

I. How it started
I entered the Rega Institute for Medical Research in August 1964, as a medical student, to start working under the guidance of Prof. Piet De Somer, then professor of microbiology at the Leuven School of Medicine. When I graduated as medical doctor (MD) in 1966, I hesitated between a clinical career in Internal Medicine or a scientific career in experimental research, the latter under the tutorship of Prof. De Somer who persuaded me to work on interferon (inducers). The discovery of the interferon-inducing capacity of double-stranded RNAs, such as poly(I)·poly(C), by Maurice Hilleman's group at Merck in 1967 would prove of key importance in my decision to engage in interferon research. In 1968 I described the induction of interferon by polyacrylic acid and polymethylacrylic acid; one year after Thomas C. Merigan at Stanford University had described interferon induction by pyran copolymer. As a postdoctoral fellow at Stanford (from 1968 till 1970), I discovered, with T.C. Merigan, several new polynucleotides as inducers of interferon. Upon my return to Leuven at the end of 1970, I picked up a new line of research, that of the reverse transcriptase (RT), which had just been discovered by Temin (and Mizutani) and Baltimore. In 1975 I then discovered suramin as a potent RT inhibitor (published in 1979), and this prompted Mitsuya and his colleagues (including R.C. Gallo and S. Broder) to evaluate suramin as a potential anti-HIV agent.
2014 (August 2014 to be precise) marks the 50th anniversary of my arrival, in 1964, as a 23-year old medical student who had just passed his 5th year medical studies (second doctorate), at the Rega Institute. I do not recall it as “une entrée joyeuse” (“blijde intrede”), but only as a “let us try and see”. That my stay at the Rega would finally last for 50 years could hardly be anticipated, at the beginning of an uneventful start of what later could be considered as an equally uneventful career. At the age of 18, right on time, I finished high school (“Oude” Grieks-Latijnse Humaniora) at the Heilige Maagd College (HEMACO) in Dendermonde. As Primus Perpetuus, being the first of my class from the age of 12 till 18th (Fig. 1), I was predestined to become a priest, certainly after having studied the classical humanities (Greco-Latin), but I did not feel like being summoned by any providence or superior force. Instead, I thought of mathematics, physics, chemistry or chemical engineering, algebra being my favored course at high school, but to further enroll at the university in engineering, I would have to follow a preparative year, which for medicine was not necessary, and my mother dreamed of seeing her only son becoming a medical doctor in general practice. She won! Mainly because the heavy emphasis on chemistry in the first undergraduate year of the medical school. And there I went! The first year at the university was partially successful. I passed the first year with “distinction”, the second year, I got “great distinction”, and the third year, as the only of my class, I climbed up to “greatest distinction”, by surprise, I should admit, as I did not feel I deserved such accolade which I rather saw as a discrimination. Meanwhile, I had started to work in the laboratory as a “free” student. This was expected only from the best students, and advised by Prof. Xavier Aubert, professor in physiology (at the future UCL) and son-in-law of Prof. Albert Dalcq (Université Libre de Bruxelles, ULB), also permanent secretary of the Royal Academy of Medicine. I was parachuted in the laboratory of a certain Prof. Raymond Devis (Laboratoire de Chimie Hormonologique). I missed the opportunity to start working in the laboratory of Christian de Duve (who in 1974 would be honored by the Nobel Prize in medicine or physiology, but this was not obvious (yet) around 1960, although de Duve's basic discovery of the lysosomes is going back till 1955). I spent a lot of time in Prof. Devis' laboratory, trying to set up an analytical test for catecholamines through some spectrophotometric techniques, but this work carried out in the period of 1960 till 1963, despite encouragements and flattering comments of the professor concerned, never resulted in any publication. Adding to the disenchantment was that I worked in a French speaking laboratory, which in the wake of the imminent separation of the university in a Flemish and French speaking section, gradually became an untenable situation for Flemish-speaking students of the then called Université Catholique de Louvain (UCL). The fact that I spent my time in a French-speaking laboratory was only a small part of the problem, the major part being it was not an inspiring environment for any significant accomplishment or prospect thereof.
Figure 1.

Photograph of class Retorica, 1958.
In 1963, at the exam of microbiology (bacteriology), Prof. Piet De Somer, who must have been informed of the fact that I was not tremendously enthralled by my “séjour” in Prof. Devis' laboratory, offered me the opportunity to come to work with him at the Rega Institute, on viruses. My first reaction was “No, thank you. I do not want to work on viruses, just chemistry”. When my fellow students and in particular, the preses of the medical students, Olav Leuridan, heard that I had declined an offer of Prof. De Somer, who at that time was already Scientific Adviser to the Rector Magnificus of the university (Monseigneur Descamps), he told me I had to be stupid to refuse such an offer. In 1964, again at the exam of microbiology (virology), Prof. De Somer asked me once more whether I had in the meantime changed my mind, and I gladly told him “I had” and that I would start working in his laboratory on “the chemistry related to viruses”, whatever that meant. For De Somer, it meant “interferon”. The discovery of interferon by Isaacs and Lindenmann in 1957, had made De Somer very enthusiastic about the prospects of interferon as a broad-spectrum antiviral agent, and he wanted to transmit this enthusiasm to myself. At the time, interferon was still an esoteric principle rather than a molecule, and many chemists I talked to even doubted that this molecule really existed, but De Somer believed in it (in 1976 he would lose his belief in “interferon”), and he could persuade me to start working in his laboratory, originally not on interferon, but on rubella, in setting up an immunofluorescent technique to detect antibodies against rubella virus, and following up on the production of these antibodies in rabbits given the rubella virus vaccine (this was the Cendehill strain vaccine that was later commercialized by RIT, and, is as of today, still part of the GSK vaccines against measles, mumps and rubella). This work gave rise to sort of a master's thesis which I must have completed in 1965. For Prof. De Somer, it was sufficient to drag me into research and start a scientific career. In July 1966 (Fig. 2), I finished my MD studies, graduating as first of the class with the greatest distinction, and Prof. De Somer was there at my graduation, to tell me he had been of some help in securing that I got the greatest honor (“maxima cum laude”). It was not in vain: being confronted with the option of starting a career in internal medicine (with Prof. Jozuë Vandenbroucke) and a career in science (with Prof. Piet De Somer), I first settled for a 50%–50% solution, which later on became 100% De Somer, and so in August 1966 I started to work, now full time, in the laboratory of Prof. De Somer. While in 1964, Prof. De Somer had tried to couple me to an older colleague Prof. Alfons Billiau, I had told him by December 1964 that I wanted to work independently, and so, by 1966, when I definitely started to work under Prof. De Somer's mentorship, he assured me that I would gain full independence under his final guidance, of course.
Figure 2.

Photograph of MD graduation, 1966.
When I started to work as an MD in Prof. De Somer's laboratory in August 1966, I was reasonably lucky in that I had the whole laboratory for myself, about 8 technicians [since at that time my colleague Dr. Billiau spent a post doc with Sam Baron at National Institutes of Health (NIH)], whom I had to supervise. I remember Anick Focant, Josette Costermans, Francine Cornette, Lieve Aelvoet, and a few others of which I have forgotten the names by now. This was a luxury I would never have again. Also Prof. Desmyter, who actually did not belong to the inner circle of Prof. De Somer, was at this time at Baylor University in Houston. I made, during August 1966, some observations which Prof. De Somer found rather exciting: (i) the fact that interferon upon being induced by Sindbis virus in rabbits, appeared into the urine1, proving that interferon had a molecular weight sufficiently low (≤20,000) to pass the kidney threshold (Prof. De Somer would present these data at a meeting of the Pan American Health Organization (PAHO) in Fort Lauderdale), (ii) the fact that interferon could be induced by synthetic polyanions such as polyacrylic acid2, 3. At about the same time, Tom Merigan at Stanford University had shown that interferon could also be induced by synthetic polyanions such as pyran copolymer. Prof. De Somer met with Dr. Tom Merigan in Fort Lauderdale and when Prof. De Somer came back from Fort Lauderdale, he suggested to me that I should go to Stanford to spend a postdoc year with Tom Merigan. As alternatives he suggested Johns Hopkins (Baltimore) with Bob Wagner and Bronx (New York) with Phil Marcus. The decision was easily made. After some deliberation in 1967 with my future wife, Lili (whom I married in 1968), we decided to go to Stanford; to go over there, I got a fellowship from Eli Lilly, including first class fare for myself (with PanAm) which I could easily convert into two economy tickets for Lili and myself, and on 4 September 1968, three days after we married, off we went from Brussels via London, to Los Angeles, San Francisco, and finally, Palo Alto, the last segment of the trip by helicopter. The stay at Stanford University was planned for one year, but we liked it so much that we finally stayed for two years, 2 months and 3 weeks before we returned to Belgium at the end of November in 1970. To support this stay at Stanford, I got a Lilly fellowship for the first year extended by a Damon Runyon fellowship for the second year. The year 1967 had been a year full of discoveries, first and most importantly, the fact that I fell in love with Lili which would result in our becoming engaged and subsequently married in 1968. From a more scientific viewpoint, a very important breakthrough in 1967 was the induction of interferon by double-stranded (ds)RNAs by Maurice Hilleman's group at Merck (Fig. 3)4, 5, 6, 7. The amounts of interferon induced by dsRNAs exceeded by far those induced by polyacrylic acid or pyran copolymer (Fig. 4), and for this discovery, as well as the vaccines he developed at Merck, Maurice Hilleman would have deserved the Nobel Prize, which, unfortunately, he never won.
Figure 3.

Structure of poly(I)·poly(C).
Figure 4.

Structures of polyacrylic acid, polymethacrylic acid and pyran copolymer.
From September 1968 till November 1970, I spent together with my wife Lili a marvelous time at Stanford University in the Department of Medicine. My host at Stanford University was Thomas Chandler Merigan. At Stanford I would focus, under the guidance of T.C. Merigan (Fig. 5), on various aspects of interferon induction. He left me free to work on whatever aspect I liked, on the condition that it led to publishable results with me and T.C. Merigan as co-authors. He showed an unbridable enthusiasm for whatever result or finding I came up with, so that after 2 years at Stanford I had managed to publish about 25 papers, all with T.C. Merigan as co-author, two papers in Nature8, 9, one in Science10, and a couple others in such distinguished journals as JCI (Journal of Clinical Investigation)11 and JMB (Journal of Molecular Biology)12. I did not have other obligations except for working hard (enjoying myself) in the laboratory and producing manuscripts, which at one occasion, after I had finished writing it, my wife typed during the night, so that when T.C. Merigan appeared in the office the next morning at 9:00 am he found the manuscript waiting for him at his desk. The paper was sent to, and published in, the Journal of Immunology13. At Stanford, I had the (scientific) luxury of attending, almost on a daily basis, lectures from the most prestigious lecturers worldwide, but those leaving the most unforgettable impression were those given by the members of the Department of Biochemistry with Arthur Kornberg, Paul Berg and a few others, who a few years later, would all be honored by a Nobel Prize in either medicine (or physiology) or chemistry. I attended their lectures as a free student, not being enrolled in any (Ph.D.) program. At Stanford, I remember working day and night, especially at night and weekends (Saturday and Sunday), because I had then the scintillation counters free for use, so that I did not have to sign up beforehand. On Monday evenings we (the students) had to give seminars attended by the staff members of the Department of Medicine, the most critical (and most feared) being a certain Bill Robinson (at the time married to Harriet Robinson) who worked on hepatitis B virus (HBV) infection. I also remember Hugh McDevitt (Immunology) and Kenneth Vosti (Bacteriology), and I became very well settled in Merigan's laboratory with Johanna Lederer as his only secretary and Janet Kulhanek as his only technician. T.C. Merigan even offered me the opportunity to stay at Stanford (Medical School) as the head of the Clinical Virology, which was then (November 1970) being set up.
Figure 5.
Photograph of E. De Clercq and T. C. Merigan (Chemical Engineerings News, June 1969, 17–18).
But, in the beginning of November 1970, Lili and I returned back home from Stanford to Leuven, not without a few lectures I gave on 2 November 1970 at NIH (with Robert Friedman as my host), on 3 November at Johns Hopkins [with W.A. (Bill) Carter and P.O.P. (Paul) Ts'O as my hosts] and on 5 November at du Pont de Nemours (with Royce Lockart Jr. as my host). Back in Leuven, I was not welcomed as the lost son who returned, except by my boss Prof. De Somer. He wished me a warm welcome, but his assistants were somehow reluctant to see me back. De Somer assigned me a laboratory that was just evacuated by Carlo Cocito who had moved to the UCL in Woluwe and he had taken with him all what was useful, so that the only materials left over was mostly broken (or useless) furniture and glassware. When I went to express my wrath to Prof. De Somer, he assured me I had to get accustomed again to the Belgian way of life. When I told him I needed help from a technician, he asked around, and found one, Mrs. Anita van Lierde, who would remain my beloved technician for the rest of my life, till her pension, at the age of 60, in 2006. The least I could say was that, except for my boss, Prof. De Somer, his assistants were not over-excited to see me back in town. There was a tremendous benefit; however, De Somer let me work on whatever I liked, and this was at that time, at the end of 1970, two things, induction of interferon by dsRNAs (double-stranded RNAs)14 and inhibition of the reverse transcriptase (RT), which had just been discovered by Howard Temin and David Baltimore15, 16. At a Saturday night working in the laboratory, I tried to repeat the experiments of Temin and Baltimore, and confirmed that it worked (I got TCA [trichloro acetic acid]-precipitable counts). I must have told my wife later that night, but she probably fell asleep when I tried to explain the significance of these findings. Since this eventful Saturday evening, I would remain for my whole life attached to RT, admiring Howard Temin for not only having discovered, but also having predicted the existence of such enzyme. While I was struggling in forgetting how nice life was at Stanford, and how miserable it was in Belgium (Leuven), I suddenly had two scientific lives in 1970, interferon induction by dsRNAs and the RT which soon after had been discovered by Temin and Baltimore, confirmed by R. (Bob) C. Gallo, and was postulated by Sol Spiegelman to be at the origin of all cancers, a tantalizing hypothesis.
Back in Leuven, upon my return from Stanford, it took me almost a year before I could get climatized again and I finally did. I went into two directions, interferon induction by synthetic double-stranded polynucleotides17, and RT inhibition by synthetic single-stranded polynucleotides. I got into contact with David Shugar (from Warsaw), who helped me from moving from the polynucleotides to the direct antiviral activity of nucleosides, but through my contacts with Bernhard Witkop18, and particularly Paul F. Torrence, I remained hooked onto the polynucleotides, i.e., poly(I)·poly(A)·poly(U)19, a triple-stranded RNA complex, that was not able to induce interferon, and therefore of only academic interest. This was one of my major research findings in the middle of the 1970s. This finding was not going to elicit any practical value. My long-standing collaboration with Torrence/Witkop on polynucleotides inducing interferon was predestined to come to a premature end. Around the 1975s, I was slowly moving from the polynucleotides and their interferon inducing potential to the nucleoside analogues and their direct antiviral effects. De Somer had indicated that he had lost his faith in interferon (as President of the university, he was too much absorbed by his many other duties) and he persuaded me to leave interferon behind, in exchange for the nucleosides/nucleotides. The meeting that I attended in Göttingen (Max Planck Institut für Biophysikalische Chemie) in May 1976 (Fig. 6) determined the further course of my life. I met there Antonín Holý, Dick Walker, and together with several other nucleoside chemists, i.e., H. Vorbrüggen, W. Pfleiderer, J. Montgomery, J. Moffatt, they re-oriented my further scientific life towards the antiviral potential of nucleosides/nucleotides. From this symposium originated a number of collaborations which would later yield a wealth of new antivirals, 2,3-dihydroxypropyladenine (DHPA), bromovinyldeoxyuridine (BVDU), (S)-9-(3-hydroxy-2-phosphonylmethoxypropyl)adenine (HPMPA), (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine (HPMPC), 9-(2-phosphonylmethoxyethyl)adenine (PMEA)…, so that Göttingen 1976 remains anchored in my memory as the year when I made my greatest discovery, not that of any compound, but the discovery of so many distinguished chemists, who would later synthesize all the compounds that revolutionized the antiviral field. In particular, the collaboration with A. Holý, which then started as East-West collaboration, would later evolve as the prototype of how chemistry and biology/medicine, when joining forces with industry, would foster successful antiviral drug development.
Figure 6.

