

Abstract
Tobacco smoke contains more than 4,000 compounds. These include phenols, carbonyls, and nitrosamines that may be irritants and carcinogens; particulate matter such as tars; volatiles and gases such as carbon monoxide; and nicotine. Many of these compounds may contribute to the adverse health effects of tobacco. For example, recent findings have shown that the angiogenic and proliferative effects of nicotine are mediated by activation of nicotinic receptors on the vascular cells. Nicotine-induced activation of vascular cells may contribute to pathological neovascularization in cancer, age-related macular degeneration, and atherosclerosis. This review focuses on how nicotine adversely affects cardiovascular health and highlights intriguing new data about nicotine's potent angiogenic and proliferative properties.
Keywords: nicotine, atherosclerosis, angiogenic effect, nicotine replacement therapy, SLURP peptides, nicotinic acetylcholine receptors, vascular endothelial growth factor

J. P. Cooke, M.D., Ph.D.
The Burden of Tobacco-Related Vascular Disease
A microlife is a unit of measurement used to communicate the effects of chronic risks on longevity.1 In counseling a young adult about the health effects of various behaviors, a microlife, or 1/1,000,000 of the remaining lifetime of the young adult, is about 30 minutes. Sitting for 2 hours in front of a television causes a loss of one microlife. Comparatively, smoking a pack of cigarettes causes a loss of 10 microlives.1 This loss of longevity from smoking is due to an increased risk for cancer, chronic obstructive pulmonary disease, and atherosclerotic disease. The latter remains the major cause of death worldwide, largely due to heart attack and stroke. Peripheral arterial disease and aortic aneurysms are also more common in smokers and cause substantial morbidity.
Multiple processes contribute to the pathobiology of tobacco-induced vascular disease. Cigarette smoke or its individual components may cause insulin resistance and dyslipidemia, promote vascular thrombosis and inflammation, induce aberrant vascular growth and angiogenesis, and impair endothelial homeostatic and regenerative functions.2–4 Uncovering the mechanisms of tobacco-related disease is complicated by the fact that smoke contains more than 4,000 different components,5 some of which are potentially injurious, such as gases (e.g., carbon monoxide), phenolics and carbonyl compounds, oxidizing agents, particulate matter, and nicotine. In addition, secondhand smoke may be more toxic, per gram of total particulate matter, than the “mainstream” cigarette smoke that a smoker inhales.6 Epidemiologic studies have indicated that passive smokers (e.g., individuals exposed to secondhand smoke) may suffer from an increased risk of respiratory and coronary artery disease.7 Recently, scientists have described the concept of “thirdhand” smoke as the vapor that comes from drapes, rugs, or other fabric in a room that was exposed to cigarette smoke. Components of the smoke are trapped by the fabric, which may act as a catalytic surface to further oxidize the smoke components. Subsequently, they can be released back into the air and may contribute to the “staleness” of a room previously occupied by a smoker.8 The adverse health effects of thirdhand smoke are now under investigation.
Nicotine is the habituating agent in tobacco smoke, and it also has adverse cardiovascular effects. Nicotine activates the sympathetic nervous system, releasing catecholamines that elevate the heart rate and blood pressure.9 Acutely, this effect increases myocardial oxygen demand, which can be dangerous in patients with coronary artery disease. Chronically, the elevation of heart rate and/or blood pressure would be expected to accelerate the progression of atherosclerosis and the risk of myocardial infarction and heart failure. In this regard, nicotine also increases platelet aggregability,4 which may increase the risk of an acute coronary syndrome. Platelets also may contribute to the gradual growth of plaque through the accretion of thrombus as well as through the release of growth factors, such as platelet-derived relaxing factor, that induce vascular smooth muscle cell proliferation. In addition, nicotine has direct actions on vascular cells. As described below, we and others have made the surprising observation that there are receptors for nicotine on the vessel wall. The activation of these vascular nicotine receptors has unexpected effects on vascular cell proliferation and growth.
