
Fig. 1.
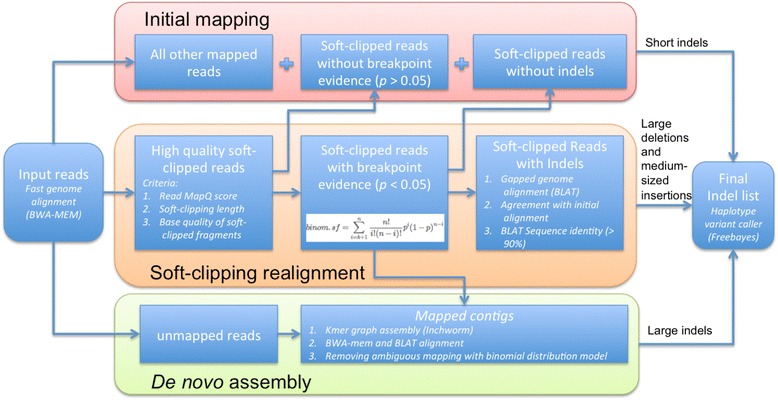
The ScanIndel workflow. ScanIndel aligns the raw read FASTQ files with a gapped NGS aligner (BWA-MEM) to detect short indels according to the initial mapping results. Soft-clipped reads with breakpoint evidence support are extracted for BLAT re-alignment to refine the CIGAR and genomic positions. Those re-aligned soft-clipped reads help to identify large deletions and medium-sized insertions. Meanwhile, ScanIndel carries out de novo assembly with the Inchworm assembler from Trinity for unmapped reads and BLAT realigned soft-clipped reads to detect large indels. All individual calling sets are merged by vcfcombine (from vcflib) to get one final VCF output containing all indel predictions