Symposium on synthetic nucleosides, nucleotides and polynucleotides, Max-Planck-Institut für Biophysikalische Chemie, Göttingen, Germany, 3–5 May 1976.
The first years, after my return from Stanford University, at the Rega Institute were explorative and uncertain as to whatever the future was yielding. A memorable day was 9 May 1975, when Raf (Rafaël), my son was born. As we married in 1968, it took 7 years before my greatest achievement ever went “into press”, but it was especially Lili's greatest achievement, and she took so extreme care of our son. This event overshadowed all my scientific endeavors, although it was already apparent at that time that my interest was slowly shifting from interferon (inducers) to nucleoside analogues and RT inhibitors. Suramin was an example of the polyanionic type of compounds which I found active as an RT inhibitor. I kept this as a little secret to myself, investigating whether suramin, to see if Sol Spiegelman was right, could be anticipated to exhibit anticancer activity. Around 1975, I anxiously looked into the activity of suramin against leukemia in mice. It had, however, no activity and I abandoned the suramin approach, until in 1978, Bob Gallo, when visiting our lab, encouraged me to publish the original results of suramin as an RT inhibitor, which I finally did in Cancer Letters (1979)20 (Fig. 7). I then forgot suramin, thereby losing my faith in RT as a possible component in the origin of cancer. My dream to ever find a cure for cancer, certainly a cure based on suramin, was obviously not going to be fulfilled, but life around me was going full speed, and in 1981, the year that the disease AIDS was identified, Prof. De Somer was elected again (his last time) as President of our university. I participated in the house party celebrating his re-election on 21 June 1981. It would be the last party ever at his mansion. One year later, on a trip to South East Asia, he fell sick with an intestinal obstruction. He recovered from the abdominal surgery, being it for only a few years, and on 17 June 1985, he rather unexpectedly died from a lung embolism complicating a second abdominal intervention. It happened that at the time I was in Czechoslovakia, with Holý (on Sunday). The tragic news reached me on Tuesday 19 June, in Bechyne, at the meeting that I then attended together with Vladimir Vonka. I could not believe that De Somer had died. For me he simply could not die, and as of today, I still think it was a nightmare from which he would ultimately resurrect.
Figure 7.

Structural formula of suramin.
The death of De Somer (17 June 1985), at the age of 67, just one month after he had hosted the visit of the Pope in Leuven (Fig. 8), was a deadly tragic event: it was a great loss for the university, but even more so, for his family, colleagues and myself. He was an “icon”, who just could not die; his death proved he was not immortal. Leaving us, his disciples, behind was simply unacceptable. At the Institute, I was quickly canonized as his successor, the youngest of his disciples, my competitors being Alfons Billiau, Hubert Vanderhaeghe, Hendrik Eyssen and Michel Vandeputte. They all wanted to be De Somer's successor at the institute, whereas I did not envy this position, but finally, my colleagues selected me as De Somer's successor. In 1985 I was parachuted in a position I did not want, I became Chairman (Director) of the institute, not knowing of the implications that were lying ahead.
Figure 8.

Picture of Prof. P. De Somer when receiving Pope John Paulus II in Leuven on 18 May 1985.
In no time I was myself surrounded by clever disciples: Piet Herdewijn, Jan Balzarini, Rudi Pauwels, Robert Snoeck, Graciela Andrei, Lieve Naesens, Johan Neyts, Dominique Schols and Christophe Pannecouque. A nice “family”, quite coherent, I thought, each excelling in their own field, which they had developed, first with my guidance, later, independently thereof. R. Pauwels left the group to start Tibotec/Virco, and later Biocartis. In leaving the family, in 1994, he made the most successful move, admittedly with the support of Dr. Paul Janssen. The others stayed on board, and still are, but in the future, they will have to fight for their own destiny. Up till my obligatory retirement in 2006, and even thereafter, I felt as the conductor (dirigent) of an ever expanding chorus of dedicated scientists, each trying to make his own way, with “a chef d'orchestre” still trying to keep his leading role, thereby witnessing how the different members of his team got dispersed in different directions: J. Balzarini in the biochemistry of the acyclic nucleoside phosphonates, and later from the 2000s, the antiviral potential of glycopeptide antibiotics; D. Schols, through the role of chemokine receptors (CXCR4, CCR5) in HIV infection, finding a new future in the microbicides for the prevention of HIV infections (an ever self-perpetuating approach and never-disappearing strategy to prevent HIV infections); J. Neyts, who quickly made the step to veterinary (virological) medicine, thereby never giving up on the potential in human medicine for those compounds targeting flaviviridae (i.e., dengue, yellow fever, hepatitis C virus [HCV]) and picornaviridae (i.e., polio and rhino), thereby tackling almost all viruses except for the human immunodeficiency virus (HIV); G. Andrei and R. Snoeck, who got united in our laboratory, working together on a number of viruses, the others did not want to work with, cytomegalovirus (CMV), varicella-zoster virus (VZV), human papilloma virus (HPV), poxviruses and, later on, polyomaviruses; L. Naesens, who always remained faithful to her predestined vocation, first human herpes virus type 6 (HHV-6), later influenza virus; and C. Pannecouque, originally educated by P. Herdewijn, but then transplanted to our laboratory to lead the screening of anti-HIV compounds. And C. Pannecouque is still doing so, even after my term as head of the laboratory elapsed (in 2006, to be exact). Looking back at what history generated, I should be proud of the progeny I left behind (Fig. 9), J. Balzarini, D. Schols, J. Neyts, G. Andrei, R. Snoeck, L. Naesens, C. Pannecouque, who stayed, and R. Pauwels, who left. My achievement? The luck for keeping them, except for Rudi Pauwels, together at the Rega Institute. There were other indelible personalities that, with immense satisfaction, I ever worked with, including my currently best Japanese friend, Masanori Baba, former Vice-Rector at Kagoshima University, and his boss, Shiro Shigeta (formerly, President of the Medical College at Fukushima), Chong-Kyo Lee at Daejeon (South Korea) and my old student from the KULAK, Zeger Debyser, and his once-upon-a time co-worker, Myriam Witvrouw, whom I remember well from the exciting days their careers began to blossom.
Figure 9.

Collaborators of Prof. E. De Clercq.
Time for reflection
I started my career with the hope to discover at least one anticancer drug, but I did not discover any. This failure was, to some extent, compensated by the discovery of a few antiviral drugs. Serendipity played a predominant role in how these compounds emerged. Aminoacyl esters of acyclovir (i.e., glycyl- and alanyl-acyclovir) were designed to increase the aqueous solubility of acyclovir; next in the series of aminoacyl esters of acyclovir was the valine ester; it later proved to be the ideal candidate prodrug of acyclovir to increase its oral bioavailability, thus substantiating its clinical use in the oral treatment of herpes simplex virus (HSV) and VZV infections. BVDU was conceived as a successor for idoxuridine (IDU) and trifluridine (TFT), the first two 5-substituted 2′-deoxyuridines ever to be launched for the topical treatment of herpetic eye infections; BVDU became the runner-up for the treatment of VZV infections (herpes zoster). With stavudine (d4T), we had at hand what was at a certain time the gold standard for the treatment of HIV infections (AIDS); however, d4T was discovered at three places at the same time (Leuven, Yale and Tokyo), which engendered some international animosity in the old days. In the meantime, other HIV inhibitors that outweighed d4T were discovered in both potency and safety, the most prominent being TDF (tenofovir disoproxil fumarate). Tenofovir has been the life-time achievement of the Holý Trinity, i.e., Antonín Holý, myself and John C. Martin (Fig. 10). For 10 years or more, it has stood up as the cornerstone for the treatment of HIV infections, and for its prevention as well, either by itself as Viread®, or in combination with other compounds, i.e., Truvada®, Atripla®, Complera®/Eviplera®, or Stribild®. TDF (Viread®) has now become the gold standard for the treatment of both HIV and HBV infections, but, in the future, it will likely to be replaced by a new tenofovir prodrug, TAF (tenofovir alafenamide), which, like its predecessor, TDF, may evolve as the drug of choice for the treatment of both HIV and HBV infections.
Figure 10.

The Holy Trinity in Olomouc (a) and the Holý Trinity representing chemistry, biomedicine and industry (b).
My comments
I was not born to be a researcher (or “scientist”), but born to be a teacher (or “professor”). What I learned from my parents were the virtues of hard working, perseverance, honesty and enthusiasm. Two scientific discoveries made me particularly enthusiastic; first, the discovery by Maurice Hilleman's group at Merck that interferon could be induced by double-stranded RNA, and second, the discovery of the reverse transcriptase by Temin and Baltimore. As a young student in medicine, I had always been dreaming of discovering a cure for cancer, and here, I had two potential leads: interferon, originally discovered because of its antiviral activity, had become extremely popular in the 1970s as a potential anticancer agent; and the reverse transcriptase which was originally (also in the 1970s) considered as the key enzyme in the origin of cancer (proposed by Sol Spiegelman), before its role was established (in the 1980s) in the origin of AIDS. In addition to perseverance and enthusiasm, flexibility may seem of intrinsic value as it would allow to re-orient the research to new frontiers (i.e., AIDS) which were not recognized initially.
II. Period 1975–2005
In 1979 I described the anti-HSV activity of BVDU (brivudin), which would later be marketed as the most potent antiviral drug ever developed for the treatment of VZV infections. In 1983 followed the amino acid esters of acyclovir, the valine ester would ultimately emerge as the successor of acyclovir for the treatment of HSV and VZV infections. d4T, discovered in 1987, would be the fourth nucleoside reverse transcriptase inhibitor (NRTI), following 3′-azido-2′,3′-dideoxythymidine (azidothymidine, AZT), 2′,3′-dideoxyinosine (ddI) and 2′,3′-dideoxycytidine (ddC), developed for the treatment of HIV infections. Then followed in 1989 and 1990, 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine (HEPT) and tetrahydro-imidazo[4,5,1-jk][1,4]-benzodiazepine-2(1H)-one and -thione (TIBO) were discovered as the first non-nucleoside reverse transcriptase inhibitors (NNRTIs), which, however, were not developed as anti-HIV drugs. Instead, a nucleotide analogue, tenofovir, originally described in 1993, as its prodrug form (TDF), became the cornerstone for HIV treatment. After being launched as Viread®in 2001, it was in combination with emtricitabine marketed as Truvada®for HIV treatment in 2004, and extended in 2006 to efavirenz (Atripla®), which was then replaced in 2011 by rilpivirine (Complera®/Eviplera®), and extended to the quadruple pill containing TDF, emtricitabine, elvitegravir and cobicistat (Stribild®) in 2012. Till 2006 when I was forced to retire, I had more than 200 collaborations worldwide, my scientific publications peaked at some 80 per year (exactly 80 in 1991), and my teaching duties amounted up to 10 hours per week, and this lasted over a period of 37 years (from 1972 to 2009).
2.1. Acyclovir
The discovery of acyclovir as an antiviral agent in 1974 at Burroughs Wellcome in the UK (Beckenham) by Peter Collins and John Bauer meant a milestone in the antivirals' saga21. The compound was only published 3 years later (in 1977) when Gertrude B. Elion and her coworkers22 in the US (Research Triangle Park) revealed in the December 1977 issue of PNAS (Proceedings of the National Academy of Sciences of the United States of America) that acyclovir owed its antiherpetic selectivity to a specific phosphorylation by the herpesvirus-encoded thymidine kinase. Schaeffer et al.23 would further describe the antiviral potential of acyclovir in the April 1978 issue of Nature. This meant a milestone in the era of antiviral drug development as acyclovir represented the first specific antiviral compound ever described. It would later emerge as the “gold standard” for the treatment of HSV infections, and its use would be extended to the treatment of VZV infections. Although not recognized in this capacity as such, acyclovir would later contribute to the Nobel Prize in physiology or medicine, attributed to Elion and Hitchings in 1988 (their Nobel Prize was formally recognized for their contributions to antimetabolites in general). Having described DHPA as a broad-spectrum antiviral agent in Science24, not acting in the same way, but yielding a similar result as acyclovir, I was one of the first witnesses to appreciate the success bestowed to acyclovir. In no time, acyclovir conquered the world of antivirals, leaving its potential competitors, fluoroiodoaracytosine (FIAC) and bromovinyldeoxyuridine (BVDU), far behind. These competitors would either disappear from the scene like FIAC or later follow a meandrous path in their development like BVDU. Acyclovir would clearly excel as the leader of the gang, gaining general acceptance in all countries where its application was deposited. One of these countries was mine, Belgium. As of today, in 2014, acyclovir has remained the preferred drug for the treatment of both HSV and VZV infections. The professor of medicinal chemistry, H. Vanderhaeghe must have foreseen this fate when back in the early 1980s, he convinced me of the potential importance of the acyclovir esters, i.e., glycinyl (glycyl) and alaninyl (alanyl) esters of acyclovir (Fig. 11) that may play a superior role to acyclovir because of their increased aqueous solubility compared to the parent compound25, 26. I thought that the derivatization of an existing compound by modifying it to its prodrug was not very innovative. I accepted his arguments in trying to sell this prodrug to the original producer of acyclovir, Burroughs Wellcome (Beckenham, UK). It made me travel, with Prof. De Somer (then rector) as my companion, to the UK to introduce both the Rega Institute and our plans, and it eventually worked out splendidly. Burroughs Wellcome (BW) in 1993 agreed to buy the rights for the acyclovir amino acyl esters for a small lump sum and even smaller prospect on potential royalties, and this was the first successful deal on any of the compounds I have ever (co)discovered. The amino acyl ester of acyclovir, valacyclovir (Valtrex®, Zelitrex®) (Fig. 11), would later supersede acyclovir because of its better oral absorption, but this advantage was not predicted by our original observations. Our original patent claims on the amino acyl esters of acyclovir were therefore not universally granted (i.e., in the US they were not), but, with my co-inventors, Prof. H. Vanderhaeghe and Dr. (later Prof.) R. Busson, we were granted patent rights on valacyclovir in the rest of the world. These rights were granted from 1995 till 2002, when valacyclovir turned generic.
Figure 11.

