

Abstract
Background and objectives
Hyperkalemia affects up to 10% of patients with CKD. Sodium polystyrene sulfonate has long been prescribed for this condition, although evidence is lacking on its efficacy for the treatment of mild hyperkalemia over several days. This study aimed to evaluate the efficacy of sodium polystyrene sulfonate in the treatment of mild hyperkalemia.
Design, setting, participants, & measurements
In total, 33 outpatients with CKD and mild hyperkalemia (5.0–5.9 mEq/L) in a single teaching hospital were included in this double–blind randomized clinical trial. We randomly assigned these patients to receive either placebo or sodium polystyrene sulfonate of 30 g orally one time per day for 7 days. The primary outcome was the comparison between study groups of the mean difference of serum potassium levels between the day after the last dose of treatment and baseline.
Results
The mean duration of treatment was 6.9 days. Sodium polystyrene sulfonate was superior to placebo in the reduction of serum potassium levels (mean difference between groups, −1.04 mEq/L; 95% confidence interval, −1.37 to −0.71). A higher proportion of patients in the sodium polystyrene sulfonate group attained normokalemia at the end of their treatment compared with those in the placebo group, but the difference did not reach statistical significance (73% versus 38%; P=0.07). There was a trend toward higher rates of electrolytic disturbances and an increase in gastrointestinal side effects in the group receiving sodium polystyrene sulfonate.
Conclusions
Sodium polystyrene sulfonate was superior to placebo in reducing serum potassium over 7 days in patients with mild hyperkalemia and CKD.
Keywords: hyperkalemia; randomized controlled trial; polystyrene sulfonic acid; kidney failure, chronic; double-blind method; humans; outpatients; polystyrenes; potassium; renal insufficiency, chronic
Introduction
Hyperkalemia affects up to 10% of patients suffering from CKD and up to 42% of patients with an eGFR<20 ml/min per 1.73 m2 (1). Hyperkalemia can cause fatal cardiac arrhythmias and has been associated with increased mortality in patients with CKD (2–4). Reduction of dietary intake, modification of contributing medications, and use of cation-exchange resins are presently the cornerstone of treatment of mild chronic hyperkalemia (5–8). Sodium polystyrene sulfonate (SPS) is a cation-exchange resin that was approved by the US Food and Drug Administration (FDA) in 1958, 4 years before the Kefauver–Harris Drug Amendments were passed to ensure drug efficacy and safety. Consequently, data on SPS in the treatment of mild hyperkalemia are insufficient, especially when used over several days in a population of patients with CKD. SPS has historically been used combined with sorbitol. In September of 2009, the FDA released a black box warning about the increased risk of intestinal necrosis with this combination (9). This new warning along with the lack of strong data on its efficacy affect clinicians’ willingness to use SPS as a first-line treatment of hyperkalemia (10).
Efficacy of SPS in the treatment of chronic hyperkalemia was evaluated in a retrospective observational study in 14 patients with CKD and cardiovascular disease taking medication affecting the renin-angiotensin system (11). The small sample size and the lack of control group made it difficult to differentiate the effect of SPS from that of other factors, such as dietary restrictions and medication changes. Our double–blind, randomized, placebo–controlled trial aimed to evaluate the use of SPS in the treatment of mild hyperkalemia in outpatients with CKD.
Materials and Methods
Study Design
The study was conducted at the nephrology and predialysis outpatient clinics of Maisonneuve-Rosemont Hospital between February and September of 2014, which was the predetermined end date. The trial was registered on ClinicalTrials.gov (NCT 02065076) and funded by the Nephrology Research Axis of Maisonneuve-Rosemont Hospital, the Dean’s Circle of the Faculty of Pharmacy of the University of Montreal, and the Department of Pharmacy of Maisonneuve-Rosemont Hospital. The placebo (carob gum and lactose; Galenova Inc.) and SPS (Kayexalate; Sanofi Aventis) powders were purchased from Galenova Inc., an independent firm, to ensure blinding. Neither products contained sorbitol. Study protocol was approved by the Institutional Scientific Committee and Research Ethics Board. All patients provided written informed consent. All authors declare their adherence to the Declaration of Helsinki.