The Direct Effect of Nicotine on Vascular Cells
The endothelium is a diaphanous tissue, a delicate monolayer of cells that exert powerful effects on vessel tone, vessel structure, and vascular interaction with circulating blood elements. The healthy endothelium produces a panoply of paracrine factors that generally induce vasodilation, inhibit platelet adherence, reduce vascular proliferation, and suppress leukocyte infiltration.10 Paradigmatic of such factors is endothelium-derived nitric oxide (NO). Thus, a healthy endothelium opposes atherogenic processes. By contrast, exposure to cardiovascular risk factors causes endothelial dysfunction. For example, in response to oxidized low-density lipoprotein (LDL) cholesterol or elevated levels of glucose, the endothelium generates less NO and more adhesion molecules and becomes more adhesive for leukocytes.11,12 These endothelial alterations contribute to the initiation and progression of atherosclerosis.
Accordingly, we hypothesized that nicotine would impair endothelial function, and we began by looking at the critical endothelial function of angiogenesis. To our surprise, we observed that nicotine enhanced key angiogenic processes—including endothelial growth, migration, and survival—at clinically relevant concentrations, i.e., concentrations to which moderate smokers are exposed.13 Nicotine also promoted capillary-like network formation in vitro. Although these findings were surprising, they are consistent with the fact that pathological neovascularization contributes to many tobacco-related diseases. For example, tumor angiogenesis and choroidal neovascularization, two diseases more common in smokers, contribute to cancer and age-related macular degeneration, respectively.14,15 Our observations were also consistent with the notion that plaque neovascularization is critically involved in plaque progression (Figure 1). Cliff Barger at Harvard first highlighted the association between coronary artery plaque and localized expansion of the vasa vasorum in human hearts postmortem16 by proposing that neovascularization of the plaque played a role in its progression. About a decade later, Judah Folkman and colleagues confirmed the association between plaque neovascularization, plaque growth, and macrophage infiltration in the aorta of hypercholesterolemic apolipoprotein (ApoE)-deficient mice. Furthermore, antiangiogenic agents reduced macrophage accumulation and slowed the progression of plaque growth.17 More evidence that angiogenesis plays a role in plaque growth was noted in the cholesterol-fed rabbit model by Celletti and colleagues. They observed that the potent angiogenic agent vascular endothelial growth factor (VEGF) could promote plaque growth in association with greater plaque neovascularization.18
Figure 1.
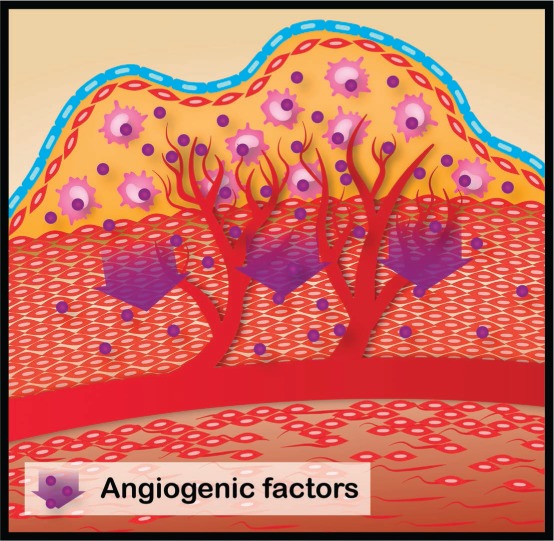
As an atherosclerotic plaque grows, cells in the interior of the lesion may become hypoxic. Cells within the lesion respond to this stress by activating the expression of genes such as vascular endothelial growth factor (VEGF). The angiogenic response to VEGF induces capillary sprouting, endothelial cell migration, proliferation, and luminogenesis to generate new capillaries. The small blood vessels that penetrate into the lesion would provide oxygen and nutrition for further proliferation of the plaque cells. These small, delicate plaque vessels may become disrupted, causing plaque hemorrhage, rapid expansion, and instability. Illustration by Matthew Landry.