Structures of (val)acyclovir.
2.2. BVDU (Brivudin)
Whereas acyclovir was originally synthesized in the US (Howard Schaeffer), and its antiviral activity originally demonstrated in the UK (Peter Collins/John Bauer), BVDU (Fig. 12) was originally synthesized in the UK (Phil Barr/Stan Jones/Dick Walker), its antiviral activity was first demonstrated in our laboratory at the Rega Institute. I divulged its antiviral potential at a meeting in Prague (10–12 July 1978), a few months after the antiviral activity of acyclovir had been described in Nature (April 1978). The full paper (sponsored by Bernhard Witkop) was published in the PNAS in 197927. As compared to acyclovir, BVDU was slightly more potent against HSV type 1, much less active against HSV type 2, but clearly much more potent against VZV. In fact, BVDU proved about 1000-fold more potent than acyclovir against VZV in vitro (cell culture). Although an anecdotal case report study published in 1980 in the British Medical Journal with four patients had already pointed to the potential efficacy of BVDU in the treatment of VZV infections (herpes zoster)28, it would take another 20 years before the compound would be officially approved in a number of European countries (Germany, Italy and Belgium); but it would never be approved in the UK or USA. The main reason for this delay is that the Company G.D. Searle, who had originally taken the license on BVDU, decided in 1984 not to further pursue its development because of its carcinogenic potential (liver tumors) if administered perorally at 1 g/kg/day for 1 year to a special breed of rats that were highly sensitive to this event (whereas humans would only need treatment with 125 mg per 70 kg per day for at most 7 days). This schedule has obviously never proven carcinogenic in humans. The fact that G.D. Searle (later Monsanto) was principally operative in the US and the UK explains why BVDU never made it to the market in these countries. That BVDU made it to the market in Germany and several other European countries was actually due to the success obtained with BVDU in the DDR (East Germany), where it had been synthesized in Berlin Buch by Peter Langen/Dieter Bärwolff, evaluated by Peter Wutzler (Erfurt) and licensed to Berlin-Chemie. After the DDR ceased to exist in 1989, the rights on BVDU would eventually be transferred from Berlin-Chemie to Menarini, a private Italian company headquatered in Florence. As mentioned above, for the treatment of herpes (oral) zoster, a daily dose of 125 mg BVDU for 7 days (that may be reduced to 5 days) suffices, whereas acyclovir and valacyclovir have to be given at 4 g or 3 g per day, respectively. BVDU can also be administered as eye drops in the treatment of herpetic cornea infections (herpetic keratitis) (work pioneered on by my good friend, Prof. Prabhat Maudgal)29, 30, 31, 32 and ointment in the treatment of cold sores of (herpes labialis), but, despite the anecdotal successes obtained in these conditions, BVDU has never been commercialized for these indications.
Figure 12.
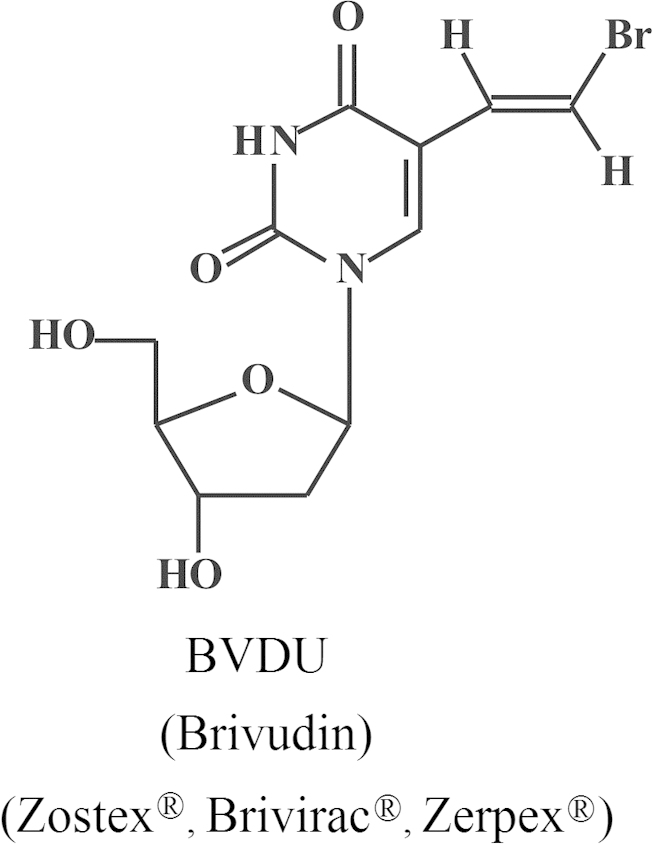
Structure of brivudin (BVDU).
2.3. d4T (2′,3′-Didehydro-dideoxythymidine)
d4T (stavudine, Zerit®) (Fig. 13) was officially discovered simultaneously at 3 different sites, Yale University, Rega Institute and Tokyo (Yamamoto's Laboratory). To be honest, we at the Rega Institute were the first to recognize d4T (which we originally called 2′,3′-didehydro-thymidinene) as an anti-HIV agent with the potential to combat HIV infections. I have never known what exactly happened in Japan, till the paper of Hamamoto et al.33 was published in the June issue of Antimicrobial Agents and Chemotherapy (1987). Piet Herdewijn had synthesized the compound by November 1985 within one month after the PNAS paper (October 1985) of Mitsuya et al.34 had appeared on the inhibition of the infectivity of HIV (then called HTLV-III/LAV) by AZT. I sent the compound overseas to J. Balzarini, who had just started from December 1985 to work in Sam Broder's laboratory at National Cancer Institute (NCI). Followed the technique developed by Hiroaki Mitsuya, using ATH8 cells, which had proven successful to demonstrate the anti-HIV activity of AZT, and later of ddC and ddI as well35 (but which for some reasons were never clarified), did not prove particularly useful to demonstrate the anti-HIV activity of d4T. From August 1986, Masanori Baba had joined our research team in Leuven and I asked him to look again into the anti-HIV activity of d4T, now using a different cell line, MT-2. He found an astonishingly potent anti-HIV activity. By November 1986, we had already completed our paper which we submitted to Dr. Sols who was the editor of Biochemical and Biophysical Research Communications (BBRC) for publication in the journal. The paper was accepted right away and finally published in the journal on 17 January 198736. The paper had also been sent (in November 1986) to our patent attorney, a certain Mr. Bruin from Arnold and Siedsma (the patent office in Den Haag), who let the paper sitting on his desk, without any due action, for 2 months in his office, before filing the patent application. In the meantime, Prusoff and Lin (from Yale) had submitted their patent application (sometime in December 1986). In these days I was often visited by Julius Vida, Director (later President) of licensing at Bristol-Myers (several years later it became Bristol-Myers Squibb [BMS]), who expressed the desire of Bristol-Myers to further develop d4T as an anti-HIV drug and to license the compound from whomever got the valid patent (Yale or Rega). I realized that fighting Yale University from a small place in a small country was like fighting David against Goliath, an “impossible mission”. We had been clearly the first to demonstrate the anti-HIV activity of d4T (as a matter of fact, Bill Prusoff never had worked with HIV in his lab); his patent was based only on the anti-murine retrovirus activity of d4T: this appeared sufficient for the patent examiners to surmise that the compound should be effective against AIDS, whereas in our case the patent examiners reasoned that activity against HIV in cell culture could not be extrapolated to activity against HIV in the patient. This was the rather enigmatic verdict of the US patent office thus assigning the patent rights to Yale, which then transferred the rights to Bristol-Myers, … and d4T turned out to be a success story, which, at a certain time, yielded yearly sales of 700,000 US dollars. I could gladly accept Bill Prusoff's success with d4T. He had missed his success with IDU (5-iodo-2′-deoxyuridine) for which he was the sole discoverer, because Yale had neglected to protect the compound. They did so for 5′-amino-5-iodo-2′-deoxyuridine (AIU), but this compound did not become a successful antiviral. Finally, d4T became a success, but for sure some Belgians and Japanese did not indulge in Yale's success. I valued my personal relationship with the Prusoff's (Bill and Brigitte) higher than any possible rewards from patent income and I consented with the adulation of Bill Prusoff as the godfather of antivirals, until he passed away at the age of 90 on 3 April 2011.
Figure 13.
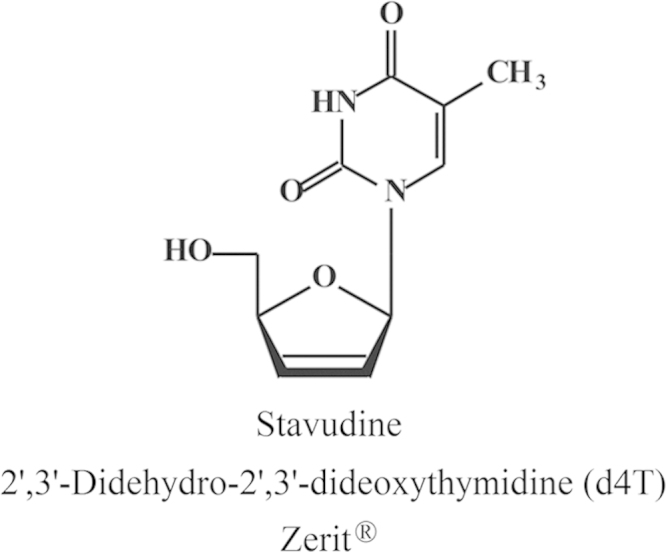
Structure of stavudine (d4T).
2.4. The dawn of the anti-HIV antivirals
In 1985, sometime in the summer, a young pharmacist, by the name of Rudi Pauwels, knocked at my door. He actually introduced himself first at the institute's reception desk where he was met and ousted by our Secretary-in-Chief, Mrs. Jane Putzeys (a first reaction from Jane to deter strangers, so that they would not get too easy access to the Sanctuary of what she considered the temple of God (Prof. Piet De Somer). Rudi, nevertheless, broke the ban, as he would do repeatedly at later occasions, penetrated into my office and expressed his desire he would like to come to work for me. Why Rudi had selected me was not immediately clear. Perhaps he had some hidden plans, but a cure for AIDS was certainly not on top of his original list (principally because HIV was barely known or appreciated at the time). I proposed originally to Rudi to work on 2-5A as mediator of the action of interferon, but he proposed that HIV would be a more attractive subject. I had my co-worker, J. Balzarini, sent to the US to learn how to handle HIV in Sam Broder's laboratory with Hiroaki Mitsuya, whereas in the meantime, Rudi Pauwels, came to work in our laboratory with the aim to develop the cure (ideal treatment) for HIV. Rudi Pauwels was a pharmacist, who had an unequalled talent for robotics and technology, besides being an entrepreneur: he became highly regarded by Paul Janssen, and I could only confirm Dr. “Paul's” insight. J. Balzarini was supposed to get some training with Thomas Kalman in Buffalo (New York, US) on the metabolism of nucleoside analogues and their mechanism of action against cancer, but the emergence of suramin as an RT inhibitor crossed these plans. Based on my observation in 1975 (published in 1979)20 that suramin appeared to be a rather potent inhibitor of RT, Sam Broder at the suggestion of R.C. Gallo with the help of H. Mitsuya tested whether suramin would inhibit the infectivity of HIV (then called HTLV-III/LAV), as the RT was assumed to play a key role in the replication of HIV. Suramin actually inhibited the replication of HIV and this was originally attributed to its inhibitory effect on the RT37 (although in later studies it was shown that suramin, being a polyanionic compound, also inhibited virus adsorption to the host cells)38. S. Broder called me at home to inform me that suramin was inhibitory to HIV infectivity (this apparently happened after he had received news from Science that his paper had been accepted37 and in this phone conversation he invited me to come over to the NCI to spend a year (sabbatical) further following-up on suramin and other RT inhibitors for their anti-HIV activity. As I felt I could not just leave the laboratory behind and interrupt my teaching in Kortrijk (at the KULAK), I declined the invitation but, instead, convinced J. Balzaini to take up this assignment and from the first December 1985 he went over for a year to work in Sam Broder's laboratory on the anti-HIV activity of new nucleoside analogues. Most of these nucleoside analogues had been synthesized by P. Herdewijn in our institute and he had started his project in November 1985 just after the paper of Mitsuya in PNAS (October issue of 1985) had been published that AZT proved inhibitory to the infectivity of HIV34. One of the compounds P. Herdewijn had first synthesized was d4T, but as J. Balzarini used the ATH8 cells routinely used by Mitsuya in Broder's laboratory, he did not find high potency with d4T (or certainly not enough activity to justify taking a patent). From the end of 1985, after J. Balzaini had left for the US, R. Pauwels started to work in our laboratory, originally on the role of 2-5A in the mode of action of interferon, but he soon switched to HIV and by the middle of 1986 he had set up the assay systems for HIV which he would later gradually perfectionize. Thus was set up the AIDS laboratory, which was later joined by D. Schols and M. Baba (both from 1986). In the AIDS laboratory they would use MT-2 cells, which M. Baba used to demonstrate the potent anti-HIV activity of d4T, and later in 1987 the anti-HIV activity of HEPT. At the end of 1987 we knew of the anti-HIV activity of HEPT, but we did not know how it really worked: as an (acyclic) nucleoside analogue, there was no precedent of any other acyclic nucleoside analogues that would act similarly, so that in the beginning I thought the HEPT sample was contaminated by a known nucleoside analogue of the AZT type, especially when we found that HEPT had only activity against HIV-1, but not -239, 40 Dough Richman had just published in Antimicrobial Agents and Chemotherapy (AAC)41 that AZT was more active against HIV-1 than -2, an observation that later on was not confirmed. At the end of 1989, we published that HEPT (Fig. 14) was a newly identified anti-HIV compound with an unknown mechanism of action. The mechanism of action would be resolved in 1990, and 1991. HEPT was found to act as an NNRTI inhibiting HIV-1 by an allosteric interaction with a non-catalytic site, just like the TIBOs would do. The TIBOs (Fig. 15) were discovered as the result of an extensive collaboration I had started in 1988 with Dr. Paul Janssen with the ambitious goal of discovering a “cure” for AIDS, which, according to Dr. Janssen, could not be that difficult. Little did he know that it would prove to be an Herculean task.
Figure 14.
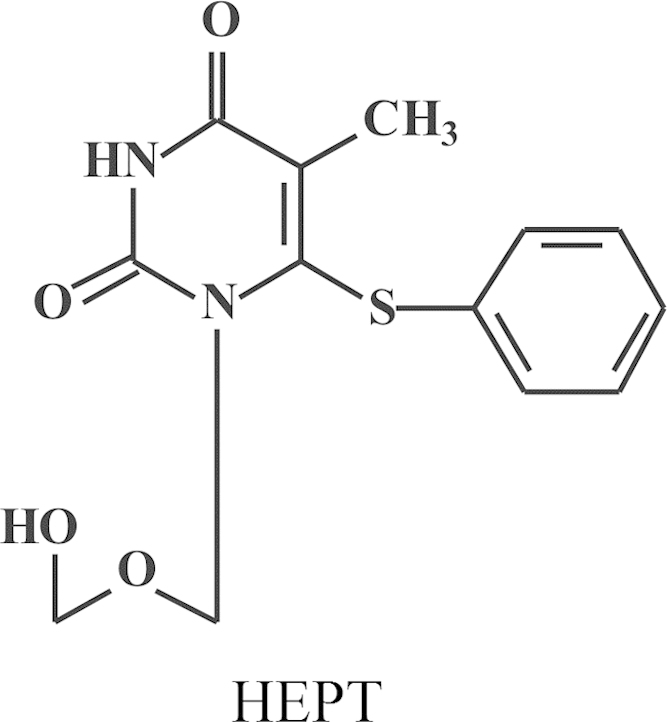
Structure of HEPT (1-[2-(hydroxyethoxy)methyl]-6-(phenylthio)thymine).
Figure 15.