Study Population
Patients ages 18 years old and older with eGFR<40 ml/min per 1.73 m2 (calculated using the Modification of Diet in Renal Disease four–variable equation) were assessed for eligibility according to their serum potassium levels measured for their usual follow-up with their nephrologist and on the day of randomization (12). Patients who had taken SPS in the 7 days before their initial visit were eligible if their serum potassium level measured for their regular follow-up was from 4.5 to 5.5 mEq/L (multiply by 1 to convert to millimoles per liter) inclusively and from 5.0 to 5.9 mEq/L inclusively after a 7-day washout period (day of randomization). Patients who had not taken SPS in the 7 days before their initial visit were eligible if their serum potassium levels measured for their regular follow-up and on the day of randomization were both from 5.0 to 5.9 mEq/L inclusively.
Patients on dialysis were not included in this study. Exclusion criteria included a change in medication affecting potassium levels within 30 days before enrolment. Exception was made for insulin, for which dose modifications occurring >7 days before randomization were accepted or if more recent modifications were not >10% of the total daily dose up to a maximum variation of five units. Patients with changes in dosage of angiotensin–converting enzyme inhibitors (ACEIs), angiotensin receptor blockers (ARBs), or aliskiren within 60 days before randomization were also excluded. For safety reasons, patients were excluded if they were pregnant or breastfeeding, had a contraindication to SPS, had a lactose intolerance, or had an episode of decompensated heart failure in the 30 days before enrolment. Patients with symptomatic hyperkalemia were not recruited. No electrocardiogram assessments were performed to objectify changes associated with hyperkalemia. Patients were ineligible if they were enrolled in another study.
Study Treatment and Procedures
Eligible patients were randomized in a 1:1 ratio in blocks of four to receive a fixed dose of 30 g SPS or placebo orally one time per day for 7 days. The random treatment arm sequence was generated by the website www.randomization.com and managed by independent pharmacists of the research sector of the pharmacy department. Patients, investigators, care providers, and statisticians were blinded for the duration of the trial and until all statistical analyses were completed. The placebo was similar in taste, texture, and appearance to SPS. Both were supplied as individual doses in tinted bottles. Patients were instructed to mix each dose with 150 ml water immediately before administration and take their study medication in the evening starting on the day of randomization. Their final follow-up was scheduled the day after their last dose.
Three days after randomization, investigators contacted patients by telephone to assess side effects using a standardized questionnaire. Seven days after randomization, patients returned for blood tests and completed the same side effects questionnaire as on day 3. For patients’ convenience, a 1-day difference was tolerated, meaning that patients could receive between six and eight doses. They were instructed to return all bottles (including empty ones) to investigators on the day of their final follow-up to allow an objective assessment of compliance, which was also evaluated with a modified version of a validated questionnaire.
Although no formal diet education was part of this trial, all patients with CKD and hyperkalemia followed in our nephrology and predialysis outpatient clinics routinely receive information on a low–potassium intake diet by a nutritionist, and patients were instructed not to modify their diet. Average dietary intake of potassium was estimated through a food diary completed by patients for 3 of 7 study days. Compilation was done in accordance with the Canadian Nutrient File from Health Canada, which was exported to a Microsoft Excel database.
Outcomes
The primary outcome was the comparison between study groups of the mean difference in serum potassium levels between the day after the last dose of treatment (day 7) and baseline (day 0). Secondary outcomes were to compare the proportion of patients achieving normokalemia, defined as a serum potassium level of at least 3.5 mEq/L and <5.0 mEq/L, at the end of the study period as well as compare the prevalence of side effects between study groups. Specifically, nausea, vomiting, constipation, and diarrhea were assessed on study days 3 and 7. Additionally, on day 7, blood tests assessed the frequency of hypernatremia, hypophosphatemia, hypomagnesemia, and hypocalcemia.
Statistical Analyses
To detect a mean difference of serum potassium of 0.5 mEq/L between study groups, a change felt to be clinically significant by investigators according to prior literature, a sample size of 38 patients (19 patients in each group) was estimated a priori to provide a power of 85% and a two–sided α-level of 0.05 (13).