These studies are consistent with our understanding that atherosclerotic plaque is not an inanimate obstruction but includes proliferating immune cells and myofibroblasts. When oxygen delivery to a tissue is reduced, the cells within that tissue begin to respond in a concerted manner orchestrated in large part by the transcriptional factor hypoxia-inducible factor-1a (HIF-1a).19 In the cells of hypoxic tissues, HIF-1a translocates to the nucleus and coordinates the ischemic response by activating the expression of genes such as VEGF. The angiogenic response to VEGF induces capillary sprouting, endothelial cell migration, proliferation, and luminogenesis to generate new capillaries. Accordingly, as an atherosclerotic plaque grows, cells inside the lesion may become hypoxic, activating HIF-1a.20 This transcriptional activation would be expected to increase the expression of angiogenic cytokines that encourage the ingrowth of capillaries derived from the vasa vasorum. These small, delicate blood vessels that penetrate into the lesion would provide oxygen and nutrition for further proliferation of the plaque cells and may in turn become disrupted, causing plaque hemorrhage, rapid expansion, and instability.
Given our discovery of the angiogenic effect of nicotine, we then investigated the effect of the drug on plaque neovascularization and growth and found that clinically relevant doses of nicotine increased plaque progression and neovascularization in the ApoE−/− mouse13; however, nicotine administration did not alter plasma lipid values. Therefore the nicotine-induced plaque progression was not due to an effect on plasma cholesterol. Notably, the effects of nicotine were reversed by coadministration of the antiangiogenic agent rofecoxib. These data suggest that nicotine increases the progression of atherosclerosis in part through pathological neovascularization. In subsequent studies, we also demonstrated that nicotine could exacerbate other forms of pathological angiogenesis in murine models of disease, including tumor angiogenesis and choroidal neovascularization, thereby contributing to the progression of cancer and macular degeneration.21,22
Nicotine Receptors in the Vessel Wall
We and others have now characterized the cholinergic receptors on endothelial and vascular smooth muscle cells. Vascular cells express two major types of cholinergic receptors: muscarinic and nicotinic.23 The muscarinic receptors are in the large family of 7-transmembrane spanning G protein–gated receptors. The nicotinic acetylcholine receptors (nAChRs) are quite different. These pentameric receptors are arranged in a barrel-like configuration to form an ion channel in the cell membrane. Of course, the nicotinic acetylcholine receptor did not evolve to detect nicotine in the bloodstream. These receptors detect endogenous acetylcholine, which is synthesized and stored in endothelial cells and acts as an autocrine factor in the vascular system.23–25 Other potential endogenous activators of the vascular nAChRs include choline and the peptides SLURP-1 and -2 (lymphocyte antigen-6/urokinase-type plasminogen activator receptor-related protein-1 and -2).26 The SLURP peptides allosterically modulate the activity of the nAChRs. It is not known if these peptides are generated in blood vessels or expressed in atherosclerosis. However, it is clear that endogenous acetylcholine activates both the muscarinic and the nicotinic acetylcholine receptors in the vessel wall. By contrast, exogenous nicotine preferentially stimulates the nAChRs. When activated by native acetylcholine or exogenous nicotine, the central channel of the nAChR becomes more permeable to cations (e.g., sodium or calcium). The sodium or calcium current that is generated by the activated nAChR then triggers further intracellular signaling.