Structures of TIBO R82150, TIBO R82913, TIBO R86183 (tetrahydroimidazo[4,5,1-jk][1,4-benzodiazepin-2(1H)]-one and -thiones).
2.5. NATO/FEBS meetings
Together with Dick Walker (University of Birmingham, UK), I organized three NATO Advanced Study Institutes (ASIs). So as to allow participants from Eastern Europe, these meetings were also FEBS (Federation of European Biochemical Societies) Advanced Courses. These NATO/FEBS meetings invariably lasted for 12 days and took place on 7–18 May 1979 in Sogeta, close to Urbino, Italy (this meeting was also co-organized by Fritz Eckstein (Göttingen, Germany)), on 19 June–2 July 1983 in Les Arcs, France, and on 10–23 May 1987 in Il Ciocco, close to Barga, Italy. These meetings were mainly focused on the design of new antiviral agents, particularly nucleoside analogues. They were limited to about 100–120 attendants, originating from either NATO countries, FEBS countries, or other countries such as Japan. NATO rules had to be implemented: that means that as many NATO countries as possible should be represented, and no NATO country should exceed 20% of the total, which was a serious problem for the US (but not for Luxemburg, of course). The money (travel, subsistence) had to be meticulously divided (NATO money to NATO participants, FEBS money to FEBS participants, and the remaining (industrial) money to the rest of the world). In these East-West (Iron Curtain) times, funds had to be carefully distributed so that, for instance, no NATO money was ever allocated to an East European, Polish or East German. I personally enjoyed puzzling with the funds thus taking care of the administration, whereas my colleague, Dick Walker, enjoyed running the programme and handling the talking (his English (British) was supposed to be better than mine). As we had to get together for the full two weeks, the mean purpose of these meetings was that they not only stimulated the exchange of scientific knowledge, but also fostered personal social contacts which quite often would later last for the rest of our lives. The 1987 Il Ciocco meeting would be the last of the three NATO meetings. From the ashes of the NATO meetings then arose, as a Phoenix, the ICAR (International Conferences on Antiviral Research) meetings, which from 1988 (Williamsburg) and 1990 (Brussels) would subsequently held annually: 1991 (New Orleans), 1992 (Vancouver), 1993 (Venice), 1994 (Charleston), 1995 (Santa Fe), 1996 (Urabandai, Japan), 1997 (Atlanta), 1998 (San Diego), 1999 (Jerusalem), 2000 (Baltimore), 2001 (Seattle), 2002 (Prague), 2003 (Savannah), 2004 (Tucson), 2005 (Barcelona), 2006 (Puerto Rico), 2007 (Palm Springs), 2008 (Montreal), 2009 (Miami), 2010 (San Francisco), 2011 (Sofia, Bulgaria), 2012 (Sapporo, Japan), 2013 (San Francisco), 2014 (Raleigh) and 2015 (Rome). The original NATO rule (no one NATO country should represent more than 20% of the attendants) was quickly abandoned with roughly 70% (two-thirds) of the attendants being US citizens. This also explains why the ICAR meetings are classically held for two years in a row in the US, alternating with one outside the US.
2.6. Francqui Prize
As a young researcher devoting his whole career to science, one is not immune to recognition by his peers, reflected by scientific prizes. In Belgium, the most prestigious is the Francqui Prize attributed to promising researchers under the age of 50 in three alternating disciplines: (i) medicine, (ii) humanities (i.e., linguistics, psychology, etc) and (iii) positive sciences (physics, mathematics, chemistry, etc). Every year the King of Belgium hands over this prize on behalf of the Francqui Foundation. This means that for each of the three prizes, until the age of 50, every third year the opportunity arises to compete for the prize. My boss, Prof. P. De Somer, alerted me to this possibility for the first time in 1978. I felt at that time too young and not sufficiently qualified, but he convinced me and I competed and lost. The winner was a certain Nihoul from the University of Liège, but the tone was set and I competed again for the 1981 prize; this time I was introduced by Prof. Walter Fiers. I lost again, at this occasion the winner was André Trouet whom was sponsored by the 1974 Nobel Prize laureate Christian de Duve. For the 1984 prize, I was nominated again by W. Fiers, but a few weeks before the jury reached their verdict, two things happened that completely annihilated my chances: (i) BVDU, the antiviral drug that I had discovered was stopped for further development by G.S. Searle, and (ii) my competitor, Désiré Collen got a breakthrough paper for the discovery of his drug, tissue plasminogen activator (TPA), in the New England Journal of Medicine42. No wonder, Désiré got the Francqui Prize in 1984. With André Trouet and Désiré Collen out of the way, I reckoned I should have a fair chance in 1987, and this time, I had secured the help of Peter Wildy, professor of pathology at Cambridge, to defend my case at the deliberations of the jury, but, as in a Greek tragedy, Peter Wildy whom I had met several times at WHO meetings, died a fortnight before the jury's meeting, and the Prize 1987 went to a certain Urbain from the Université Libre de Bruxelles for the discovery of the anti-idiotypic antibodies (a discovery actually made by Jerne, for which Jerne got honored by the Nobel Prize). I then put all my hope to the 1990 Francqui Prize, which that year coincided with the NFWO Prizes given every 5 years (so that both Prizes coincide only every 15 years). I had counted on the help of Prof. Hans Rosenthal (Charité, Berlin). He would defend me in the two juries which assembled on two consecutive days, just before I organized the 3rd International Conference on Antiviral Research (ICAR) in Brussels on 22–27 April 1990. Hans Rosenthal called me at home to inform me that I had missed both prizes (the Francqui 1990 Prize went to Thierry Boon-Lateur, who was once again the disciple of Christian de Duve and the NFWO 1990 Prize was awarded to my colleague A. Billiau). I broke into tears when learning the double-sad news, just a few hours before I had to confront my colleagues attending the ICAR meeting. Because of the age limitation, I could never compete again for the Francqui Prize. The NFWO Prize, then renamed FWO (Fonds voor Wetenschappelijk Onderzoek) Prize, I would eventually get in 2000. The jury member defending my case was Hugh J. Field from Cambridge, after I had been introduced by Paul Janssen and Maurice Hilleman.
2.7. Antonín Holý
At the Symposium on Synthetic Nucleosides, Nucleotides and Polynucleotides, Max-Planck-Institut für Biophysikalische Chemie in Göttingen, which was held on 3–5 May 1976, I made the biggest discovery of my life, the discovery of a pleiade of nucleoside chemists, the primus inter pares being Antonín Holý from the Czechoslovak Academy of Sciences, active at the Institute of Organic Chemistry and Biochemistry (IOCB) at Flemingovo námestí in Prague. It was hardly predictable that this apparently accidental encounter marked the beginning of an almost four decades' long collaboration that would lead to almost ten marketed products: (i) DHPA (Duviragel®) for the treatment of herpes labialis, (ii) cidofovir [(S)-HPMPC] (Vistide®) for the treatment of various DNA virus infections, (iii) adefovir (PMEA), in its prodrug form, adefovir dipivoxil (Hepsera®), for the treatment of HBV infections; (iv) tenofovir [(R)-PMPA], in its prodrug form, tenofovir disoproxil fumarate (Viread®, TDF) (Fig. 16) for the treatment of both HBV and HIV infections, (v) TDF in combination with emtricitabine (Truvada®) (Fig. 17) for both the therapy and prophylaxis of HIV infections, (vi) TDF in combination with emtricitabine and efavirenz (Fig. 18) (Atripla®) for the therapy of HIV infections, (vii) TDF in combination with emtricitabine and rilpivirine (Fig. 19) (Complera®, Eviplera®) for the therapy of HIV infections, (viii) TDF in combination with emtricitabine, elvitegravir and cobicistat (Fig. 20), again for the treatment of HIV infections, and, forthcoming, (ix) tenofovir alafenamide (TAF), in combination with emtricitabine, elvitegravir and cobicistat, for the treatment of HIV infections. This made (R)-PMPA and/or its oral prodrug, TDF, the most successful drug ever developed for the treatment of AIDS, and it owed its success to the ingenuity of the chemist, A. Holý, who ever synthesized the compound, but also to the foresight of the industrialist, by chance also a chemist, John C. Martin, who brought it to the market, and the medical doctor (myself) who served as the go-in-between. Based on this triangle, which I rightfully compared to the Holý Trinity, inspired by the Holy Trinity monument in Olomouc, as one of the favorite cities of A. Holý, the Holý Trinity stands as an example of the successful interaction in (antiviral) drug development, the chemist, the biologist, and the industrialist. Unfortunately, the Holý Trinity lost on 16 July 2012, with the death of Antonín Holý, the basic reason for its existence, but perhaps not really if we still continue to commemorate the spiritual existence of the Holý Trinity.
Figure 16.

Structures of (R)-PMPA (tenofovir) and tenofovir disoproxil fumarate (TDF).
Figure 17.
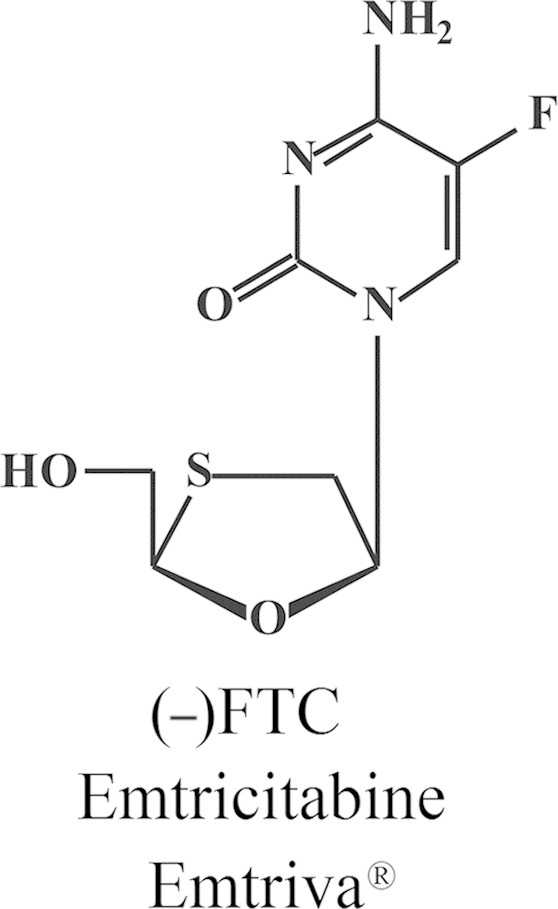
Structure of emtricitabine (Emtriva®).
Figure 18.
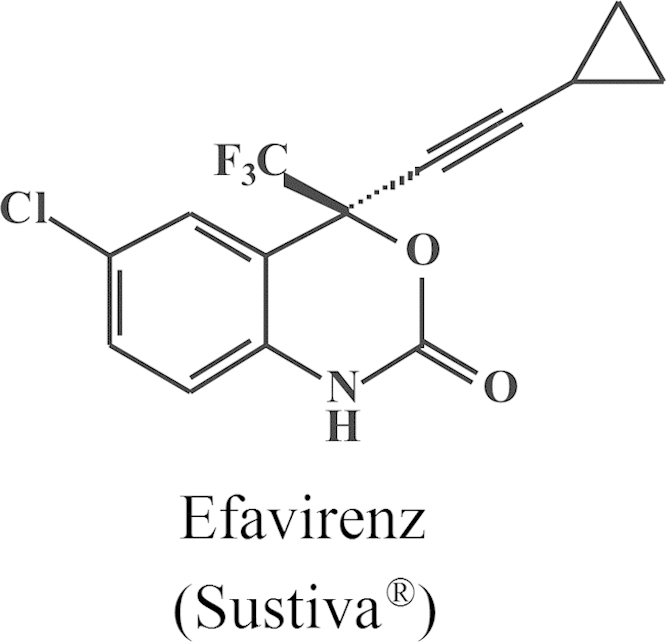
Structure of efavirenz (Sustiva®).
Figure 19.
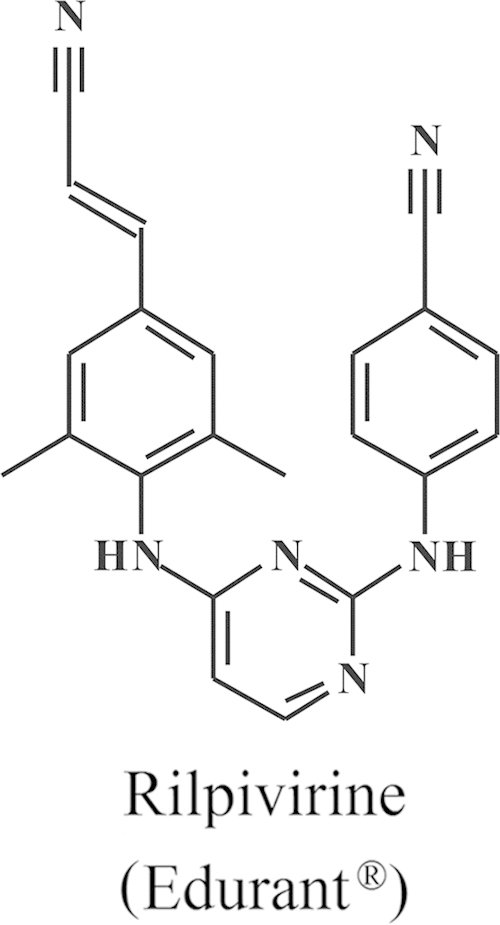
Structure of rilpivirine (Edurant®).
Figure 20.