Continuous descriptive baseline data are presented as means and SDs. For the analysis, patients were kept in the group assigned by randomization, whatever treatment they received. Two patients (one in each group) had to be excluded from the main analysis, because no serum potassium value at day 7 was available. These two patients were still included in the adverse events analysis. For the primary outcome, means of change in serum potassium level were compared between study groups using a t test. For the secondary outcomes, proportions of patients achieving normokalemia and presenting side effects were compared between study groups using a chi-squared test or a Fisher exact test in the presence of cells with fewer than five observations. As a sensitivity analysis, baseline characteristics that were clinically different between groups were entered in multivariate linear regression (for the primary outcome) models to investigate their influence on the main results. No interim analyses were performed. All analyses were conducted with the use of SAS software, version 9.4 (SAS Institute, Cary, NC).
Results
Patients
Of 329 eligible patients on the basis of their serum potassium levels measured for their usual follow-up, 106 declined to participate, and 190 were excluded, leaving 33 patients to be randomized into two groups (Figure 1); 16 patients received SPS, whereas 17 patients received placebo. Recruitment ended after the predetermined 7-month study period, despite the fact that the initially calculated sample size was not attained. Baseline characteristics were generally similar between the two study groups (Table 1), except that a higher proportion of patients was using insulin in the control group compared with the SPS group (58.8% versus 25.0%). Two patients had no measure of serum potassium levels at the end of the study period (day 7) and therefore, could not be included in the analysis. One patient allocated to the SPS group left the study on day 3 for important gastrointestinal side effects and refused to come to the follow-up visit on day 7. One patient receiving placebo had unplanned bloodwork done through a different laboratory, which revealed an elevated serum potassium level of 6.2 mEq/L. On the basis of the recommendation of the patient’s regular physician, the patient left the study to receive unblinded treatment. In both patients, blinding of study treatment was maintained.
Figure 1.
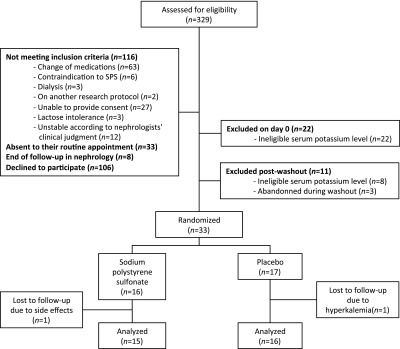
Consolidated Standards of Reporting Trials (CONSORT) flow diagram. SPS, sodium polystyrene sulfonate.
Table 1.
Characteristics of patients at baseline
| Characteristics | SPS (n=16) | Placebo (n=17) |
|---|---|---|
| Sex, no. (%) | ||
| Men | 10 (62.5) | 13 (76.5) |
| Age, yr | 72.7 (±11.6) | 71.9 (±9.6) |
| Serum creatinine, mg/dla | 3.0 (±0.8) | 3.6 (±1.3) |
| eGFR (MDRD), ml/min per 1.73 m2 | 20.0 (±7.2) | 17.7 (±6.6) |
| CKD stage, no. (%) | ||
| 3 (30–59 ml/min per 1.73 m2) | 2 (12.5) | 1 (5.9) |
| 4 (15–29 ml/min per 1.73 m2) | 10 (62.5) | 9 (52.9) |
| 5 (<15 ml/min per 1.73 m2) | 4 (25.0) | 7 (41.2) |
| Cause of CKD, no. (%) | ||
| Vascular (includes diabetic nephropathy) | 14 (87.5) | 12 (70.6) |
| GN | 0 | 2 (11.8) |
| Polycystic | 1 (6.3) | 0 |
| Other | 1 (6.3) | 3 (17.6) |
| Body mass index, kg/m2 | 27.4 (±6.4) | 29.4 (±6.0) |
| Comorbidities, no. (%) | ||
| Dyslipidemia | 13 (81.3) | 16 (94.1) |
| Hypertension | 14 (87.5) | 15 (88.2) |
| Diabetes | 11 (68.8) | 13 (76.5) |
| Coronary artery disease | 8 (50.0) | 11 (64.7) |
| History of stroke | 1 (6.3) | 4 (23.5) |
| Arrhythmia | 0 | 1 (5.9) |
| Congestive heart failure | 3 (18.8) | 0 |
| Medication, no. (%) | ||
| Any medication affecting potassium | 15 (93.8) | 16 (94.1) |
| Insulin | 4 (25.0) | 10 (58.8) |
| β-Blockers | 12 (75.0) | 9 (52.9) |
| Loop diuretics | 10 (62.5) | 8 (47.1) |
| ACEIs or ARBsb | 13 (81.3) | 10 (58.8) |
| Thiazide diuretics | 2 (12.5) | 2 (11.8) |
| Potassium-sparing diuretics | 0 | 2 (11.8) |
| Laxatives | 2 (12.5) | 2 (11.8) |
| NSAIDsc | 0 | 0 |
| BP, mmHg | ||
| Systolic | 150.0 (±21.3) | 141.9 (±13.4) |
| Diastolic | 69.1 (±8.1) | 68.1 (±8.8) |
| Use of SPS immediately before study enrolment, no. (%) | 4 (25.0) | 5 (29.4) |
SPS, sodium polystyrene sulfonate; MDRD, Modification of Diet in Renal Disease four–variable equation; ACEI, angiotensin–converting enzyme inhibitor; ARB, angiotensin receptor blocker; NSAID, nonsteroidal anti–inflammatory drug.
