
Fig. 1.
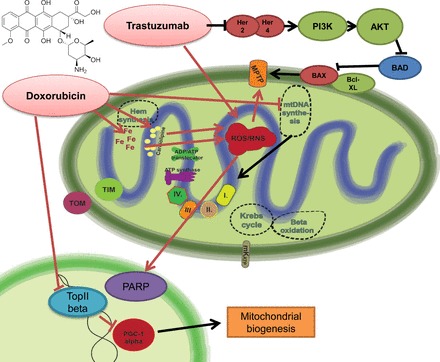
Doxorubicin-induced mitochondrial dysfunction and the effect of trastuzumab on mitochondria-related survival pathways. Doxorubicin leads to marked induction of mitochondrial reactive oxygen species (ROS) production. It shows specific binding activity to the mitochondrial abundant cardiolipin, leading to selective mitochondrial accumulation. Doxorubicin is prone to redox cycling, thereby promoting ROS and reactive nitrogen species (RNS) production. There is also increased intramitochondrial free iron accumulation after doxorubicin exposition, giving rise to additional nonenzymatic ROS production by the Haber-Weiss reaction. The major consequences of uncontrolled ROS/RNS production are mitochondrial permeability transition pore (MPTP) opening and poly(ADP-ribose) polymerase (PARP) activation, converging to the propagation to cell death signaling mechanisms. In parallel with ROS/RNS induction, doxorubicin also interferes with the action of topoisomerase-2β, being involved in the regulation of peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) and thereby mitochondrial biogenesis- and metabolism-related pathways. Altogether, these doxorubicin-induced alterations profoundly alter both mitochondrial structure and function. The release of neuregulin-1 by the coronary endothelium activates human epidermal growth factor receptor (Her)4 (ErbB4) to dimerize with Her2 (ErbB2). The Her4/Her2 (ErbB4/ErbB2) dimer activates cardioprotective signaling pathways, including phosphatidyinositol 3-kinase (PI3K)/protein kinase B (Akt), ERK1/2, and focal adhesion kinase (FAK), which promote cell survival upon cellular stress. Trastuzumab blocks Her2 signaling and disrupts this cardioprotective machinery, resulting in loss of cytoprotective mechanisms. Furthermore, trastuzumab may trigger cellular oxidative stress and induce the expression and activation of proapoptotic proteins [e.g., Bcl-2-associated X protein (BAX)]. These events result in mitochondrial defects, leading to the opening of the MPTP and the activation of cell death pathways that precipitate myocardial dysfunction. BAD, Bcl-2-associated death promoter; mKATP, mitochondrial ATP-sensitive potassium channel.