Structures of elvitegravir and cobicistat (Tybost®).
2.8. Collaborations
Antonín Holý and John C. Martin excelled among the numerous collaborations I had built up during the course of my career. Most of these collaborators were (inorganic) chemists, spread over the 5 continents of the world. It started with Fritz Eckstein (Göttingen, Germany) when I was still working in Merigan's laboratory at Stanford, but it would quickly extend upon my return to Leuven to David Shugar (Warsaw, Poland), Paul F. Torrence and Bernhard Witkop (NIH, Bethesda), Morio Ikehara (Osaka, Japan), Shiro Shigeta (Fukushima, Japan) and many others. At the height of these collaborations I had developed a network of more than 200 collaborations, spreading over the following countries (in alphabetical order): Argentina, Armenia, Australia (G. Holan), Austria [H. Griengl (Graz); G. Heinisch/G. Pürstinger (Innsbruck)], Belgium [A. Vlietinck (Antwerp)], Belarussia [I.A. Mikhailopulo (Minsk)], Canada [V.S. Gupta (Saskatoon), J.R. Dimmock (Saskatoon), G. Henson, M.J. Abrams and G. Bridger (AnorMed at Langley, British Columbia), L.I. Wiebe and E.E. Knaus (Edmonton)], Croatia [M. Mintas and S. Raic-Malic (Zagreb), L.J. Tusek-Bozic (Zagreb)], Egypt [E.-S. El-Ashry (Alexandria)], France [A. Gueiffier (Tours), G. Guillerm (Reims), S. Kirkiacharian (Paris-Sud), B. Meunier (Toulouse), M. Reboud-Ravaux (Paris), J.-P. Roque (Montpellier)], Germany [W. Pfleiderer (Konstanz), F. Seela/H. Rosemeyer (Osnabrück), P. Wutzler (Erfurt)], Greece [N. Kolocouris (Athens), D. Papaioannou/G. Balayiannis (Patras)], Hungary [L. Ötvös/G. Sagi (Budapest), F. Sztaricskai (Debrecen)], India [S.S. Karki (Bangalore), S.B. Katti (Lucknow), T. Pathak (Pune), A.K. Prasad (Delhi), K.N. Singh (Varanasi)], Ireland [M. Lewis (Trinity College, Dublin)], Italy [A. Chimirri (Messina), S. Manfredini/P. Baraldi (Ferrara), G. Natile (Bari), M. Roberti (Bologna), G. Romeo/U. Chiacchio (Messina)], Japan [M. Baba (Kagoshima), T. Maruyama (Tokushima), A. Matsuda (Sapporo), H. Tanaka (Tokyo), M. Ubasawa (Yokohama)], Korea [Chong-Kyo Lee (Taejon), Jong Chan Son (Taejon)], Mauritius [S. Jhaumeer-Laulloo/S.R. Ramadas (Reduit)], Morocco [H.B. Lazrek (Marrakech)], The Netherlands [A.D.M.E. Osterhaus (Rotterdam), H. Timmerman (Amsterdam)], Norway [M.L. Sandvold/F. Myhren (Porsgrunn)], People's Republic of China [Xinyong Liu (Shandong University, Jinan), Fen-Er Chen (Fudan University, Shangai)], Poland [J. Boryski/B. Golankiewicz (Poznan), T. Kulikowski/D. Shugar (Warsaw), W.J. Stec (Lodz)], Russia [A.A. Ozerov/M.S. Novikoff (Volgograd), M. Preobrazhenskaya (Moscow)], Spain [M.-J. Camarasa (Madrid), F. Fernandez-Gonzalez (Santiago de Compostela), M. Nogueras (Jaén), S. Vega (Madrid)], Switzerland [J.M.J. Tronchet (Geneva)], Taiwan (Jih-Rhu (Ruben) Hwu], Turkey [I. Küçükgüzel (Istanbul)], United Kingdom [S.J. Archibald (Hull), C. McGuigan (Cardiff), P.J. Sadler (Warwick)] and the United States of America [R.T. Borchardt (Lawrence, Kansas), M. Cushman (Purdue University, West Lafayette, Indiana), M.K. Lakshman (New York), V. Marquez (Bethesda), M.J. Miller (Notre Dame, Indiana), V. Nair (Athens, Georgia), M.J. Robins (Provo, Utah), S.W. Schneller (Auburn, Alabama), C.E. Stephens (Augusta, Georgia), S.F. Wnuk (Miami) and P.F. Torrence (Flagstaff, Arizona)]. I look back at these and many other collaborations with utmost nostalgic feelings, because once upon a time they launched high expectations at both sides. Most of these collaborations died quietly because of the protagonists disappeared or retired (like myself), but for those that still lasted as of today, I will keep cherishing. Both those that are gone and those that are still remaining testify as to the paradigm I have always defended as emblematic for the triumph of science, the innate link between chemistry and medicine.
2.9. Writers’ cramp
In 1993 I featured in the issue Vol. 259 (January 1993) of Science under the editorials on page 180 the listing of the most productive (prolific) science authors of 1991 (Table 1). I ranked No. 4 worldwide after No. 1 Thomas Starzl (from Pittsburgh), a transplant surgeon, No. 2 Yury T. Struchkov, and No. 3. K. Ploog, with some 80 publications per year. Starzl had 155 publications for 1991. I wondered how he did achieve this quasi Herculean task. Little did I know that in January 2005, I would meet him in person at a rather small meeting of the Philosophical Society in Philadelphia, in honor of Maurice Hilleman, a few months before Maurice passed away from pancreatic cancer. At the speakers' party the night before the meeting, I was sitting next to Starzl, but I don't remember we talked about publications. This is the only time I have ever met Starzl. The meeting honored Maurice Hilleman, who for all these years had stayed my hero (also present at the meeting were Tony Fauci, Bob Gallo, Stanley Plotkin, Hilary Koprowski, Roy Vagelos, and from the European side only Ehrling Norrby, Britta Wahren, and myself). It was at this meeting that Roy Vagelos epitaphed that “M. Hilleman was a bastard from the outside, but from the inside, he was still a bastard”. Bob Gallo had hoped to honor Hilleman again in April 2006, tentatively in Taormina (Sicily), but Maurice's death on 11 April 2005 trespassed these plans. My publication record leveled off at 100 per year, before it started to sharply decline with my retirement in 2006. University rules in Belgium dictate that from the age of 65, any professor is discharged from all his functions: administration, teaching and research. While I readily accepted this rule for the administrative duties, I found it totally unacceptable for research and teaching. I bitterly resented and objected to this obligatory retirement, but even almost 10 years after the verdict, I still feel it hard to accept and consider the obligatory abdication of my research and teaching duties as unjustified, penalizing and discriminatory. University authorities (rectors and the like) consistently admitted it was a great pity that I could not continue my job, but nobody undertook any action, or (to use soccer terms) passed an assist to any other responsible (and responsive) person to overrule the ordeal. The sole attempt to try to break this rule came from Antonín Holý. In a handwritten letter of 2 pages in 2004, which I have kept as a relic. He pointed out the possibility of leaving Leuven and join him at the IOCB in Prague. Although this transition was never formalized, it made me to come over to the Czech Republic more often. I became an honorary doctor, thanks to Holý, at Charles University in Prague, and after Holý had brought me into contact with his good friend, Libor Grubhoffer in České Budějovice, both Holý and I were together promoted to honorary doctors in České Budějovice in 2009, and from 2007 till today I am still teaching the course of “Chemistry at the Service of Medicine” at Jihočeská University in České Budějovice, and I must confess I immensely appreciate this opportunity to continue teaching, which I hope to continue as long as my capabilities permit to do so.
Table 1.
Listing of the most productive science authors of 1991 (Science 1993; 259:180).
| Scientific papers: top producers of 1991 | |||
|---|---|---|---|
| Rank | Researcher | Papers | Authors per paper |
| 1. | Thomas E. Starzl, transplant surgery | 155 | 7.8 |
| U. Pittsburgh | |||
| 2. | Yury T. Struchkov, chemistry/crystallography | 83 | 6.0 |
| Inst. Organoelemental Compounds, Moscow | |||
| 3. | K. Ploog, condensed-matter physics | 81 | 5.0 |
| Max Planck Institut für Festkörperforschung, Stuttgart, Germany | |||
| 4. | Eric De Clercq, virology | 80 | 6.0 |
| Catholic University of Louvain, Belgium | |||
| 5. | John J. Fung, transplant surgery | 72 | 8.1 |
| U. Pittsburgh | |||
| 6. | Pierre Braquet, immunology/pharmacology | 61 | 5.4 |
| Institute Henri Beaufour, Le Plessis Robinson, France | |||
| 7. | Virgil Percec, organic and polymer chemistry | 56 | 2.7 |
| Case Western Reserve University, Cleveland | |||
| 8. | Allan H. White, physical and inorganic chemistry | 51 | 4.9 |
| U. Western Australia, Nedlands | |||
| Stephen J. Pearton, condensed-matter physics | 51 | 5.2 | |
| AT&T Bell Labs, Murray Hill, New Jersey | |||
| 9. | Hans Georg von Schnering, chemistry/crystallography | 50 | 4.7 |
| Max Planck Institut für Festkörperforschung | |||
| Carlo A. Maggi, pharmacology | 50 | 4.8 | |
| A. Menarini Pharmaceuticals, Florence, Italy | |||
| Genrikh A. Tolstikov, chemistry/crystallography | 50 | 5.3 | |
| Inst. Chemistry, Academy of Sciences, Ufa, Russia | |||
2.10. Teaching
In 1965 an affiliated campus to Leuven, the KULAK, was started with some faculties in the humanities (linguistics, law, philosophy). In 1971, the Faculty of Medicine was added (this was just after my return from Stanford), and Prof. P. De Somer, already endowed with the function of rector, invited me to teach biochemistry in the 2nd and 3rd year of medicine, together with a colleague of mine, Marcel Joniau. Joniau was a real biochemist, who was interested in medicine, whereas I was a medical doctor interested in biochemistry. It was hard to predict that I would continue teaching in Kortrijk at the KULAK for 37 years, from 1972 to 2009 (Joniau would stop teaching when he had to retire in 2004). Together we formed an ideal couple, teaching and examining the students for their insights in (bio)chemistry especially as related to medicine. In the more than 30 years when we were teaching the course, I don't remember any quarrel or dispute. As the years passed by, we felt each other's needs, capacities and attitudes even better, and when Marcel Joniau had to abandon his teaching, I felt like I had lost my “compagnon de route in teaching”. It was the prelude of my own farewell to the teaching in Kortrijk. At the KULAK I met wonderful colleagues, invariably from other faculties (Mgr. G. Maertens, Prof. Baron Louis Suetens, Louis Vos, Jean Goossens, Christophe Waelkens, Madeleine Sergeant, Odon Leys, Luc Draye, Michel Cloet, Jos Monballyu, etc. From Mgr. Maertens I remember an anecdote when he had observed that P.C. Paardekoper when sunbathing with his torso naked, that this was not decent to appear like this, in front of the students, adding to his statement that his remarks were not from ethical but esthetical nature (Mgr. Maertens taught the course of ethics to the medical students). Yet, the KULAK did not seem to recognize or appreciate my 37 year service of teaching (including the examinations of circa 5000 students). When I stopped teaching in Kortrijk after 37 years, there was not even any token of recognition, let stand, gratitude. I left Kortrijk in 2009 the same way as I had entered it in 1972, unnoticed.
Conclusion: retrospectives
Over the whole period of my academic career, I have always divided my time for research, teaching and organizational activities. Highlights in research were the discovery (in 1978) of BVDU as a highly selective anti-HSV-1 and anti-VZV agent, that was eventually approved for the systemic (oral) treatment of herpes zoster. In the early 1980s followed the acyclovir aminoacyl esters, the prototype of which, the valine ester of acyclovir, would succeed acyclovir for the treatment of HSV and VZV infections. In the wake of the AIDS epidemic, followed in 1986 the discovery of d4T, for which our laboratory got the scientific credit, but not the financial one. The year 1986 also marked the birth of the acyclic nucleoside phosphonates (ANPs) with (S)-HPMPA and PMEA (adefovir), which in 1987 would be followed by (S)-HPMPC (cidofovir) and in 1993 by (R)-PMPA (tenofovir). The birth of the NNRTIs dated back from the late 1980s with the discovery of HEPT (in 1987) and TIBO (in 1988).
This research found its “raison d'être” in the contacts, appreciation and encouragements I continuously had from the collaborations with at their height about 200 chemists worldwide, foremost among them are, Antonín Holý (Prague) and John C. Martin (Gilead Sciences). The collaboration with Holý started in 1976 (and ended with his death in 2012), that with Martin started in 1986 (and still continues). My research duties never hampered my teaching (mainly biochemistry and virology) which I had started in 1972 at the KULAK in Kortrijk, where I continued teaching till 2009. For the courses of virology (which I had to relinquish in 2006) I was also appointed from the early 1990s in Leuven. Both in Leuven and Kortrijk my teaching only involved medical students (Medical School). Teaching was fun, and so were the three NATO ASIs I ever organized, with R.T. Walker as co-director, in Sogesta (Italy) in 1979 (which was also co-organized by F. Eckstein), Les Arcs (France) in 1983 and Il Ciocco (Italy) in 1987. From these NATO meetings emanated from 1988, the ICAR meetings, which have continued ever since on an annual basis, and of which the 28th was held on 11–15 May 2015 in Rome (Italy).
My comments
Highlights in antiviral drug research are serendipity, collegiality, collaborative ties and making competitors happy. Acyclovir, the gold standard among the antiviral drugs, was discovered by chance, or serendipitously. It was originally intended as an inhibitor of adenosine deaminase, to potentiate the antiviral activity of a known antiviral compound, vidarabine. BVDU (brivudin) was also discovered serendipitously, when found active against HSV-1, simply because HSV-1 together with vaccinia virus was part of our antiviral screening assay. BVDU would later be commercialized for the treatment of VZV infections. The discovery of d4T should be viewed as an example of collegiality or friendship among colleagues which should prevail over the millions of dollars that could be generated. The NATO/FEBS meetings that I organized fostered collaborative ties over the whole world (US, West and East Europe, Japan and Australia), which through the research results they generated, proved beneficial to mankind, and, as I competed (and lost) 5 times when competing for the Francqui Prize (sometimes called the Belgian Nobel Prize) I made 5 competitors happy, which should be viewed as a consolation for those who participated (and lost) in the contest.
III. From 2006 to 2015
In 2006 I was obliged to retire from all duties, administrative, educational (teaching) and scientific (research). Although the teaching at my own university was drastically reduced in 2006 and finally stopped in 2009, my love for teaching was better appreciated in the Czech Republic where from 2007 till today I was given the opportunity to continue teaching my favored course “Chemistry at the service of Medicine” in Ceské Budějovice, in a program co-sponsored by Jihočeská University and Kepler University in Linz (Austria). Meanwhile, Truvada®was licensed (on 16 July 2012) for the prophylaxis of HIV infections, the first anti-HIV drug approved by the US FDA for this purpose. The successor of BVDU, the valine ester of Cf1743 (FV-100) is still under clinical development for the treatment of VZV infections, and AMD3100 that was originally discovered as an anti-HIV agent is now on the market (as Mozobil®) for the mobilization of (hematopoietic) stem cells. TAF is scheduled to replace TDF in future tenofovir-based drug combinations such as Stribild®. Holý's legacy yielded a number of original compounds such as 1-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]-5-azacytosine (HPMP-5-azaC), and various 6-(3-hydroxy-2-(phosphonylmethoxy)propoxy)- (HPMPO-), 6-(2-(phosphonylmethoxy)ethoxy)- (PMEO-) and 6-(2-(phosphonylmethoxy)propoxy)-2,4-diaminopyrimidine (PMPO-DAPy) derivatives, for which the future clinical development looks uncertain. At the international pharmaceutical drug scene, much interest has shifted from HIV to HCV, where the prospects for a real cure have become realistic with the advent of sofosbuvir (Sovaldi®) and ledipasvir, and the combination thereof (Harvoni®).
3.1. 2006, the hoax
I knew beforehand that the year 2006 would be disastrous, because I would turn 65 years old on 28 March 2006, and this should be the inevitable end of my active career. A committee, existing of the Vice-Rector, Mark Waer, the General Commissioner, Koenraad Debackere, the Dean of the Medical Faculty, Bernard Himpens, and a certain Bamelis from the Board of the KU Leuven Directors, was installed, in my honor, to streamline the transition from the active professorship to the retired emeritus state. Not all retiring professors get such distinguished funeral. This is reserved for the happy few. It did not prevent the then acting Rector, Mark Vervenne, from sending me a formal letter, separately from the Committee's decision that as of October first, 2006, I would be relieved from all my functions, administration, teaching and research. No alternatives were offered, as if it could be presumed that retirement would be quickly followed, possibly by a causal relationship, by death. In any case the Committee had by November 2006 prepared a text, written by Bernard Himpens, where they complimented me, still alive, for the nice legacy I had left. They must have referred to my exemplary teaching (up to 11 hours per week), publication record (then exceeding 2000, now more than 2,500, publications in peer-reviewed journals resulting in ten thousands of citations), and about ten of my pupils who succeeded me as professors. As Prof. Marcel Janssens (Linguistics), who was succeeded by six professors, once said in his farewell speech, “I must have been a very cheap employee at the University, as they now have to pay six times more for the same job done”. The money I generated for the university, in terms of royalties, became redeemed only from 2008 onwards, and this brought the KU Leuven in the top ten ranking of the universities over the world. The management of the university has been well aware of these royalties (received from Gilead Sciences), but the origin of the money remained a rather well-kept secret, as if it had fallen, like manna, from Heaven (at a Catholic university, miracles may have been considered likely to happen!). Some of my good friends (as Prof. Frank Seela from Osnabrück, now at Münster, in Germany) had warned me that, according to his own experience, I had not to expect much clemency or gratitude from the university. At least I could keep my parking space in front of the institute's entrance which appeared very handy now that my walking abilities have become problematic and my seat at the secretary's desk which allows me to sit down, instead of standing up, when going through my correspondence and reviewing my publications at the service of the university.
3.2. Honorary doctorates
My first honorary doctor's degree in 1997 was from the University of Ghent. The eloge (“laudatio”) was pronounced by Prof. Denis De Keukeleire, worldwide known as beer-expert (“zytologist”), equivalent to a wine expert (“oenologist”). Denis had once visited the NIH library in Bethesda, and when looking for how many times his name was cited, his eyes fell on a certain De Clercq, whose name was listed just (or a few places) before his name. Exploring my CV in some further detail, and after consulting Piet Herdewijn, Denis introduced me for an honorary doctorate in pharmacy, as it was the annual turn for this discipline. I was proud with this honor from Ghent, as it bestowed me with the same distinction in Ghent as my mentor, Prof. P. De Somer, once got. That same year (1997) followed an honorary doctor's degree, again in pharmacy in Athens, with Prof. Nicolas Kolocouris as my promoter. Then followed in 2000, an honorary degree, again in pharmacia, at the University of Ferrara, with Stefano Manfredini as my promoter (and Pier Giovanni Baraldi (as co-promoter, and promoter of Dale Boger who got at the same occasion also an honorary doctor's degree from Ferrara). Incidentally, Copernicus and Paracelsus are also honorary doctors from Ferrara. Then followed in 2005, an honorary doctorate (actually professorship) from Shandong University, Jinan (with Xinyong Liu as promoter) From this university I got the nicest robe “toga”, all in red, and so many gifts my host provided me with so that I had to get an additional suitcase to take it all home. On 7 March 2007, I was awarded a doctor's degree, this time in medicine, from Charles University, Prague, certainly with the help of Antonín Holý. In the spring of 2007, I also gave a course of 15 hours on antivirals at Charles University, and in the fall of 2007 I started my teaching in České Budějovice (I had to identify the location and exact pronunciation of České Budějovice), where together with Antonín Holý, I was awarded an honorary degree on 4 June 2009. Our promoter in České Budějovice was Prof. Libor Grubhoffer. Two additional honorary degrees would follow: in 2010 in Tours (France) and in 2011 in Hull (United Kingdom), respectively, with Alain Gueiffier and Steven Archibald as my promoters. This would conclude my honorary doctorships. On 8 June 2013 I would be honored by an honorary citizenship from my native town, Hamme, which at least partially contradicts the general saying that “one is never recognized at one's own place”. The honorary citizenship in Hamme, which I cherish immensely, testifies as to the opposite.
3.3. Tsai et al. (1995)
In 1995, I appeared on the Belgian TV (VTM) to support the importance of lamivudine (Epivir®) from GSK, as an anti-HIV drug, with equally remarkable activity against HBV. I told the journalist, I was delighted to do this public relations (PR) job for GSK, but that I would even be more delighted to do this over again for my own product, (R)-PMPA (or tenofovir) for which I had heard that Tsai et al.43 had found remarkable activity against SIV (simian immunodeficiency virus), far superior to that of AZT (azidothymidine). These results would be communicated in the 17 November 1995 issue of Science. I informed the Belgian TV (VTM) that this was going to happen and told them I would not mind to appear on TV again to talk about my own product44 that had been licensed to Gilead for further development. At the TV they told me that they would be most interested to interview me on Friday, 17 November 1995, but, when I told them, this would not work out because Fridays were reserved for my teaching at the KULAK in Kortrijk, I had hoped they would call it off, but on the contrary, they came over to Kortrijk to interview me, in front of the students (who very much enjoyed such exceptional event in the otherwise rather boring KULAK or Kortrijk environment). Hence, my performance was put on TV (for which the Campus Rector Vic Nachtergaele, whom I did not ask for permission, congratulated me later wholeheartedly). The interview went very well, until the TV reporter suddenly asked whether I had now discovered the “morning after” pill for the treatment of AIDS. This question was not foreseen in a life edition (prime time) on TV, so that, taken by surprise, I must have answered, this may have been the “morning after”, but it still was not a “pill” yet. The appearance on TV in Kortrijk, may certainly have added to my prestige in the Texas (Dallas) part of Belgium, but remained largely unnoticed in Leuven. In looking back at the Tsai et al. paper43 (Fig. 21), it indeed marked the use of the “morning pill” of (R)-PMPA, later formulated as tenofovir disoproxil fumarate, in combination with emtricitabine (Truvada®), which had in 2004 been commercialized for the treatment of AIDS, and eight years later, on 16 July 2012, was approved by the US FDA for the prevention of AIDS. That same day coincided with the death of Antonín Holý, co-inventor of what I had announced on 17 November in 1995 on television as the “morning after” pill for AIDS.
Figure 21.