To convert creatinine levels from milligrams per deciliter to millimoles per liter, multiply by 88.4.
No patient was using an ACEI combined with an ARB.
Excludes aspirin at 80 mg daily.
Outcomes
Patients were followed for an average of 6.9 days. Comparisons of outcomes between groups are presented in Table 2. Serum potassium levels decreased by a mean (±SD) of 1.25±0.56 mEq/L in patients treated with SPS compared with 0.21±0.29 mEq/L in the group receiving placebo (difference of −1.04 mEq/L; 95% confidence interval, −1.37 to −0.71; P<0.001). At the end of the study period, 11 patients receiving SPS (73%) compared with 6 patients (38%) in the placebo group (P=0.07) had normokalemia, which was predefined as a serum potassium level of at least 3.5 mEq/L and <5.0 mEq/L. However, 14 patients receiving SPS (93%) compared with 6 patients (38%) in the placebo group attained a serum potassium level of <5 mEq/L (P=0.002). As a sensitivity analysis, adding insulin use in multivariate regression models did not change the results for the difference in change of serum potassium levels (difference estimate of −1.04; P<0.001).
Table 2.
Efficacy outcomes
| Outcomes | SPS (n=15) | Placebo (n=16) | Difference of Mean Serum Potassium Change (95% CI) | P Value |
|---|---|---|---|---|
| Serum potassium level at baseline, mEq/La | 5.26±0.22 | 5.23±0.22 | 0.73 | |
| Serum potassium level at final follow-up, mEq/La | 3.99±0.56 | 5.03±0.34 | <0.001 | |
| Variation of serum potassium, mEq/La | −1.25±0.57 | −0.21±0.29 | −1.04 (−1.37 to −0.71) | <0.001 |
| Normokalemia, no. (%) | 11 (73.0) | 6 (38.0) | 0.07 |
SPS, sodium polystyrene sulfonate; 95% CI, 95% confidence interval.
To convert potassium levels from milliequivalents per liter to millimoles per liter, multiply by 1.
Estimated dietary intake of potassium during the study was similar between both groups. Patients in the SPS group had a mean dietary potassium intake of 53.6±12.3 mEq/d compared with 59.3±22.7 mEq/d in the placebo group (P=0.40). Two patients in each study group did not complete their food diary.
Safety
Prevalence of gastrointestinal side effects was similar in both study groups. There was an increase in constipation, nausea, and vomiting in patients receiving SPS and an increased prevalence of diarrhea in the placebo group. No patients with colonic necrosis were observed in this trial. In the SPS group, there was a nonstatistically significant increase in the number of patients with hypokalemia (three versus zero; P=0.23), hypomagnesemia (five versus one; P=0.17), and hypocalcemia (three versus zero; P=0.23) at the end of their 7-day study period. There were no patients with hypernatremia or hypophosphatemia in either group. Electrolytic disturbances were generally minor. The most severe hypokalemia observed in the SPS group was of 3.0 mEq/L. The lowest level of magnesium observed was 1.12 mEq/L (0.56 mmol/L) after adjusted for hypoalbuminemia in a patient receiving placebo. In five patients receiving SPS with hypomagnesemia, one already had hypomagnesemia at baseline. None of the safety outcomes were statistically significant (Table 3).