The nAChRs were first described in excitable cells and were later detected in many other cell types including vascular and immune cells.13,27,28 Existing as pentameric complexes, these receptors may consist entirely of one isoform to form a homomeric channel or may consist of combinations of the 16 different isoforms (α1-α10, β1-β4, δ, γ, and ϵ) to form heteromeric channels. There is a diversity of nAChRs due to various combinations of α- and β- subunits, which generate receptors that have different affinity for ligands, selectivity for cations, and preference for signaling pathways.29,30
The surprising effect of nicotine enhancing angiogenic processes seems to be mediated primarily by the homomeric α7-nAChR, which is a pentamer composed entirely of α7 subunits. Pharmacological antagonism (by alpha-bungarotoxin), genetic knockout, or siRNA knockdown of the α7-nAChR significantly inhibits nicotine-induced activation of angiogenic processes in endothelial cells.13,21
A Role for the “Muscle Type” nAChR in Atherosclerosis
The nAChR in skeletal muscle is a specific assembly of five polypeptide subunits (α1, β1, δ, and ϵ in a 2:1:1:1 ratio). This nicotinic receptor resides in the neuromuscular junction and is known as the “muscle-type” nAChR. Recently, it has been described in other cell types including endothelial and vascular smooth muscle cells.31 In vascular smooth muscle cells, activation of this receptor increases cell proliferation and migration in vitro. This observation is consistent with prior work showing that nicotine can induce the elaboration of fibroblast growth factor and metalloproteinase by vascular smooth muscle cells.32 These findings are relevant to atherosclerosis, as vascular smooth muscle cell proliferation and extracellular matrix generation contribute to plaque growth. During the development of a vascular lesion, proliferating vascular smooth muscle cells (or a VSMC progenitor) from the adventitia or media migrate into the intima and undergo phenotypic modulation into myofibroblasts and osteoblast-like cells. There, they elaborate extracellular matrix (collagen and osteopontin) and even transdifferentiate into macrophage-like foam cells.33
It is of interest that the “muscle type” nAChR is upregulated significantly in the hypercholesterolemic mouse. Silencing of the α1 subunit using siRNA reduces vascular smooth muscle proliferation in vitro. Most intriguingly, when administered to hypercholesterolemic mice, siRNA against the α1 subunit substantially reduces accumulation of extracellular matrix (collagen and osteopontin), attenuates calcification, and markedly reduces the progression of atherosclerotic plaque.31
Immune cells in the atherosclerotic lesion may also express the “muscle-type” nAChR, which is linked to macrophage calpain activity and inflammation. On the other hand, macrophages are also known to express the homomeric α7-nAChR.34,35 This receptor might act to oppose the proatherogenic effects of nicotine, as its activation down-regulates the synthesis of proinflammatory cytokines and mitigates inflammation in murine models of sepsis. The macrophage α7-nAChR is a critical element in the anti-inflammatory effects of vagus nerve stimulation.
Clinical Ramifications of this Work
Clearly, nicotine is vasoactive. By increasing the activity of the sympathetic nervous system, nicotine indirectly increases vascular resistance. However, we and others have now shown that there are direct and powerful effects of nicotine on the cells of the vessel wall. The accumulating data suggest that nicotine's effects on the endothelium and the vascular smooth muscle promote the progression of atherosclerosis. In this regard, it is of interest that human genomic data are revealing that individual variations in the nAChRs may increase the risk for vascular disease. For example, genome-wide association studies suggest that the risk of peripheral arterial disease (PAD) is increased by a sequence variant in the cluster of genes on chromosome 15 that encode nicotinic acetylcholine receptors.36 The increased risk of PAD in these subjects may be due to a direct alteration in the activity of vascular nicotine receptors and/or to an increased susceptibility to nicotine dependence.
Our observations regarding the vascular effects of nicotine are relevant to the long-term safety of nicotine replacement therapies (NRTs) for smokers. Medicinal nicotine is a mainstay in tobacco cessation programs. Whereas short-term use of NRTs has been shown to be safe and effective in tobacco cessation, there are little clinical data on the safety and efficacy of long-term use of NRTs. The preclinical data discussed above reveals that nicotine is a potent angiogenic agent and raises the concern that the angiogenic component of tobacco-related diseases (cancer, age-related macular degeneration, and atherosclerosis) may be promoted by medicinal nicotine. Clearly tobacco smoke, with its potpourri of carcinogens, toxins, and particulate matter, is more hazardous than medicinal nicotine. Furthermore, there is no evidence from observational studies that individuals taking NRTs for greater than the recommended periods are at greater risk for adverse events related to pathological angiogenesis. For this reason, and because of the importance of tobacco cessation, the U.S. Food and Drug Administration has softened its stance on the long-term use of medicinal nicotine.37 However, in the absence of randomized clinical trials on the long-term use of nicotine, the astute clinician might consider additional approaches toward tobacco cessation so as to reduce the long-term use of medicinal nicotine. Although the effect of secondhand smoke is dependent upon many factors, including the intensity of cigarette use and the air exchange in a room, a recent meta-analysis of secondhand tobacco smoke studies indicated that plasma nicotine levels are often high enough to have biological effects.38
Conflict of Interest Disclosure: The authors have completed and submitted the Methodist DeBakey Cardiovascular Journal Conflict of Interest Statement and none were reported. Dr. Cooke is an inventor on patents related to therapeutic modulation of angiogenesis by agonists or inhibitors of the nicotinic receptors.
