dynamics at sites of DNA damage was investigated through live cell imaging of Venus-Ku70 accumulation at sites of laser-induced DNA damage. This method has been previously used to trigger Ku70/80 and XRCC4 accumulation on chromatin in the irradiated areas (36). Laser irradiation was performed in engineered RPE-1 cells, hemizygous for Ku70 and expressing from the sole gene copy a functional Venus-Ku70 fusion protein that was largely restricted to the nucleus (Figure 4A). Without LNA ssO, live-cell imaging showed retention of Venus-Ku70 at the irradiated sites, with a fast accumulation followed by a slower increase up to the latest time point registered (Figure 4B). In the presence of LNA ssO however, Venus-Ku70 retention at damaged sites was attenuated when compared to control laser-irradiated cells (Figure 4A and B), despite the relative Ku recruitment kinetics was identical to that in control conditions (Supplementary Figure S5). Together, these data show that ssO compromise accumulation of Ku at DSBs, thereby supporting the ability of ssO to hijack Ku also in cells.
Figure 4.
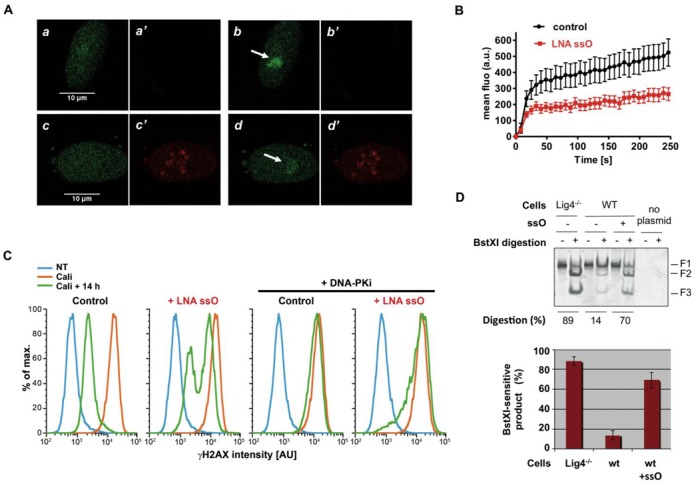
Effect of ssDNA oligonucleotides on end-joining at DSBs in cells. (A) Venus-Ku70 behavior after 800 nm pulsed-laser nuclear irradiation assessed in RPE-1 cells by live cell-imaging at 0 s (a, a’, c, c’ panels) and 256 s post-irradiation (b, b’, d, d’ panels), 4 h after lipofection without (a, a’, b, b’ panels) or with LNA ssO (c, c’, d, d’ panels). a, b, c, d and a’, b’, c’, d’ panels correspond to images in the Venus and Cy5 fluorescence channels, respectively. The white arrows mark irradiated areas. (B) Dynamics of Venus-Ku70 at laser-induced damage sites in RPE-1 cells transfected or not with LNA ssO (100 pmol per well). Images were obtained at 7.75 s intervals from 40 s time point post laser-irradiation and fluorescence intensities at the damage sites were quantified. Mean values of fluorescence intensity with SEM were calculated by averaging data obtained in individual cells in several experiments performed on different days (total number of cells = 23 for each control and +LNA ssO conditions). (C) DSB repair efficiency in RPE-1 cells transfected or not with LNA ssO. RPE-1 hemizygous for Ku70 mock transfected (control) or transfected with LNA ssO (150 pmol per well) were untreated (NT) or treated with Cali and post-incubated 14 h in medium with or without DNA-PK inhibitor NU7441 (DNA-PKi). After fixation, flow cytometry was used to monitor in G0/G1 phase of the cell cycle the appearance and disappearance of γH2AX as a readout of DNA damage induction and repair. (D) Junction characterization after end-joining of linearized pDVG94 plasmid transfected in wild-type or Lig4−/− HCT116 cells, in the presence or not of ssO as indicated. Plasmid and ssO amount per well were 0.2 pmol and 100 pmol, respectively. Cells were disrupted 24 h after transfection, plasmids were purified, then the junction was PCR-amplified and digested by BstXI. Digestion products were separated by acrylamide gel electrophoresis followed by DNA staining. Quantification of ligation experiments as in C (mean ± SD, n = 3).