Table 3.
Safety outcomes
| Outcomes | SPS (n=16) | Placebo (n=16) | P Value |
|---|---|---|---|
| Nausea, no. (%) | 4 (25.0) | 2 (12.5) | 0.65 |
| Vomiting, no. (%) | 2 (12.5) | 1 (6.3) | >0.99 |
| Constipation, no. (%) | 6 (37.5) | 4 (25.0) | 0.70 |
| Diarrhea, no. (%) | 4 (25.0) | 8 (50.0) | 0.27 |
| Electrolyte disturbances, no. (%)a | |||
| Hypokalemia (<3.5 mEq/L) | 3 (18.8) | 0 | 0.23 |
| Hypernatremia (>140 mEq/L) | 0 | 0 | N/A |
| Hypophosphatemia (<2.17 mg/dl) | 0 | 0 | N/A |
| Hypocalcemia (<8.48 mg/dl) | 3 (18.8) | 0 | 0.23 |
| Hypomagnesemia (<1.4 mEq/L) | 5 (31.2) | 1 (6.3) | 0.17 |
| Any adverse event, no. (%) | 12 (75.0) | 10 (58.8) | 0.47 |
Patients who had received at least one dose of the study drug were included in the safety analysis. One patient was lost to follow-up, and no safety data were collected. All gastrointestinal adverse events presented were specifically questioned on follow-up days 3 and 7. SPS, sodium polystyrene sulfonate; N/A, not applicable.
To convert sodium levels from milliequivalents per liter to millimoles per liter, multiply by 1. To convert potassium levels from milliequivalents per liter to millimoles per liter, multiply by 1. To convert phosphorus levels from milligrams per deciliter to millimoles per liter, multiply by 0.32. To convert calcium levels from milligrams per deciliter to millimoles per liter, multiply by 0.25. To convert magnesium levels from milliequivalents per liter to millimoles per liter, multiply by 0.5.
Two patients receiving SPS needed an increase in their doses of laxatives during the study period, whereas none were necessary in patients receiving placebo.
Discussion
The findings of this trial show that SPS is an effective potassium–lowering agent for a 7-day treatment of mild hyperkalemia in outpatients with CKD. A daily dose of 30 g SPS given during 7 days resulted in an absolute decrease of serum potassium levels of 1.25 mEq/L, with a statistically significant mean difference of 1.04 mEq/L compared with placebo. To our knowledge, this trial is the first to show the efficacy of SPS in a controlled setting, while maintaining stable medication that affects serum potassium; 73% of evaluated patients receiving SPS had normal serum potassium levels at the end of study compared with 38% of evaluated patients in the placebo group, but this outcome did not reach statistical significance, possibly because of the small sample size.
These findings are consistent with results obtained in the only observational study evaluating the use of repeated doses of SPS in patients with CKD (11). Chernin et al. (11) observed that serum potassium levels fell from 6.4±0.3 to 4.6±0.6 mEq/L (P<0.01) over a median follow-up period of 14.5 months. However, as previously stated, its lack of a control group made it difficult to differentiate the effect of SPS from other confounders.
For patients receiving a renin-angiotensin-aldosterone system blocker and developing hyperkalemia, guidelines suggest that one clinical intervention would be dose reduction or discontinuation of the renin-angiotensin-aldosterone system blocker (8). However, the clinical benefits of continuing an ARB or an ACEI as long as possible have prompted us in our practice to keep the doses stable while adding SPS and insisting on low-potassium intake in patients with mild hyperkalemia. Also, in this study, ARBs and ACEIs, although contributing to hyperkalemia, were not thought to be the principal cause, because those medications were at a stable dose for a minimum of 60 days.
Alternatives to SPS are presently developed: patiromer and zirconium cyclosilicate. Recent phase 3 trials using these new drugs have shown reductions in serum potassium levels at 48 or 72 hours ranging from 0.46 to 1.1 mEq/L, a magnitude of reduction similar to our results (14–16). However, the designs of these studies are very different and limit the comparison.