Acknowledgments
Funding/Support: This work was supported in part by grants from the National Institutes of Health (U01HL100397 and R0-1 EY02060901) and by internal funding from the Houston Methodist Research Institute.
References
- 1.Spiegelhalter D. Using speed of ageing and “microlives” to communicate the effects of lifetime habits and environment. BMJ. 2012 Dec 14;345:e8223. doi: 10.1136/bmj.e8223. [DOI] [PubMed] [Google Scholar]
- 2.Benowitz NL. Cigarette smoking and cardiovascular disease: pathophysiology and implications for treatment. Prog Cardiovasc Dis. 2003 Jul–Aug;46(1):91–111. doi: 10.1016/s0033-0620(03)00087-2. [DOI] [PubMed] [Google Scholar]
- 3.Heiss C, Amabile N, Lee AC et al. Brief secondhand smoke exposure depresses endothelial progenitor cells activity and endothelial function: sustained vascular injury and blunted nitric oxide production. J Am Coll Cardiol. 2008 May 6;51(18):1760–71. doi: 10.1016/j.jacc.2008.01.040. [DOI] [PubMed] [Google Scholar]
- 4.Flouris AD, Vardavas CI, Metsios GS, Tsatsakis AM, Koutedakis Y. Biological evidence for the acute health effects of secondhand smoke exposure. Am J Physiol Lung Cell Mol Physiol. 2010 Jan;298(1):L3–L12. doi: 10.1152/ajplung.00215.2009. [DOI] [PubMed] [Google Scholar]
- 5.Préfontaine D, Morin A, Jumarie C, Porter A. In vitro bioactivity of combustion products from 12 tobacco constituents. Food Chem Toxicol. 2006 May;44(5):724–38. doi: 10.1016/j.fct.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 6.Schick S, Glantz S. Philip Morris toxicological experiments with fresh sidestream smoke: more toxic than mainstream smoke. Tob Control. 2005 Dec;14(6):396–404. doi: 10.1136/tc.2005.011288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Novak K. Passive smoking: out from the haze. Nature. 2007 Jun 28;447(7148):1049–51. doi: 10.1038/4471049a. [DOI] [PubMed] [Google Scholar]
- 8.Becquemin MH, Bertholon JF, Bentayeb M et al. Third-hand smoking: indoor measurements of concentration and sizes of cigarette smoke particles after resuspension. Tob Control. 2010 Aug;19(4):347–8. doi: 10.1136/tc.2009.034694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Middlekauff HR, Park J, Moheimani RS. Adverse effects of cigarette and noncigarette smoke exposure on the autonomic nervous system: mechanisms and implications for cardiovascular risk. J Am Coll Cardiol. 2014 Oct 21;64(16):1740–50. doi: 10.1016/j.jacc.2014.06.1201. [DOI] [PubMed] [Google Scholar]
- 10.Cooke JP, Ghebremariam YT. DDAH Says NO to ADMA. Arterioscler Thromb Vasc Biol. 2011 Jul;31(7):1462–4. doi: 10.1161/ATVBAHA.111.228833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Böger RH, Bode-Böger SM, Szuba A et al. Asymmetric dimethylarginine (ADMA): a novel risk factor for endothelial dysfunction: its role in hypercholesterolemia. Circulation. 1998 Nov 3;98(18):1842–7. doi: 10.1161/01.cir.98.18.1842. [DOI] [PubMed] [Google Scholar]
- 12.Lin KY, Ito A, Asagami T et al. Impaired nitric oxide synthase pathway in diabetes mellitus: role of asymmetric dimethylarginine and dimethylarginine dimethylaminohydrolase. Circulation. 2002 Aug 20;106(8):987–92. doi: 10.1161/01.cir.0000027109.14149.67. [DOI] [PubMed] [Google Scholar]