In cells competent for C-NHEJ, A-EJ activity is maintained at a low level (37). Among other C-NHEJ factors, Ku is mainly responsible for A-EJ limitation under normal conditions through its avid binding to dsDNA ends (23,30,38). Indeed, A-EJ is more active under conditions of Ku deficiency than in cells defective for DNA-PKcs, XRCC4 or Lig4 (21,38–40). Thus, in order to assess the effect of ssO on DSB repair we used flow cytometry to analyze the disappearance of γH2AX after treatment of RPE-1 cells with Calicheamicin-γ1 (Figure 4C). This analysis was performed on confluent cells that accumulate in G0/G1 cells to exclude DSB repair by HR. In these cells, efficient DNA repair was observed 14 h after treatment with Cali and mainly relied on C-NHEJ since it was abolished upon DNA-PK inhibition (Figure 4C, compare first and third panels). In contrast, a partial γH2AX persistence was observed in the presence of ssO (Figure 4C, compare first and second panels). Strikingly, the remaining DSB repair activity was partially resistant to DNA-PK inhibition (Figure 4C, compare third and fourth panels). These data showed that ssO strongly prevent C-NHEJ and promote a DNA-PK independent repair activity that is expressed outside S/G2, thus most likely corresponding to A-EJ.
Despite leading to different joining efficiencies, it has been demonstrated that any defect in DNA-PKcs, XRCC4 or Lig4 C-NHEJ factors promoted a strong trend toward microhomology-mediated repair at DSBs detected in plasmid-rejoining assays (18,21). Thus, to assess the effect of ssO on A-EJ activity in cells, we transfected the A-EJ reporter plasmid described above (Figure 1D) into HCT116 cells. After plasmid recovery and amplification of the junction by PCR, microhomology usage was quantified by measuring BstXI sensitivity. Microhomology usage was restrained to ∼10% of repair junctions in parental HCT116 cells but increased up to ∼90% in Lig4−/− mutant cells (Figure 4D), as previously reported (21). Strikingly, cotransfection of modified ssO in parental cells promoted a nearly seven-fold increase in microhomology usage at plasmid junctions when compared to conditions without ssO (Figure 4D). These data demonstrate that ssO switch repair from C-NHEJ to microhomology-mediated end-joining at DSBs in cells.
DISCUSSION
We report here the unexpected property of ssO to act both in vitro and in cells as activators of A-EJ at the expense of C-NHEJ through hijacking Ku protein. The low affinity of Ku for single-stranded oligonucleotides has previously been reported (15,16) and some sequence-specific RNAs have been selected that bound Ku and prevented DNA-PKcs activation in vitro (41). However, the effect of ssO on NHEJ has not previously been assessed.
We discovered that a free DNA end is needed for optimal Ku binding to ssDNA, suggesting that it does not rely on a secondary structure, that otherwise was not predicted by standard algorithms in the ssO used here (data not shown). In addition, we found that ssDNA required a 10 nt minimal length with an optimal 25 nt length for Ku binding and subsequent effects on end-joining. These values are reminiscent of early EMSA and footprinting results showing that >14 bp were required for detectable Ku binding to dsDNA probe and that optimally ∼25 bp were covered, further confirmed by the structure of Ku on DNA (15,42,43). Also, the requirement of a free 5′-end for optimal binding of Ku to ssO indicates a preferred geometry of the protein toward ssDNA, again reminding of the strongly favored orientation previously reported by elegant cross-linking experiments on Ku–dsDNA complexes (44) and later confirmed by Ku structure resolution (45). Together, our data favor a model in which ssDNA engages through its 5′-end into the channel present in the ring-shaped Ku heterodimer structure that is large enough to accommodate dsDNA, albeit with higher affinity (45). Further structural experiments would be required to definitively establish this model.
We showed that the addition of purified Ku counteracted the effect of ssO on end-joining and that ssO is efficient in the absence of DNA-PKcs or Lig4. This indicated that Ku is the main target of ssO and confirmed that Ku is the main blockade to A-EJ mechanism (23,38). We found that, unlike Ku, PARP-1 and Lig3 proteins were refractory to ssO binding. Indeed, our pull-down experiments revealed that ssO prevent Ku binding to DSBs whilst PARP-1 and Lig3 were still recruited. Biochemical experiments with purified proteins have provided evidence of a competition between Ku and PARP-1 for binding to dsDNA-ends (30). However, the binding mode of Ku and PARP-1 to DNA is different since the former is based on a ring-based structure (45) while the latter binds through zinc finger domains (46). In addition, a zinc-finger domain in Lig3α has also been shown to mediate its XRCC1 and PARP-1-independent mode of recruitment to DNA breaks (47). These features may support the preferential binding of ssO to Ku over PARP-1 and Lig3 proteins that we observed.