Protein immunoblot analysis of SIV-specific antibody response in macaques 20 weeks post infection with SIV. Mock-treated control macaques (n=10) and macaques treated with PMPA (tenofovir) starting at 24 h post infection (n=5) are presented. Antibodies to env glycoprotein gp120, gag proteins p28, p16 and p8 and vpx protein p14 were detected in all of the control macaques. None of the macaques treated with PMPA starting 24 h post infection showed SIV-specific antibodies (Tsai et al.43).
3.4. Nature reviews drug discovery/biochemical pharmacology
In 2000 I was awarded the Otto Krayer Award from ASPET (American Society of Pharmacology and Experimental Therapeutics). The award was handed over by Sam Enna in Orlando (at the end of March) and a few days later I gave the commemorative lecture later published in the Journal of Pharmacology and Experimental Therapeutics45. Present in the audience was a certain Mr. Smith, Editor of a new journal (Nature Reviews Drug Discovery, NRDD) who invited me to write a review article for NRDD, which was published in 200246. In 2003 I published a perspective article on AMD3100 (see section on AMD3100) in NRDD47, which summarized the saga on the discovery of AMD3100 from its early days when it was recognized as an anti-HIV agent until its recognition as an hematopoietic stem cell mobilizer. Additional, more recent review articles would follow on the stem cell mobilization of AMD3100 after it had been marketed as Mozobil48, 49. In 2004, I wrote a review article for Nature Reviews Microbiology50, and in 2005, I returned to NRDD51 with a review article on the acyclic nucleoside phosphonates with Antonín Holý as co-author: it covered our collaboration over a 20-year period, from 1985 till 2005. Although I was not actively involved in experimental work with influenza or antivirals against influenza, I wrote purely from scientific interest a review article on influenza in NRDD52, simultaneously with a more chemically oriented paper that I co-authored with Irène Lagoja in Medicinal Research Reviews53. In 2007 I would write my last paper for NRDD54. It was worth a double paper, as it covered both HIV and HCV, highlighting the similarities (and dissimilarities) between anti-HIV and anti-HCV agents and their mechanism(s) of action. Eventually I would start writing a paper on cellular targets for antivirals, intended for publication in NRDD, but the paper was never completed, and has thus remained unpublished. Instead I kept writing commentaries for Biochemical Pharmacology, on a wide variety of antiviral compounds, strategies, and perspectives55, 56, 57, 58, 59, 60, most recently focused on Ebola virus (therapeutic strategies)61.
3.5. The successor of BVDU
BVDU appeared to be the most potent anti-VZV agent, ever described until 2000, far exceeding the acyclic nucleoside analogues acyclovir (valacyclovir), ganciclovir and penciclovir (famciclovir) in anti-VZV potency. Then, in 2000, McGuigan et al. described a new 2′-deoxynucleoside analogue, bicyclic nucleoside analogue (BCNA), actually a bicyclic furo [2,3-d]pyrimidine derivative (Cf 1743, Cf standing for Cardiff)62, which was still about 10-fold more potent than BVDU. This type of molecule contains a highly apolar tail which must function as a ligand to fit snugly into a cleft of the VZV thymidine kinase (TK). Although the BCNA is structurally unrelated to the bicyclam AMD3100, it is curious to note that for both sorts of compounds the replacement of an aliphatic by an aromatic bridge in the bicyclams, when moving from JM2763 to JM310063, 64, and the introduction of an aromatic phenyl moiety into an entirely aliphatic side chain of the BCNA62, 65, led to a quantum jump in potency against either HIV or VZV, respectively. Whereas BVDU was active against HSV-1, VZV and some herpesviruses of veterinary importance, Cf 1743 was uniquely active against VZV. The exquisite potency of Cf 1743 against VZV has been convincingly demonstrated by Andrei et al.66 It clearly depends on a specific interaction of the compound with the viral TK. This interaction is necessary but not sufficient to explain the anti-VZV activity of the compound: for example, it has not been resolved whether the compound has to be phosphorylated to either the mono-, di- or triphosphate to be antivirally active. In analogy with the conversion of acyclovir to valacyclovir, Cf 1743 (Fig. 22) was also converted to its 5′-valine ester (termed FV-100, FV standing for Fermavir)67. Migliore68 called FV-100 the most potent and selective anti-VZV agent reported to date. The compound is not only very active, it can also be guaranteed to be safe, and unlike 1-β-d-Arabinofuranosyl-(E)-5-(2-bromovinyl)uracil (BVaraU), another potent anti-VZV compound, Cf 1743 (or FV-100) is devoid of any potential interaction with the catabolism of the anticancer drug 5-fluorouracil. Phase II clinical trials with FV-100 in the treatment of herpes zoster look very encouraging69. In the unique specificity of FV-100 against VZV lie both its strength and weakness, the latter being due to its limited therapeutic activity spectrum (VZV only).
Figure 22.

Structures of Cf1743 and FV-100.
3.6. AMD3100→Mozobil™
AMD3100 started as an impurity present in a preparation (JM1600) sent from Johnson Matthey (JM) to be checked for its anti-HIV activity at the Rega Institute. This sort of compounds, the bicyclams, had never been accredited with any antiviral properties, let stand anti-HIV activity, so there were no precedents. Its mode of action was attributed to a possible interference with HIV entry into the cells (then interpreted as “uncoating”). The paper was published in the PNAS in 199263 sponsored by the Nobel Prize winner, Max Perutz. Starting from JM1600 as the model compound, JM2763 was described as an anti-HIV agent63 and in 199464 we came up with an even more potent anti-HIV agent, JM3100, having anti-HIV activity in the nanomolar concentration range, the name was changed to AMD3100 (Fig. 23) when AnorMed was created as a spin off from JM. In AMD3100 the aliphatic bridge tethering the cyclam moieties is replaced by an aromatic (i.e., phenylene bis-methyl) bridge. The activity and specificity of AMD3100 was such that it surpassed all known anti-HIV agents in both potency and selectivity. Together with my JM colleagues (Don Picker, Mike Abrams, Geoff Henson and Gary Bridger, whom I unrespectfully sometimes referred to as the four “Daltons”), I went on a road tour, in an attempt to get pharmaceutical companies (JM being a chemical company) interested. We visited about 15 companies, to whom I could tell the story, and, finally, Sandoz (Basel) took it over. Sandoz (with Rolf Datema) actively pursued the compound until, on a blue Monday sometime in November 1998, Sandoz merged with Ciba-Geigy to become Novartis, and consequently they dropped their entire anti-HIV program encompassing not only JM3100 (AMD3100), but also a cyclosporin derivative and a protease inhibitor, which if combined may have allowed Sandoz (or Novartis) at the end of the 1990s to become the leader in the field. Meanwhile, in 1997, we had discovered the reason for the anti-HIV activity of AMD3100, that is a specific interaction with CXCR4, the co-receptor for T-lymphotropic strains of HIV to enter the cells (the co-receptor for the M (macrophage) tropic strains being CCR5)70, 71. Under the auspices of AnorMed, they started at Johns Hopkins the first clinical trials, where Craig Hendrix and his colleagues72 found that AMD3100 at 8–10 hours upon intravenous (or subcutaneous) injection caused a remarkable increase in the white blood cell (WBC) counts (Fig. 24), which was just the opposite of what to be expected from cytotoxic compounds. These white blood cells, at closer inspection, appeared to carry the CD34 marker, which identified them as belonging to the hematopoietic stem cell (HSC) type49. AMD3100 thus qualified as a stem cell mobilizer, and while it was never commercialized as an anti-HIV drug for which it was originally intended, it was finally commercialized as a stem cell mobilizer (Mozobil™) by Genzyme after they had taken over AnorMed (Genzyme is now part of an even larger company, Sanofi). Mozobil is used for autologous transplantation of hematopoietic stem cells in patients with non-Hodgkin's lymphoma (NHL) or multiple myeloma (MM). It should be further explored for its stem cell mobilization in other pathophysiologic conditions, including malignant and rheumatic diseases, and tissue regeneration.
Figure 23.
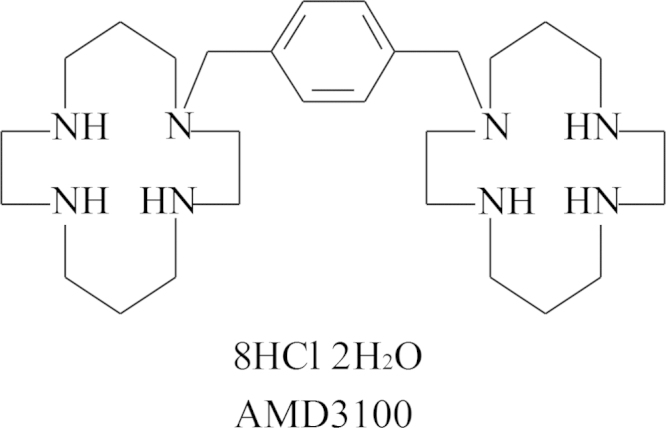
Structure of AMD3100.
Figure 24.

WBC ratio versus time compared to AMD3100 concentration versus time following single-dose intravenous AMD3100 administration (Hendrix et al.72).
3.7. Paul Janssen and John C. Martin united: Complera/Eviplera
The year 1986 was crucial in our search for anti-HIV agents. This year we described (with Antonín Holý) the broad-spectrum anti-DNA virus activity of (S)-HPMPA as the prototype of the ANPs73. On 6 of November 1986 I visited Paul Janssen, with whom I spent about 8 hours together (including lunch and dinner) in Beerse. We discussed the feasibility to join our forces to discover the “ideal” (miraculous) drug for the treatment of AIDS, which had now become a full-fledged globally feared epidemic, for which at the end of 1987 we would discover HEPT, and in 1988, the TIBO compounds. In this period I often traveled to Bristol-Myers at Wallingford, where my host was John C. Martin. At one of the diners preceding my formal lecture the next morning John would interrogate me (as he always did). One of his penetrating questions was which compound I would prefer for the treatment of AIDS, an ANP (the ANP he meant was PMEA) or TIBO. I told him he probably needed both, to hit vigorously with an ANP (running the risk for toxicity without much risk for resistance development), and smoothly (elegantly) with TIBO (no toxicity but risk for resistance). This distinction was perhaps not entirely correct, but emblematically it predicted the combination that more than twenty years later became one of the most successful regimes for the treatment of AIDS, the combination of TDF as an ANP derivative and rilpivirine as a TIBO derivative (now generally referred to as a NNRTI). This combination has been dubbed Complera® (in the US) and Eviplera® (in the EU)56. It contains tenofovir, which we described as (R)-PMPA44 and rilpivirine, which Dr. Paul considered his “miraculous” (or at least the ideal) drug for the treatment of AIDS74.
3.8. TAF
In the original agreement signed by the inventors, Antonín Holý and myself, and Bristol-Myers (the agreement was later taken over by Gilead Sciences), it was agreed that to compensate for the rather symbolic downpayment fee received for our invention, we should receive royalties on the prodrugs that would ever be synthesized from the original compounds such as tenofovir. The original patent on tenofovir [(R)-PMPA] dates from 1992 (published in 199344), but the patent on its prodrug TDF is going back till 199775, 76, so that if the 20-year protection time is respected, the protection of TDF is expiring in 2017. However, in the meantime, a new prodrug of tenofovir, GS-7340 or 9-[(R)-2-[[[[(S)-1-(isopropoxycarbonyl)ethyl]amino]phenoxy-phosphinyl]methoxy]propyl]adenine (Fig. 25), dubbed TAF, has been synthesized77, 78, the unique propensity of GS-7340 to be preferentially taken up by the lymphatic tissue was published in 2005 by Lee et al79. (patented in 2004 so that it remains protected until 2024). In HIV-1-infected patients, 10-day monotherapy with 25 mg TAF demonstrated superior efficacy, due to its higher uptake by the lymphoid cells, than TDF administered at 300 mg80. GS-7340 effected high and persistent levels of (R)-PMPApp in peripheral blood mononuclear cells following oral administration of TAF81. It is to be anticipated that in the future TDF may be replaced by TAF in all combinations containing TDF (i.e., Truvada®, Atripla®, Complera®/Eviplera®, Stribild®), and that other combinations containing TAF (i.e., with darunavir) may be launched.
Figure 25.
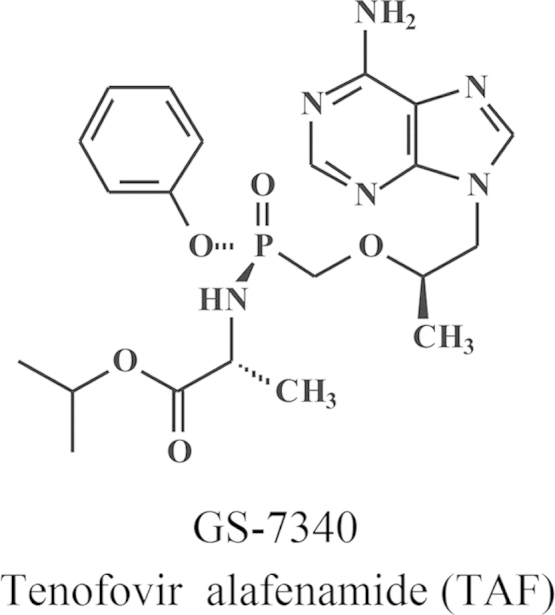
Structure of tenofovir alafenamide (TAF).
3.9. Antonín Holý legacy
While an impressive number of Holý's compounds reached the market, an even more impressive number of valuable (potential) candidate drugs did not, the first being (S)-HPMPA73 but also its diamino counterpart (S)-HPMPDAP82. Neither did the diamino counterpart of adefovir (PMEA), 9-(2-phosphonylmethoxyethyl)-2,6-diaminopurine (PMEDAP). This must have something to do with the prejudice that nature did not select 2,6-diaminopurine as a natural base in its “repertoire” of purines (adenine, guanine and hypoxanthine) and pyrimidines (cytosine, thymine and uracil). Additional considerations may be the fact that no antivirals are known to be built upon a 2,6-diaminopurine as heterocycle, that 2,6-diaminopurine skeleton may be quickly metabolized to a guanine moiety, and if it were to be incorporated as such into DNA, it may lead to (transition) mutations. Of the guanine derivatives, PMEG [9-(2-phosphonomethoxyethyl)guanine] has been known since 198783, but considered to be too (cyto)toxic to be further pursued for its antiviral potential. Instead, it has been further explored for its antitumor potential as its prodrug cPrPMEDAP [9-(2-phosphonomethoxyethyl)-N6-cyclopropyl-2,6-diaminopurine], from which two proprodrugs were prepared: GS-9191 (Fig. 26), which is further explored by Graceway Pharmaceuticals (Bristol, TN) for the topical treatment of genital warts (in humans); and GS-9219 (Fig. 27), which is evaluated by Vet DC (Fort Collins, CO) for the intravenous treatment of NHL in dogs. A series of compounds for which Antonín Holý had high hopes are the so-called O-DAPy derivatives (R)-HPMPO-DAPy, PMEO-DAPy and (R)-PMPO-DAPy (DAPy standing for 2,4-diaminopyrimidine, the oxygen linking the C-6 of the pyrimidine with the acyclic side chain). The prototype of these compounds had been described by Holý in 200284. Although the 6-[2-(phosphonomethoxy)alkoxy]-2,4-diaminopyrimidines are pyrimidine derivatives (Fig. 28), they act as purine nucleotide mimetics. Their spectrum and mode of action, and especially their therapeutic potential, remain largely unexplored85. Following a tradition initiated at the IOCB in the 1960s by Dr. Holý's boss, Dr. František Šorm86, Holý, in the last years of his life, turned his attention to replacing the pyrimidine moiety of (S)-HPMPC by a triazine moiety, 5-azacytosine, thus synthesizing (S)-HPMP-5-azaC, its cyclic derivative, (S)-cHPMP-5-azaC and several alkoxyalkyl ester prodrugs thereof (Fig. 29)87. The 5-azacytosine counterpart of (S)-HPMPC (and its prodrugs) may be worth further evaluating now that the intellectual property protection period of (S)-HPMPC has expired.
Figure 26.
Structure of GS-9191.
Figure 27.
Structure of GS-9219.
Figure 28.