As mentioned before, the small sample size of the study does not permit us to draw conclusions about the safety of SPS. There was a trend toward higher rates of electrolytic disturbances and an increase in gastrointestinal side effects in the group receiving SPS. No adverse effects requiring hospitalization occurred throughout the study, although one patient in the SPS group stopped after 3 days of treatment because of important emesis. The most common side effects were nausea, constipation, hypomagnesemia, hypocalcemia, and hypokalemia. Although the newer agent patiromer is believed to have a better gastrointestinal side effect profile than SPS, it was associated with slight increases in constipation, diarrhea, and nausea (16). In recent trials, the gastrointestinal safety profile was similar between zirconium cyclosilicate and placebo (14,15).
Of note, a limiting factor in recruitment was the variability of potassium levels. Several patients meeting eligibility criteria were later excluded because of lower potassium levels on the day of their appointment without modification of their medication or diet. This high fluctuation suggests that controlling a high serum potassium level may be an appropriate first step before initiating any specific interventions for hyperkalemia.
This trial had several limitations. Serum potassium levels were measured disregarding fasting state because of logistic restraints, but the variation of potassium levels caused by this phenomenon would be balanced between groups and should not influence the results. In the placebo group, no patients were fasting (last meal >8 hours) on day 0, and one patient was fasting on day 7. In the SPS group, four patients were fasting on days 0 and 7. Changes in dietary intake of potassium were not measured. However, randomization and blinding of patients would limit the effect of diet on the results. Moreover, dietary intake of potassium during the study period was similar between study groups. Although most characteristics were well balanced between the two groups, use of insulin was more frequent in patients receiving placebo. However, insulin doses were stable during the study period in both groups, and adding insulin use in multivariate models did not change the results in the sensitivity analysis. This study does not address how SPS reduces serum potassium levels, for which urine collections may have been useful. Our small sample size limited our capacity of detecting rare side effects or a statistically significant difference in common side effects. In fact, to detect a difference of 10% in safety outcomes, at least 144 patients would be needed per treatment arm. For our actual sample size, a difference of six patients between groups (a 37.5% difference) was needed to attain statistical significance. Colonic necrosis, a rare but fatal side effect of SPS and the object of a black box warning from the FDA, was not observed in our trial (9). Risk of colonic necrosis may be related to high content in sorbitol, the reason why SPS diluted with water was used in our study (10). As a single-center study, the external validity is limited. However, the study population is representative of the general population of patients before starting dialysis in terms of age, body mass index, comorbidities, medication, and eGFR (11,17). Unfortunately, no black patients were randomized in this trial. However, there is no evidence that the effect of SPS would be different among blacks. The results of this trial cannot be extrapolated to use in acute treatment. Use >7 days was not evaluated; therefore, interpretation of these results in the context of chronic use over several weeks should be done with caution.
SPS is superior to placebo for the treatment of mild hyperkalemia in patients with CKD when used over a short period. Its use requires monitoring of gastrointestinal symptoms and electrolyte disorders. Colonic necrosis, although not seen in this trial, remains a concern. Because of its efficacy and low cost, SPS may remain an interesting first–line therapy for potassium lowering in patients who can tolerate it. Forthcoming potassium–lowering agents should be compared with SPS, and additional trials of comparative effectiveness research will be necessary (3). Finally, studies evaluating longer use of SPS are needed.
Disclosures
S.P. has a research chair from Sanofi Canada on drug use but has not received any funding for this specific project. All other authors declare no conflict of interest.