Several reports have substantiated a role for PARP-1 (28,30,48,49) and Lig3 (28,29,50–52) in A-EJ. Therefore, the unique property of ssO to target preferentially Ku and not PARP-1 and Lig3 in the presence of ds DSB ends as shown here may account for its strong potency to promote A-EJ at DSBs both in vitro and in cells. In addition, the trend of Ku-independent end-joining promoted by ssO for microhomology pairing at DSBs ends is in full agreement with this general trait reported for A-EJ (10,49,53–56).
Although the conditions used here exceeded the ssO versus DSB ratio that may be expected in cells, they have helped to reveal unanticipated ssO properties as potential DSB repair regulators. Interestingly, end resection activity associated with MRE11 at DSBs has been shown to produce short oligonucleotides able to boost ATM activation (5). It is conceivable that in some instances, these short oligonucleotides locally contribute to modulate the balance between Ku-independent end-joining and dominant Ku-dependent repair that only allows very limited resection (57). For example, this may be particularly relevant for the repair of some complex breaks, such as breaks with blocked DNA ends that particularly rely on resection (58–61); at these breaks sites, local ss oligonucleotides production associated with resection could favor A-EJ at the expense of C-NHEJ and Ku diversion may thus prevent abortive attempts of C-NHEJ. Suppression of Ku binding to DNA ends is also required in situations such as repair and restart at stalled DNA replication forks during S-phase (62). Interestingly, a role for A-EJ has been proposed in the repair of one-ended DSBs following hydroxyurea-mediated replication fork arrest (37); so it is possible that ss oligonucleotides produced following MRE11-dependent resection also contribute to DSBs repair at arrested forks through inhibition of Ku binding.
We and others have shown that PARP-1 inhibition in cells defective for C-NHEJ worsens defects in DSBs repair and survival following ionizing irradiation (28,49,63). In addition, it has been reported that after ionizing irradiation, PARP-1 dependent A-EJ preferentially mediates DSBs repair in some tumor cell lines that could be radio-sensitized through PARP-1 inhibition (64). Our present results reinforce previous evidence that Ku is the main factor that counteracts A-EJ (23,38). We thus speculate that the striking property of ssO as strong A-EJ stimulator that we report here establishes a rationale for the development of Ku-targeting approaches aimed at extending the use of PARP inhibitors to cancer cells beyond those defective in homologous recombination.
Supplementary Material
Acknowledgments
We are indebted to P.R. Hamann (Wyeth Research) for the gift of calicheamicin γ1, to Pr Eric A. Hendrickson (University of Minnesota Medical School, Minneapolis, USA) for the gift of HCT116 Lig4−/− cell line, to Dr Dik C. van Gent (Cancer Genomics Center, Rotterdam, The Netherlands) for the gift of pDVG94 plasmid, to Dr Muriel Golzio (IPBS, Toulouse, France) for the gift of Cy5-LNA ssO and to Rachael V. Walker (Babraham Institute, Cambridge, UK) for helping with the generation of RPE-1 hTERT XRCC6+/− and RPE-1 hTERT XRCC6Venus/−.
We dedicate this work to the memory of our colleague and friend Pr Christophe Cazaux who died accidentally in early August 2015. We would like to acknowledge his great contribution as professor in genetic and cell biology, investigator in genomic instability and coordinator of both cancer research funding and popularization in Toulouse.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ligue Nationale Contre le Cancer (Equipe labellisée 2013), Electricité de France (Programme Conseil de Radioprotection) (to P.C. laboratory); China Scholarship Council doctoral grant (to Y.Y.); Ligue Nationale Contre le Cancer post-doctoral grant (to S.B.); Région Midi-Pyrénées (CPER), Grand Toulouse community, Association pour la recherche contre le cancer (ARC Equipement No 8505), CNRS, EU through the FEDER program (to TRI Optical Imaging Platform at IPBS (Genotoul, Toulouse, France)); Cancer Research UK programme [C6/A11224], European Research Council, European Community Seventh Framework Programme [HEALTH-F2–2010-259893 (DDResponse)], CRUK [C6946/A14492], Wellcome Trust [WT092096] (to S.P.J laboratory); University of Cambridge, UK, supplemented by CRUK (for S.P.J. salary); INSERM (for P.C. salary). Funding for open access charge: University of Cambridge.