Structures of the O-DAPys.
Figure 29.

Structures of (S)-HPMP-5-azaC and derivatives thereof.
3.10. Sofosbuvir
HCV is fundamentally different from HIV and HBV: HIV is a retrovirus that transcribes its RNA by the virus-associated reverse transcriptase (RT) into DNA which is then integrated by the virus-associated integrase into the chromosomal DNA. HBV is an hepadnavirus, a DNA virus, which can persist in the HBV-infected cell as cccDNA (ccc=covalently closed circular). HCV, on the other hand, is an RNA virus (i.e., hepacivirus, belonging to the flaviviridae). Like RNA viruses in general it does not replicate through a DNA intermediate. It can thus be eradicated, and this is the current aim of the treatment, that is to eliminate it as soon as possible. For the last decade, standard treatment of HCV infections existed of pegylated (PEG) interferon combined with ribavirin. This standard of care (SOC) will soon belong (or already belongs) to the past. There are now plenty of direct-acting antivirals (DAAs) at hand, which inhibit the replication of HCV: they are mainly targeted at specific viral proteins (NS3/4A protease, NS5A protein, NS5B polymerase (nucleoside (N)-type of inhibitors), NS5B polymerase (non-nucleoside (NN) type of inhibitors)). Some (i.e. cyclosporins) are targeted at a cellular target (i.e. cyclophilin). Personally I have been involved with some of the NN type of NS5B polymerase inhibitors (i.e. tegobuvir (GS-9190)) and cyclosporin inhibitors of both HIV and HCV. The most promising DAA, however, is sofosbuvir (Sovaldi®) (Fig. 30). It is a highly decorated uracil derivative with high potency against all HCV genotypes (1–6), devoid of any significant side effects, and not leading to resistance development within the proposed treatment duration time of 12 weeks60, 88. Sofosbuvir will likely become the cornerstone for the treatment of hepatitis C, offering the tantalizing perspective that it will be the first antiviral drug ever shown to be effective in eliminating (or curing) an established virus infection. To this end, sofosbuvir may be combined with one or more other DAAs, such as ledipasvir, a NS5A inhibitor. This combination strategy has already been shown to achieve a sustained viral response (synonymous for cure) after a treatment period of 8–12 weeks. In the future, treatment of hepatitis C may even be further shortened with sofosbuvir used in the appropriate combination with other DAAs (such as ledipasvir).
Figure 30.