Acknowledgments
This work was funded by the Nephrology Research Axis of Maisonneuve-Rosemont Hospital, the Dean’s Circle of the Faculty of Pharmacy of the University of Montreal, and the Department of Pharmacy of Maisonneuve-Rosemont Hospital.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
References
- 1.Arora P, Vasa P, Brenner D, Iglar K, McFarlane P, Morrison H, Badawi A: Prevalence estimates of chronic kidney disease in Canada: Results of a nationally representative survey. CMAJ 185: E417–E423, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hayes J, Kalantar-Zadeh K, Lu JL, Turban S, Anderson JE, Kovesdy CP: Association of hypo- and hyperkalemia with disease progression and mortality in males with chronic kidney disease: The role of race. Nephron Clin Pract 120: c8–c16, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kovesdy CP: Management of hyperkalaemia in chronic kidney disease. Nat Rev Nephrol 10: 653–662, 2014 [DOI] [PubMed] [Google Scholar]
- 4.Einhorn LM, Zhan M, Hsu VD, Walker LD, Moen MF, Seliger SL, Weir MR, Fink JC: The frequency of hyperkalemia and its significance in chronic kidney disease. Arch Intern Med 169: 1156–1162, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hudson JQ: Chronic kidney disease: Management of complications. In: Pharmacotherapy: A Pathophysiologic Approach, 7th Ed., edited by Dipiro JT, Talbert RL, Yee GC, Matzke GR, Wells BG, Posey LM, New York, McGraw-Hill Medical, 2008, pp 765–791 [Google Scholar]
- 6.Evans KJ, Greenberg A: Hyperkalemia: A review. J Intensive Care Med 20: 272–290, 2005 [DOI] [PubMed] [Google Scholar]
- 7.Alfonzo AV, Isles C, Geddes C, Deighan C: Potassium disorders--clinical spectrum and emergency management. Resuscitation 70: 10–25, 2006 [DOI] [PubMed] [Google Scholar]
- 8.Kidney Disease Outcomes Quality Initiative (K/DOQI) : K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am J Kidney Dis 43[Suppl 1]: S1–S290, 2004 [PubMed] [Google Scholar]
- 9.US Food and Drug Administration : Kayexalate (Sodium Polystyrene Sulfonate) Powder. Available at: http://www.fda.gov/Safety/MedWatch/SafetyInformation/ucm240075.htm. Accessed November 8, 2015
- 10.Sterns RH, Rojas M, Bernstein P, Chennupati S: Ion-exchange resins for the treatment of hyperkalemia: Are they safe and effective? J Am Soc Nephrol 21: 733–735, 2010 [DOI] [PubMed] [Google Scholar]
- 11.Chernin G, Gal-Oz A, Ben-Assa E, Schwartz IF, Weinstein T, Schwartz D, Silverberg DS: Secondary prevention of hyperkalemia with sodium polystyrene sulfonate in cardiac and kidney patients on renin-angiotensin-aldosterone system inhibition therapy. Clin Cardiol 35: 32–36, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levey AS, Coresh J, Greene T, Stevens LA, Zhang YL, Hendriksen S, Kusek JW, Van Lente F, Chronic Kidney Disease Epidemiology Collaboration : Using standardized serum creatinine values in the modification of diet in renal disease study equation for estimating glomerular filtration rate. Ann Intern Med 145: 247–254, 2006 [DOI] [PubMed] [Google Scholar]
- 13.Fordjour KN, Walton T, Doran JJ: Management of hyperkalemia in hospitalized patients. Am J Med Sci 347: 93–100, 2014 [DOI] [PubMed] [Google Scholar]
- 14.Kosiborod M, Rasmussen HS, Lavin P, Qunibi WY, Spinowitz B, Packham D, Roger SD, Yang A, Lerma E, Singh B: Effect of sodium zirconium cyclosilicate on potassium lowering for 28 days among outpatients with hyperkalemia: The HARMONIZE randomized clinical trial. JAMA 312: 2223–2233, 2014 [DOI] [PubMed] [Google Scholar]
- 15.Packham DK, Rasmussen HS, Lavin PT, El-Shahawy MA, Roger SD, Block G, Qunibi W, Pergola P, Singh B: Sodium zirconium cyclosilicate in hyperkalemia. N Engl J Med 372: 222–231, 2015 [DOI] [PubMed] [Google Scholar]
- 16.Weir MR, Bakris GL, Bushinsky DA, Mayo MR, Garza D, Stasiv Y, Wittes J, Christ-Schmidt H, Berman L, Pitt B, OPAL-HK Investigators : Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N Engl J Med 372: 211–221, 2015 [DOI] [PubMed] [Google Scholar]
- 17.Sarafidis PA, Blacklock R, Wood E, Rumjon A, Simmonds S, Fletcher-Rogers J, Ariyanayagam R, Al-Yassin A, Sharpe C, Vinen K: Prevalence and factors associated with hyperkalemia in predialysis patients followed in a low-clearance clinic. Clin J Am Soc Nephrol 7: 1234–1241, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]