Conflict of interest statement. None declared.
REFERENCES
- 1.Marechal A., Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013;5:a012716. doi: 10.1101/cshperspect.a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Symington L.S. End resection at double-strand breaks: mechanism and regulation. Cold Spring Harb. Perspect. Biol. 2014;6:a016436. doi: 10.1101/cshperspect.a016436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kemp M.G., Reardon J.T., Lindsey-Boltz L.A., Sancar A. Mechanism of release and fate of excised oligonucleotides during nucleotide excision repair. J. Biol. Chem. 2012;287:22889–22899. doi: 10.1074/jbc.M112.374447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neale M.J., Pan J., Keeney S. Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature. 2005;436:1053–1057. doi: 10.1038/nature03872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jazayeri A., Balestrini A., Garner E., Haber J.E., Costanzo V. Mre11-Rad50-Nbs1-dependent processing of DNA breaks generates oligonucleotides that stimulate ATM activity. EMBO J. 2008;27:1953–1962. doi: 10.1038/emboj.2008.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Waters C.A., Strande N.T., Wyatt D.W., Pryor J.M., Ramsden D.A. Nonhomologous end joining: a good solution for bad ends. DNA Repair (Amst) 2014;17:39–51. doi: 10.1016/j.dnarep.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ochi T., Blackford A.N., Coates J., Jhujh S., Mehmood S., Tamura N., Travers J., Wu Q., Draviam V.M., Robinson C.V., et al. DNA repair. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science. 2015;347:185–188. doi: 10.1126/science.1261971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xing M., Yang M., Huo W., Feng F., Wei L., Jiang W., Ning S., Yan Z., Li W., Wang Q., et al. Interactome analysis identifies a new paralogue of XRCC4 in non-homologous end joining DNA repair pathway. Nat. Commun. 2015;6:6233. doi: 10.1038/ncomms7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gottlieb T.M., Jackson S.P. The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell. 1993;72:131–142. doi: 10.1016/0092-8674(93)90057-w. [DOI] [PubMed] [Google Scholar]
- 10.Frit P., Barboule N., Yuan Y., Gomez D., Calsou P. Alternative end-joining pathway(s): Bricolage at DNA breaks. DNA Repair (Amst) 2014;17:81–97. doi: 10.1016/j.dnarep.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 11.Leuther K.K., Hammarsten O., Kornberg R.D., Chu G. Structure of DNA-dependent protein kinase: implications for its regulation by DNA. EMBO J. 1999;18:1114–1123. doi: 10.1093/emboj/18.5.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hammarsten O., DeFazio L.G., Chu G. Activation of DNA-dependent protein kinase by single-stranded DNA ends. J. Biol. Chem. 2000;275:1541–1550. doi: 10.1074/jbc.275.3.1541. [DOI] [PubMed] [Google Scholar]
- 13.Pawelczak K.S., Andrews B.J., Turchi J.J. Differential activation of DNA-PK based on DNA strand orientation and sequence bias. Nucleic Acids Res. 2005;33:152–161. doi: 10.1093/nar/gki157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pawelczak K.S., Turchi J.J. A mechanism for DNA-PK activation requiring unique contributions from each strand of a DNA terminus and implications for microhomology-mediated nonhomologous DNA end joining. Nucleic Acids Res. 2008;36:4022–4031. doi: 10.1093/nar/gkn344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Falzon M., Fewell J.W., Kuff E.L. EBP-80, a transcription factor closely resembling the human autoantigen Ku, recognizes single- to double-strand transitions in DNA. J. Biol. Chem. 1993;268:10546–10552. [PubMed] [Google Scholar]