- 13.Heeschen C, Jang JJ, Weis M et al. Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nat Med. 2001 Jul;7(7):833–9. doi: 10.1038/89961. [DOI] [PubMed] [Google Scholar]
- 14.Zhu BQ, Heeschen C, Sievers RE et al. Second hand smoke stimulates tumor angiogenesis and growth. Cancer Cell. 2003 Sep;4(3):191–6. doi: 10.1016/s1535-6108(03)00219-8. [DOI] [PubMed] [Google Scholar]
- 15.Thornton J, Edwards R, Mitchell P, Harrison RA, Buchan I, Kelly SP. Smoking and age-related macular degeneration: a review of association. Eye (Lond) 2005 Sep;19(9):935–44. doi: 10.1038/sj.eye.6701978. [DOI] [PubMed] [Google Scholar]
- 16.Barger AC, Beeuwkes R, 3rd, Lainey LL, Silverman KJ. Hypothesis: vasa vasorum and neovascularization of human coronary arteries. A possible role in the pathophysiology of atherosclerosis. N Engl J Med. 1984 Jan 19;310(3):175–7. doi: 10.1056/NEJM198401193100307. [DOI] [PubMed] [Google Scholar]
- 17.Moulton KS, Heller E, Konerding MA, Flynn E, Palinski W, Folkman J. Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation. 1999 Apr 6;99(13):1726–32. doi: 10.1161/01.cir.99.13.1726. [DOI] [PubMed] [Google Scholar]
- 18.Celletti FL, Hilfiker PR, Ghafouri P, Dake MD. Effect of human recombinant vascular endothelial growth factor165 on progression of atherosclerotic plaque. J Am Coll Cardiol. 2001 Jun 15;37(8):2126–30. doi: 10.1016/s0735-1097(01)01301-8. [DOI] [PubMed] [Google Scholar]
- 19.Semenza GL. Hypoxia-inducible factor 1 and cardiovascular disease. Annu Rev Physiol. 2014;76:39–56. doi: 10.1146/annurev-physiol-021113-170322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lim CS, Kiriakidis S, Sandison A, Paleolog EM, Davies AH. Hypoxia-inducible factor pathway and diseases of the vascular wall. J Vasc Surg. 2013 Jul;58(1):219–30. doi: 10.1016/j.jvs.2013.02.240. [DOI] [PubMed] [Google Scholar]
- 21.Heeschen C, Weis M, Aicher A, Dimmeler S, Cooke JP. A novel angiogenic pathway mediated by non-neuronal nicotinic acetylcholine receptors. J Clin Invest. 2002 Aug;110(4):527–36. doi: 10.1172/JCI14676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kiuchi K, Matsuoka M, Wu JC et al. Mecamylamine suppresses Basal and nicotine-stimulated choroidal neovascularization. Invest Opthalmol Vis Sci. 2008 Apr;49(4):1705–11. doi: 10.1167/iovs.07-0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wessler I, Kirkpatrick CJ, Racké K. The cholinergic ‘pitfall’: acetylcholine, a universal cell molecule in biological systems, including humans. Clin Exp Pharmacol Physiol. 1999 Mar;26(3):198–205. doi: 10.1046/j.1440-1681.1999.03016.x. [DOI] [PubMed] [Google Scholar]
- 24.Kawashima K, Oohata H, Fujimoto K, Suzuki T. Extraneuronal localization of acetylcholine and its release upon nicotinic stimulation in rabbits. Neurosci Lett. 1989 Oct 9;104(3):336–9. doi: 10.1016/0304-3940(89)90599-5. [DOI] [PubMed] [Google Scholar]
- 25.Lindstrom J. Neuronal nicotinic acetylcholine receptors. Ion Channels. 1996;4:377–450. doi: 10.1007/978-1-4899-1775-1_10. [DOI] [PubMed] [Google Scholar]