- 16.Krasner D.S., Daley J.M., Sung P., Niu H. Interplay between Ku and RPA in the restriction of Exo1-mediated DNA break end resection. J. Biol. Chem. 2015;290:18806–18816. doi: 10.1074/jbc.M115.660191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bombarde O., Boby C., Gomez D., Frit P., Giraud-Panis M.J., Gilson E., Salles B., Calsou P. TRF2/RAP1 and DNA-PK mediate a double protection against joining at telomeric ends. EMBO J. 2010;29:1573–1584. doi: 10.1038/emboj.2010.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verkaik N.S., Esveldt-van Lange R.E., van Heemst D., Bruggenwirth H.T., Hoeijmakers J.H., Zdzienicka M.Z., van Gent D.C. Different types of V(D)J recombination and end-joining defects in DNA double-strand break repair mutant mammalian cells. Eur. J. Immunol. 2002;32:701–709. doi: 10.1002/1521-4141(200203)32:3<701::AID-IMMU701>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 19.Britton S., Dernoncourt E., Delteil C., Froment C., Schiltz O., Salles B., Frit P., Calsou P. DNA damage triggers SAF-A and RNA biogenesis factors exclusion from chromatin coupled to R-loops removal. Nucleic Acids Res. 2014;42:9047–9062. doi: 10.1093/nar/gku601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pastwa E., Somiari R.I., Malinowski M., Somiari S.B., Winters T.A. In vitro non-homologous DNA end joining assays–the 20th anniversary. Int. J. Biochem. Cell Biol. 2009;41:1254–1260. doi: 10.1016/j.biocel.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fattah F., Lee E.H., Weisensel N., Wang Y., Lichter N., Hendrickson E.A. Ku regulates the non-homologous end joining pathway choice of DNA double-strand break repair in human somatic cells. PLoS Genet. 2010;6:e1000855. doi: 10.1371/journal.pgen.1000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruis B.L., Fattah K.R., Hendrickson E.A. The catalytic subunit of DNA-dependent protein kinase regulates proliferation, telomere length, and genomic stability in human somatic cells. Mol. Cell. Biol. 2008;28:6182–6195. doi: 10.1128/MCB.00355-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng Q., Barboule N., Frit P., Gomez D., Bombarde O., Couderc B., Ren G.S., Salles B., Calsou P. Ku counteracts mobilization of PARP1 and MRN in chromatin damaged with DNA double-strand breaks. Nucleic Acids Res. 2011;39:9605–9619. doi: 10.1093/nar/gkr656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dynan W.S., Yoo S. Interaction of Ku protein and DNA-dependent protein kinase catalytic subunit with nucleic acids. Nucleic Acids Res. 1998;26:1551–1559. doi: 10.1093/nar/26.7.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ryder S.P., Recht M.I., Williamson J.R. Quantitative analysis of protein-RNA interactions by gel mobility shift. Methods Mol. Biol. 2008;488:99–115. doi: 10.1007/978-1-60327-475-3_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Calsou P., Delteil C., Frit P., Drouet J., Salles B. Coordinated assembly of Ku and p460 subunits of the DNA-dependent protein kinase on DNA ends is necessary for XRCC4-ligase IV recruitment. J. Mol. Biol. 2003;326:93–103. doi: 10.1016/s0022-2836(02)01328-1. [DOI] [PubMed] [Google Scholar]
- 27.Chan D.W., Lees-Miller S.P. The DNA-dependent protein kinase is inactivated by autophosphorylation of the catalytic subunit. J. Biol. Chem. 1996;271:8936–8941. doi: 10.1074/jbc.271.15.8936. [DOI] [PubMed] [Google Scholar]
- 28.Audebert M., Salles B., Calsou P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J. Biol. Chem. 2004;279:55117–55126. doi: 10.1074/jbc.M404524200. [DOI] [PubMed] [Google Scholar]
- 29.Wang H., Rosidi B., Perrault R., Wang M., Zhang L., Windhofer F., Iliakis G. DNA ligase III as a candidate component of backup pathways of nonhomologous end joining. Cancer Res. 2005;65:4020–4030. doi: 10.1158/0008-5472.CAN-04-3055. [DOI] [PubMed] [Google Scholar]
- 30.Wang M., Wu W., Wu W., Rosidi B., Zhang L., Wang H., Iliakis G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34:6170–6182. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Srivastava M., Nambiar M., Sharma S., Karki S.S., Goldsmith G., Hegde M., Kumar S., Pandey M., Singh R.K., Ray P., et al. An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression. Cell. 2012;151:1474–1487. doi: 10.1016/j.cell.2012.11.054. [DOI] [PubMed] [Google Scholar]
- 32.Spitzer S., Eckstein F. Inhibition of deoxyribonucleases by phosphorothioate groups in oligodeoxyribonucleotides. Nucleic Acids Res. 1988;16:11691–11704. doi: 10.1093/nar/16.24.11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orio J., Bellard E., Baaziz H., Pichon C., Mouritzen P., Rols M.P., Teissie J., Golzio M., Chabot S. Sub-cellular temporal and spatial distribution of electrotransferred LNA/DNA oligomer. J. RNAi Gene Silencing. 2013;9:479–485. [PMC free article] [PubMed] [Google Scholar]
- 34.Quanz M., Chassoux D., Berthault N., Agrario C., Sun J.S., Dutreix M. Hyperactivation of DNA-PK by double-strand break mimicking molecules disorganizes DNA damage response. PLoS One. 2009;4:e6298. doi: 10.1371/journal.pone.0006298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bouquet F., Muller C., Salles B. The loss of gammaH2AX signal is a marker of DNA double strand breaks repair only at low levels of DNA damage. Cell Cycle. 2006;5:1116–1122. doi: 10.4161/cc.5.10.2799. [DOI] [PubMed] [Google Scholar]
- 36.Mari P.O., Florea B.I., Persengiev S.P., Verkaik N.S., Bruggenwirth H.T., Modesti M., Giglia-Mari G., Bezstarosti K., Demmers J.A., Luider T.M., et al. Dynamic assembly of end-joining complexes requires interaction between Ku70/80 and XRCC4. Proc. Natl. Acad. Sci. U.S.A. 2006;103:18597–18602. doi: 10.1073/pnas.0609061103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Truong L.N., Li Y., Shi L.Z., Hwang P.Y., He J., Wang H., Razavian N., Berns M.W., Wu X. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2013;110:7720–7725. doi: 10.1073/pnas.1213431110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mansour W.Y., Borgmann K., Petersen C., Dikomey E., Dahm-Daphi J. The absence of Ku but not defects in classical non-homologous end-joining is required to trigger PARP1-dependent end-joining. DNA Repair (Amst) 2013;12:1134–1142. doi: 10.1016/j.dnarep.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 39.Guirouilh-Barbat J., Rass E., Plo I., Bertrand P., Lopez B.S. Defects in XRCC4 and KU80 differentially affect the joining of distal nonhomologous ends. Proc. Natl. Acad. Sci. U.S.A. 2007;104:20902–20907. doi: 10.1073/pnas.0708541104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schulte-Uentrop L., El-Awady R.A., Schliecker L., Willers H., Dahm-Daphi J. Distinct roles of XRCC4 and Ku80 in non-homologous end-joining of endonuclease- and ionizing radiation-induced DNA double-strand breaks. Nucleic Acids Res. 2008;36:2561–2569. doi: 10.1093/nar/gkn094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoo S., Dynan W.S. Characterization of the RNA binding properties of Ku protein. Biochemistry. 1998;37:1336–1343. doi: 10.1021/bi972100w. [DOI] [PubMed] [Google Scholar]
- 42.Mimori T., Hardin J.A. Mechanism of interaction between Ku protein and DNA. J. Biol. Chem. 1986;261:10375–10379. [PubMed] [Google Scholar]
- 43.Yaneva M., Kowalewski T., Lieber M.R. Interaction of DNA-dependent protein kinase with DNA and with Ku: biochemical and atomic-force microscopy studies. EMBO J. 1997;16:5098–5112. doi: 10.1093/emboj/16.16.5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoo S., Kimzey A., Dynan W.S. Photocross-linking of an oriented DNA repair complex. Ku bound at a single DNA end. J. Biol. Chem. 1999;274:20034–20039. doi: 10.1074/jbc.274.28.20034. [DOI] [PubMed] [Google Scholar]
- 45.Walker J.R., Corpina R.A., Goldberg J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature. 2001;412:607–614. doi: 10.1038/35088000. [DOI] [PubMed] [Google Scholar]
- 46.Ali A.A., Timinszky G., Arribas-Bosacoma R., Kozlowski M., Hassa P.O., Hassler M., Ladurner A.G., Pearl L.H., Oliver A.W. The zinc-finger domains of PARP1 cooperate to recognize DNA strand breaks. Nat. Struct. Mol. Biol. 2012;19:685–692. doi: 10.1038/nsmb.