- 26.Moriwaki Y, Yoshikawa K, Fukuda H, Fujii YX, Misawa H, Kawashima K. Immune system expression of SLURP-1 and SLURP-2, two endogenous nicotinic acetylcholine receptor ligands. Life Sci. 2007 May 30;80(24–25):2365–8. doi: 10.1016/j.lfs.2006.12.028. [DOI] [PubMed] [Google Scholar]
- 27.Villablanca AC. Nicotine stimulates DNA synthesis and proliferation in vascular endothelial cells in vitro. J Appl Physiol (1985) 1998 Jun;84(6):2089–98. doi: 10.1152/jappl.1998.84.6.2089. [DOI] [PubMed] [Google Scholar]
- 28.Richman DP, Arnason BG. Nicotinic acetylcholine receptor: evidence for a functionally distinct receptor on human lymphocytes. Proc Natl Acad Sci U S A. 1979 Sep;76(9):4632–5. doi: 10.1073/pnas.76.9.4632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Changeux JP. Thudichum Medal Lecture. The acetylcholine receptor: a model for allosteric membrane proteins. Biochem Soc Trans. 1995 May;23(2):195–205. doi: 10.1042/bst0230195. [DOI] [PubMed] [Google Scholar]
- 30.Conti-Tronconi BM, McLane KE, Raftery MA, Grando SA, Protti MP. The nicotinic acetylcholine receptor: structure and autoimmune pathology. Crit Rev Biochem Mol Biol. 1994;29(2):69–123. doi: 10.3109/10409239409086798. [DOI] [PubMed] [Google Scholar]
- 31.Zhang G, Marshall AL, Thomas AL et al. In vivo knockdown of nicotinic acetylcholine receptor α1 diminishes aortic atherosclerosis. Atherosclerosis. 2011 Mar;215(1):34–42. doi: 10.1016/j.atherosclerosis.2010.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carty CS, Soloway PD, Kayastha S et al. Nicotine and cotinine stimulate secretion of basic fibroblast growth factor and affect expression of matrix metalloproteinases in cultured human smooth muscle cells. J Vasc Surg. 1996 Dec;24(6):927–34. doi: 10.1016/s0741-5214(96)70038-1. [DOI] [PubMed] [Google Scholar]
- 33.Feil S, Fehrenbacher B, Lukowski R et al. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ Res. 2014 Sep 12;115(7):662–7. doi: 10.1161/CIRCRESAHA.115.304634. [DOI] [PubMed] [Google Scholar]
- 34.Wang H, Yu M, Ochani M et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003 Jan 23;421(6921):384–8. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- 35.Huston JM, Ochani M, Rosas-Ballina M et al. Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med. 2006 Jul 10;203(7):1623–8. doi: 10.1084/jem.20052362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thorgeirsson TE, Geller F, Sulem P et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008 Apr 3;452(7187):638–42. doi: 10.1038/nature06846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Silver Spring, MD: U.S. Food and Drug Administration; c2015. U.S. Food and Drug Administration [Internet] FDA consumer health information: nicotine replacement therapy labels may change, 2015 [cited 2015 Apr 13]. Available from: http://www.fda.gov/downloads/ForConsumers/ConsumerUpdates/UCM346012.pdf. [Google Scholar]
- 38.Okoli TCC, Kelly T, Hahn EJ. Secondhand smoke and nicotine exposure: a brief review. Addictive Behaviors. 2007 Oct 1;32(10):1977–1988. doi: 10.1016/j.addbeh.2006.12.024. [DOI] [PubMed] [Google Scholar]