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abdou I., Poirier G.G., Hendzel M.J., Weinfeld M. DNA ligase III acts as a DNA strand break sensor in the cellular orchestration of DNA strand break repair. Nucleic Acids Res. 2014;43:875–892. doi: 10.1093/nar/gku1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Robert I., Dantzer F., Reina-San-Martin B. Parp1 facilitates alternative NHEJ, whereas Parp2 suppresses IgH/c-myc translocations during immunoglobulin class switch recombination. J. Exp. Med. 2009;206:1047–1056. doi: 10.1084/jem.20082468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mansour W.Y., Rhein T., Dahm-Daphi J. The alternative end-joining pathway for repair of DNA double-strand breaks requires PARP1 but is not dependent upon microhomologies. Nucleic Acids Res. 2010;38:6065–6077. doi: 10.1093/nar/gkq387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Della-Maria J., Zhou Y., Tsai M.S., Kuhnlein J., Carney J., Paull T., Tomkinson A. hMre11/hRad50/Nbs1 and DNA ligase III{alpha}/XRCC1 act together in an alternative non-homologous end joining pathway. J. Biol. Chem. 2011;286:33845–33853. doi: 10.1074/jbc.M111.274159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liang L., Deng L., Nguyen S.C., Zhao X., Maulion C.D., Shao C., Tischfield J.A. Human DNA ligases I and III, but not ligase IV, are required for microhomology-mediated end joining of DNA double-strand breaks. Nucleic Acids Res. 2008;36:3297–3310. doi: 10.1093/nar/gkn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oh S., Harvey A., Zimbric J., Wang Y., Nguyen T., Jackson P.J., Hendrickson E.A. DNA ligase III and DNA ligase IV carry out genetically distinct forms of end joining in human somatic cells. DNA Repair (Amst) 2014;21:97–110. doi: 10.1016/j.dnarep.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boboila C., Alt F.W., Schwer B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv. Immunol. 2012;116:1–49. doi: 10.1016/B978-0-12-394300-2.00001-6. [DOI] [PubMed] [Google Scholar]
- 54.Decottignies A. Alternative end-joining mechanisms: a historical perspective. Front Genet. 2013;4:48. doi: 10.3389/fgene.2013.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deriano L., Roth D.B. Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annu. Rev. Genet. 2013;47:433–455. doi: 10.1146/annurev-genet-110711-155540. [DOI] [PubMed] [Google Scholar]
- 56.McVey M., Lee S.E. MMEJ repair of double-strand breaks (director's cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–538. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Betermier M., Bertrand P., Lopez B.S. Is non-homologous end-joining really an inherently error-prone process. PLoS Genet. 2014;10:e1004086. doi: 10.1371/journal.pgen.1004086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alvarez-Quilon A., Serrano-Benitez A., Ariel Lieberman J., Quintero C., Sanchez-Gutierrez D., Escudero L.M., Cortes-Ledesma F. ATM specifically mediates repair of double-strand breaks with blocked DNA ends. Nat. Commun. 2014;5:3347. doi: 10.1038/ncomms4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Averbeck N.B., Ringel O., Herrlitz M., Jakob B., Durante M., Taucher-Scholz G. DNA end resection is needed for the repair of complex lesions in G1-phase human cells. Cell Cycle. 2014;13:2509–2516. doi: 10.4161/15384101.2015.941743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barton O., Naumann S.C., Diemer-Biehs R., Kunzel J., Steinlage M., Conrad S., Makharashvili N., Wang J., Feng L., Lopez B.S., et al. Polo-like kinase 3 regulates CtIP during DNA double-strand break repair in G1. J. Cell Biol. 2014;206:877–894. doi: 10.1083/jcb.201401146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schipler A., Iliakis G. DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res. 2013;41:7589–7605. doi: 10.1093/nar/gkt556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chapman J.R., Taylor M.R., Boulton S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 63.Loser D.A., Shibata A., Shibata A.K., Woodbine L.J., Jeggo P.A., Chalmers A.J. Sensitization to radiation and alkylating agents by inhibitors of poly(ADP-ribose) polymerase is enhanced in cells deficient in DNA double-strand break repair. Mol. Cancer Ther. 2010;9:1775–1787. doi: 10.1158/1535-7163.MCT-09-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kotter A., Cornils K., Borgmann K., Dahm-Daphi J., Petersen C., Dikomey E., Mansour W.Y. Inhibition of PARP1-dependent end-joining contributes to Olaparib-mediated radiosensitization in tumor cells. Mol. Oncol. 2014;8:1616–1625. doi: 10.1016/j.molonc.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.