Structure of sofosbuvir (Sovaldi®).
Conclusion: perspectives
With BVDU being generic, standing out as the optimal choice for the treatment of VZV infections (shingles, herpes zoster) is FV-100, the 5′-valine ester of Cf1743. Although FV-100 is the most potent and selective anti-VZV agent ever reported, it is questionable whether it would ever be developed and commercialized because of its limited usefulness (VZV only).
Mozobil, originally discovered as an anti-HIV agent, has through a highly convoluted development pathway been eventually marketed as a stem cell mobilizer. Its future lies in various aspects of stem cell mobilization including tissue regeneration.
With TDF turning generic in 2017, its successor will be TAF, which is about 10–30 times more potent than TDF and preferentially be taken up by the lymphatic tissue. In future combinations based on TDF, TAF will take over its place, i.e., in combination with emtricitabine and rilpivirine, or in combination with emtricitabine and elvitegravir and cobicistat, or in combination with protease inhibitors such as darunavir. Except for tenofovir (TAF), it does not seem obvious that other compounds from Holý's legacy, i.e., the so-called O-DAPy derivatives (i.e., (R)-HPMPO-DAPy, PMEO-DAPy or (R)-PMPO-DAPy), or the 5-azaC derivatives, would be developed or marketed for human use.
While for HIV, the prospects for a cure have continued to remain enigmatic, there is more hope for a cure for HBV. For HCV, however, success for a cure with sofosbuvir (Sovaldi®) with or without other DAAs such as ledipasvir, can be guaranteed for circa 90% of the patients within a time period of 12 weeks. In the future, this time period may be shortened to 8 or even 6 weeks, and be guaranteed for all patients, irrespective of their (1–6) genotype.
My comments
There are a few messages I want to convey: (i), teaching and research are complementary in that they cross-fertilize each other: teaching helps resolving problems that could help in successfully planning research, and research should help formulating how to teach; (ii) collaboration between three disciplines (chemistry, biology, industrial exploitation) is mandatory in drug development: the chemist should contact the biologist to find out what is important for a new chemical entity; and for both, the contact with industry should be essential on how to best commercialize their finding in the benefit of mankind. My personal story on the “morning after pill”, first announced on television in November 1995, and culminating on 16 July 2012 with the approval of Truvada® for the prevention of HIV infections, reflects on the importance of perseverance. Convergence of similar principles, i.e., replacement of an aliphatic by an aromatic moiety as in the bicyclams and BCNAs (bicyclic nucleoside analogues), respectively, may lead to new leads for anti-HIV and anti-VZV activity. And, finally, two fundamentally different new approaches, that of the NRTIs (tenofovir) and NNRTIs (rilpivirine), may, if brought together, yield the ideal combination for the treatment of HIV infections.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
References
- 1.De Somer P, De Clercq E, Billiau A, Schonne E. Urinary excretion of interferon in rabbits. In: Proceedings of the first international conference on vaccines against viral and rickettsial diseases of man. Fort Lauderdale, FL, USA: Pan American Health Organization (PAHO); 1967. p. 650–2.
- 2.De Somer P., De Clercq E., Billiau A., Schonne E., Claesen M. Antiviral activity of polyacrylic and polymethacrylic acids. I. Mode of action in vitro. J Virol. 1968;2:878–885. doi: 10.1128/jvi.2.9.878-885.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Somer P., De Clercq E., Billiau A., Schonne E., Claesen M. Antiviral activity of polyacrylic and polymethacrylic acids. II. Mode of action in vivo. J Virol. 1968;2:886–893. doi: 10.1128/jvi.2.9.886-893.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lampson G.P., Tytell A.A., Field A.K., Nemes M.M., Hilleman M.R. Inducers of interferon and host resistance. I. Double-stranded RNA from extracts of Penicillium funiculosum. Proc Natl Acad Sci U S A. 1967;58:782–789. doi: 10.1073/pnas.58.2.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Field A.K., Tytell A.A., Lampson G.P., Hilleman M.R. Inducers of interferon and host resistance. II. Multistranded synthetic polynucleotide complexes. Proc Natl Acad Sci U S A. 1967;58:1004–1010. doi: 10.1073/pnas.58.3.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tytell A.A., Lampson G.P., Field A.K., Hilleman M.R. Inducers of interferon and host resistance. 3. Double-stranded RNA from reovirus type 3 virions (reo 3-RNA) Proc Natl Acad Sci U S A. 1967;58:1719–1722. doi: 10.1073/pnas.58.4.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Field A.K., Lampson G.P., Tytell A.A., Nemes M.M., Hilleman M.R. Inducers of interferon and host resistance, IV. Double-stranded replicative form RNA (MS2-Ff-RNA) from E. coli infected with MS2 coliphage. Proc Natl Acad Sci U S A. 1967;58:2102–2108. doi: 10.1073/pnas.58.5.2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Clercq E., Merigan T.C. Requirement of a stable secondary structure for the antiviral activity of polynucleotides. Nature. 1969;222:1148–1152. doi: 10.1038/2221148a0. [DOI] [PubMed] [Google Scholar]
- 9.De Clercq E., Wells R.D., Merigan T.C. Increase in antiviral activity of polynucleotides by thermal activation. Nature. 1970;226:364–366. doi: 10.1038/226364a0. [DOI] [PubMed] [Google Scholar]
- 10.De Clercq E., Eckstein F., Merigan T.C. Interferon induction increased through chemical modification of a synthetic polyribonucleotide. Science. 1969;165:1137–1139. doi: 10.1126/science.165.3898.1137. [DOI] [PubMed] [Google Scholar]
- 11.De Clercq E., Nuwer M.R., Merigan T.C. The role of interferon in the protective effect of a synthetic double-stranded polyribonucleotide against intranasal vesicular stomatitis virus challenge in mice. J Clin Invest. 1970;49:1565–1577. doi: 10.1172/JCI106374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Clercq E., Wells R.D., Grant R.C., Merigan T.C. Thermal activation of the antiviral activity of synthetic double-stranded polyribonucleotides. J Mol Biol. 1971;56:83–100. doi: 10.1016/0022-2836(71)90086-6. [DOI] [PubMed] [Google Scholar]
- 13.De Clercq E., Merigan T.C. An active interferon inducer obtained from Hemophilus influenzae type B. J Immunol. 1969;103:899–906. [PubMed] [Google Scholar]
- 14.De Clercq E., De Somer P. Antiviral activity of polyribocytidylic acid in cells primed with polyriboinosinic acid. Science. 1971;173:260–262. doi: 10.1126/science.173.3993.260. [DOI] [PubMed] [Google Scholar]
- 15.Temin HM, Mizutani S. Viral RNA-dependent DNA polymerase: RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature. 1970;226:1211–1213. doi: 10.1038/2261211a0. [DOI] [PubMed] [Google Scholar]
- 16.Baltimore D. Viral RNA-dependent DNA polymerase: RNA-dependent DNA polymerase in virions of RNA tumour viruses. Nature. 1970;226:1209–1211. doi: 10.1038/2261209a0. [DOI] [PubMed] [Google Scholar]
- 17.Carter W.A., De Clercq E. Viral infection and host defense. Science. 1974;186:1172–1178. doi: 10.1126/science.186.4170.1172. [DOI] [PubMed] [Google Scholar]
- 18.De Clercq E., Torrence P.F., Witkop B. Interferon induction by synthetic polynucleotides: importance of purine N-7 and strandwise rearrangement. Proc Natl Acad Sci U S A. 1974;71:182–186. doi: 10.1073/pnas.71.1.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Clercq E., Torrence P.F., De Somer P., Witkop B. Biological, biochemical and physicochemical evidence for the existence of the polyadenylic-polyuridylic-polyinosinic acid triplex. J Biol Chem. 1975;250:2521–2531. [PubMed] [Google Scholar]
- 20.De Clercq E. Suramin: a potent inhibitor of the reverse transcriptase of RNA tumor viruses. Cancer Lett. 1979;8:9–22. doi: 10.1016/0304-3835(79)90017-x. [DOI] [PubMed] [Google Scholar]
- 21.Field H.J., De Clercq E. Antiviral drugs––a short history of their discovery and development. Microbiol Today. 2004;31:58–61. [Google Scholar]
- 22.Elion G.B., Furman P.A., Fyfe J.A., de Miranda P., Beauchamp L., Schaeffer H.J. Selectivity of action of an antiherpetic agent, 9-(2-hydroxyethoxymethyl) guanine. Proc Natl Acad Sci U S A. 1977;74:5716–5720. doi: 10.1073/pnas.74.12.5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schaeffer H.J., Beauchamp L., de Miranda P., Elion G.B., Bauer D.J., Collins P. 9-(2-hydroxyethoxymethyl) guanine activity against viruses of the herpes group. Nature. 1978;272:583–585. doi: 10.1038/272583a0. [DOI] [PubMed] [Google Scholar]
- 24.De Clercq E., Descamps J., De Somer P., Holý A. (S)-9-(2, 3-Dihydroxypropyl) adenine: an aliphatic nucleoside analog with broad-spectrum antiviral activity. Science. 1978;200:563–565. doi: 10.1126/science.200.4341.563. [DOI] [PubMed] [Google Scholar]
- 25.Colla L., De Clercq E., Busson R., Vanderhaeghe H. Synthesis and antiviral activity of water-soluble esters of acyclovir [9-[(2-hy-droxyethoxy) methyl] guanine] J Med Chem. 1983;26:602–604. doi: 10.1021/jm00358a029. [DOI] [PubMed] [Google Scholar]
- 26.Maudgal P.C., De Clercq E., Descamps J., Missotten L. Topical treatment of experimental herpes simplex keratouveitis with 2′-O-glycylacyclovir. A water-soluble ester of acyclovir. Arch Ophthalmol. 1984;102:140–142. doi: 10.1001/archopht.1984.01040030118049. [DOI] [PubMed] [Google Scholar]
- 27.De Clercq E., Descamps J., De Somer P., Barr P.J., Jones A.S., Walker R.T. (E)-5-(2-Bromovinyl)-2′-deoxyuridine: a potent and selective anti-herpes agent. Proc Natl Acad Sci U S A. 1979;76:2947–2951. doi: 10.1073/pnas.76.6.2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Clercq E., Degreef H., Wildiers J., de Jonge G., Drochmans A., Descamps J. Oral (E)-5-(2-bromovinyl)-2′-deoxyuridine in severe herpes zoster. Br Med J. 1980;281:1178. doi: 10.1136/bmj.281.6249.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maudgal P.C., De Clercq E., Descamps J., Missotten L., De Somer P., Busson R. (E)-5-(2-Bromovinyl)-2′-deoxyuridine in the treatment of experimental herpes simplex keratitis. Antimicrob Agents Chemother. 1980;17:8–12. doi: 10.1128/aac.17.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maudgal P.C., De Clercq E., Descamps J., Missotten L., Wijnhoven J. Experimental stromal herpes simplex keratitis influence of treatment with topical bromovinyldeoxyuridine and trifluridine. Arch Ophthalmol. 1982;100:653–656. doi: 10.1001/archopht.1982.01030030655027. [DOI] [PubMed] [Google Scholar]
- 31.Maudgal P.C., Uyttebroeck W., De Clercq E., Missotten L. Oral and topical treatment of experimental herpes simplex iritis with bromovinyldeoxyuridine. Arch Ophthalmol. 1982;100:1337–1340. doi: 10.1001/archopht.1982.01030040315025. [DOI] [PubMed] [Google Scholar]
- 32.Maudgal P.C., De Clercq E. Evaluation of bromovinyldeoxyuridine-related compounds in the treatment of experimental herpes simplex keratitis. Arch Ophthalmol. 1985;103:1393–1397. doi: 10.1001/archopht.1985.01050090145051. [DOI] [PubMed] [Google Scholar]
- 33.Hamamoto Y., Nakashima H., Matsui T., Matsuda A., Ueda T., Yamamoto N. Inhibitory effect of 2′,3′-didehydro-2′,3′-dideoxynucleosides on infectivity, cytopathic effects, and replication of human immunodeficiency virus. Antimicrob Agents Chemother. 1987;31:907–910. doi: 10.1128/aac.31.6.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mitsuya H., Weinhold K.J., Furman P.A., St, Clair M.H., Lehrman S.N., Gallo R.C. 3′-Azido-3′-deoxythymidine (BW A509U): an antiviral agent that inhibits the infectivity and cytopathic effect of human T-lymphotropic virus type III/lymphadenopathy-associated virus in vitro. Proc Natl Acad Sci U S A. 1985;82:7096–7100. doi: 10.1073/pnas.82.20.7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mitsuya H., Broder S. Inhibition of the in vitro infectivity and cytopathic effect of human T-lymphotrophic virus type III/lymphadenopathy-associated virus (HTLV-III/LAV) by 2′,3′-dideoxynucleosides. Proc Natl Acad Sci U S A. 1986;83:1911–1915. doi: 10.1073/pnas.83.6.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baba M., Pauwels R., Herdewijn P., De Clercq E., Desmyter J., Vandeputte M. Both 2′,3′-dideoxythymidine and its 2′, 3′-unsaturated derivative (2′,3′-dideoxythymidinene) are potent and selective inhibitors of human immunodeficiency virus replication in vitro. Biochem Biophys Res Commun. 1987;142:128–134. doi: 10.1016/0006-291x(87)90460-8. [DOI] [PubMed] [Google Scholar]
- 37.Mitsuya H., Popovic M., Yarchoan R., Matsushita S., Gallo R.C., Broder S. Suramin protection of T cells in vitro against infectivity and cytopathic effect of HTLV-III. Science. 1984;226:172–174. doi: 10.1126/science.6091268. [DOI] [PubMed] [Google Scholar]
- 38.De Clercq E. Suramin in the treatment of AIDS: mechanism of action. Antiviral Res. 1987;7:1–10. doi: 10.1016/0166-3542(87)90034-9. [DOI] [PubMed] [Google Scholar]
- 39.Baba M., Tanaka H., De Clercq E., Pauwels R., Balzarini J., Schols D. Highly specific inhibition of human immunodeficiency virus type 1 by a novel 6-substituted acyclouridine derivative. Biochem Biophys Res Commun. 1989;165:1375–1381. doi: 10.1016/0006-291x(89)92756-3. [DOI] [PubMed] [Google Scholar]
- 40.Miyasaka T., Tanaka H., Baba M., Hayakawa H., Walker R.T., Balzarini J. A novel lead for specific anti-HIV-1 agents: 1-[(2-hydroxyethoxy) methyl]-6-(phenylthio) thymine. J Med Chem. 1989;32:2507–2509. doi: 10.1021/jm00132a002. [DOI] [PubMed] [Google Scholar]
- 41.Richman D.D. Dideoxynucleosides are less inhibitory in vitro against human immunodeficiency virus type 2 (HIV-2) than against HIV-1. Antimicrob Agents Chemother. 1987;31:1879–1881. doi: 10.1128/aac.31.12.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van de Werf F., Ludbrook P.A., Bergmann S.R., Tiefenbrunn A.J., Fox K.A., de Geest H. Coronary thrombolysis with tissue-type plasminogen activator in patients with evolving myocardial infarction. N Engl J Med. 1984;310:609–613. doi: 10.1056/NEJM198403083101001. [DOI] [PubMed] [Google Scholar]
- 43.Tsai C.-C., Follis K.E., Sabo A., Beck T.W., Grant R.F., Bischofberger N. Prevention of SIV infection in macaques by (R)-9-(2-phosphonylmethoxypropyl) adenine. Science. 1995;270:1197–1199. doi: 10.1126/science.270.5239.1197. [DOI] [PubMed] [Google Scholar]
- 44.Balzarini J., Holý A., Jindrich J., Naesens L., Snoeck R., Schols D. Differential antiherpesvirus and antiretrovirus effects of the (S) and (R) enantiomers of acyclic nucleoside phosphonates: potent and selective in vitro and in vivo antiretrovirus activities of (R)-9-(2-phosphonomethoxypropyl)-2, 6-diaminopurine. Antimicrob Agents Chemother. 1993;37:332–338. doi: 10.1128/aac.37.2.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Clercq E. ASPET Otto Krayer award lecture. Molecular targets for antiviral agents. J Pharmacol Exp Ther. 2001;297:1–10. [PubMed] [Google Scholar]
- 46.De Clercq E. Strategies in the design of antiviral drugs. Nat Rev Drug Discov. 2002;1:13–25. doi: 10.1038/nrd703. [DOI] [PubMed] [Google Scholar]
- 47.De Clercq E. The bicyclam AMD3100 story. Nat Rev Drug Discov. 2003;2:581–587. doi: 10.1038/nrd1134. [DOI] [PubMed] [Google Scholar]
- 48.De Clercq E. The AMD3100 story: the path to the discovery of a stem cell mobilizer (Mozobil) Biochem Pharmacol. 2009;77:1655–1664. doi: 10.1016/j.bcp.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 49.De Clercq E. Recent advances on the use of the CXCR4 antagonist plerixafor (AMD3100, Mozobil™) and potential of other CXCR4 antagonists as stem cell mobilizers. Pharmacol Ther. 2010;128:509–518. doi: 10.1016/j.pharmthera.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 50.De Clercq E. Antivirals and antiviral strategies. Nat Rev Microbiol. 2004;2:704–720. doi: 10.1038/nrmicro975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Clercq E., Holý A. Acyclic nucleoside phosphonates: a key class of antiviral drugs. Nat Rev Drug Discov. 2005;4:928–940. doi: 10.1038/nrd1877. [DOI] [PubMed] [Google Scholar]
- 52.De Clercq E. Antiviral agents active against influenza A viruses. Nat Rev Drug Discov. 2006;5:1015–1025. doi: 10.1038/nrd2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lagoja I.M., De Clercq E. Anti-influenza virus agents: synthesis and mode of action. Med Res Rev. 2008;28:1–38. doi: 10.1002/med.20096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.De Clercq E. The design of drugs for HIV and HCV. Nat Rev Drug Discov. 2007;6:1001–1018. doi: 10.1038/nrd2424. [DOI] [PubMed] [Google Scholar]
- 55.De Clercq E. The clinical potential of the acyclic (and cyclic) nucleoside phosphonates. The magic of the phosphonate bond. Biochem Pharmacol. 2011;82:99–109. doi: 10.1016/j.bcp.2011.03.027. [DOI] [PubMed] [Google Scholar]
- 56.De Clercq E. Where rilpivirine meets with tenofovir, the start of a new anti-HIV drug combination era. Biochem Pharmacol. 2012;84:241–248. doi: 10.1016/j.bcp.2012.03.024. [DOI] [PubMed] [Google Scholar]
- 57.De Clercq E. Antivirals: past, present and future. Biochem Pharmacol. 2013;85:727–744. doi: 10.1016/j.bcp.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 58.De Clercq E. Dancing with chemical formulae of antivirals: a personal account. Biochem Pharmacol. 2013;86:711–725. doi: 10.1016/j.bcp.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 59.De Clercq E. Dancing with chemical formulae of antivirals: a panoramic view (Part 2) Biochem Pharmacol. 2013;86:1397–1410. doi: 10.1016/j.bcp.2013.09.010. [DOI] [PubMed] [Google Scholar]
- 60.De Clercq E. Current race in the development of DAAs (direct-acting antivirals) against HCV. Biochem Pharmacol. 2014;89:441–452. doi: 10.1016/j.bcp.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 61.De Clercq E. Ebola virus (EBOV) infection: therapeutic strategies. Biochem Pharmacol. 2015;93:1–10. doi: 10.1016/j.bcp.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McGuigan C., Barucki H., Blewett S., Carangio A., Erichsen J.T., Andrei G. Highly potent and selective inhibition of varicella-zoster virus by bicyclic furopyrimidine nucleosides bearing an aryl side chain. J Med Chem. 2000;43:4993–4997. doi: 10.1021/jm000210m. [DOI] [PubMed] [Google Scholar]
- 63.De Clercq E., Yamamoto N., Pauwels R., Baba M., Schols D., Nakashima H. Potent and selective inhibition of human immunodeficiency virus (HIV)-1 and HIV-2 replication by a class of bicyclams interacting with a viral uncoating event. Proc Natl Acad Sci U S A. 1992;89:5286–5290. doi: 10.1073/pnas.89.12.5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.De Clercq E., Yamamoto N., Pauwels R., Balzarini J., Witvrouw M., de Vreese K. Highly potent and selective inhibition of human immunodeficiency virus by the bicyclam derivative JM3100. Antimicrob Agents Chemother. 1994;38:668–674. doi: 10.1128/aac.38.4.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McGuigan C., Yarnold C.J., Jones G., Velázquez S., Barucki H., Brancale A. Potent and selective inhibition of varicella-zoster virus (VZV) by nucleoside analogues with an unusual bicyclic base. J Med Chem. 1999;42:4479–4484. doi: 10.1021/jm990346o. [DOI] [PubMed] [Google Scholar]
- 66.Andrei G., Sienaert R., McGuigan C., De Clercq E., Balzarini J., Snoeck R. Susceptibilities of several clinical varicella-zoster virus (VZV) isolates and drug-resistant VZV strains to bicyclic furano pyrimidine nucleosides. Antimicrob Agents Chemother. 2005;49:1081–1086. doi: 10.1128/AAC.49.3.1081-1086.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McGuigan C., Pathirana R.N., Migliore M., Adak R., Luoni G., Jones A.T. Preclinical development of bicyclic nucleoside analogues as potent and selective inhibitors of varicella zoster virus. J Antimicrob Chemother. 2007;60:1316–1330. doi: 10.1093/jac/dkm376. [DOI] [PubMed] [Google Scholar]
- 68.Migliore M. FV-100: the most potent and selective anti-varicella zoster virus agent reported to date. Antivir Chem Chemother. 2010;20:107–115. doi: 10.3851/IMP1472. [DOI] [PubMed] [Google Scholar]
- 69.Pentikis H.S., Matson M., Atiee G., Boehlecke B., Hutchins J.T., Patti J.M. Pharmacokinetics and safety of FV-100, a novel oral anti-herpes zoster nucleoside analogue, administered in single and multiple doses to healthy young adult and elderly adult volunteers. Antimicrob Agents Chemother. 2011;55:2847–2854. doi: 10.1128/AAC.01446-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schols D., Esté J.A., Henson G., De Clercq E. Bicyclams, a class of potent anti-HIV agents, are targeted at the HIV coreceptor Fusin/CXCR-4. Antiviral Res. 1997;35:147–156. doi: 10.1016/s0166-3542(97)00025-9. [DOI] [PubMed] [Google Scholar]
- 71.Schols D., Struyf S., Van Damme J., Esté J.A., Henson G., De Clercq E. Inhibition of T-tropic HIV strains by selective antagonization of the chemokine receptor CXCR4. J Exp Med. 1997;186:1383–1388. doi: 10.1084/jem.186.8.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hendrix C.W., Flexner C., MacFarland R.T., Giandomenico C., Fuchs E.J., Redpath E. Pharmacokinetics and safety of AMD-3100, a novel antagonist of the CXCR-4 chemokine receptor, in human volunteers. Antimicrob Agents Chemother. 2000;44:1667–1673. doi: 10.1128/aac.44.6.1667-1673.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.De Clercq E., Holý A., Rosenberg I., Sakuma T., Balzarini J., Maudgal P.C. A novel selective broad-spectrum anti-DNA virus agent. Nature. 1986;323:464–467. doi: 10.1038/323464a0. [DOI] [PubMed] [Google Scholar]
- 74.Janssen P.A., Lewi P.J., Arnold E., Daeyaert F., de Jonge M., Heeres J. In search of a novel anti-HIV drug: multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1E)-2-cyanoethenyl]-2, 6-dimethylphenyl]amino]-2-pyrimidinyl]amino]-benzonitrile (R278474, rilpivirine) J Med Chem. 2005;48:1901–1909. doi: 10.1021/jm040840e. [DOI] [PubMed] [Google Scholar]
- 75.Robbins B.L., Srinivas R.V., Kim C., Bischofberger N., Fridland A. Anti-human immunodeficiency virus activity and cellular metabolism of a potential prodrug of the acyclic nucleoside phosphonate 9-R-(2-phosphonomethoxypropyl)adenine (PMPA), bis(isopropyloxymethylcarbonyl)-PMPA. Antimicrob Agents Chemother. 1998;42:612–617. doi: 10.1128/aac.42.3.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Naesens L., Bischofberger N., Augustijns P., Annaert P., van den Mooter G., Arimilli M.N. Antiretroviral efficacy and pharmacokinetics of oral bis(isopropyloxycarbonyloxymethyl)-9-(2-phosphonylmethoxypropyl)adenine in mice. Antimicrob Agents Chemother. 1998;42:1568–1573. doi: 10.1128/aac.42.7.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Eisenberg E.J., He G.X., Lee W.A. Metabolism of GS-7340, a novel phenyl monophosphoramidate intracellular prodrug of PMPA, in blood. Nucleosides Nucleotides Nucleic Acids. 2001;20:1091–1098. doi: 10.1081/NCN-100002496. [DOI] [PubMed] [Google Scholar]
- 78.Chapman H., Kernan M., Rohloff J., Sparacino M., Terhorst T. Purification of PMPA amidate prodrugs by SMB chromatography and X-ray crystallography of the diastereomerically pure GS-7340. Nucleosides Nucleotides Nucleic Acids. 2001;20:1085–1090. doi: 10.1081/NCN-100002495. [DOI] [PubMed] [Google Scholar]
- 79.Lee W.A., He G.X., Eisenberg E., Cihlar T., Swaminathan S., Mulato A. Selective intracellular activation of a novel prodrug of the human immunodeficiency virus reverse transcriptase inhibitor tenofovir leads to preferential distribution and accumulation in lymphatic tissue. Antimicrob Agents Chemother. 2005;49:1898–1906. doi: 10.1128/AAC.49.5.1898-1906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ruane P, DeJesus E, Berger D, Markowitz M, Bredeek F, Callebaut C, et al. GS-7340 25 mg and 40 mg demonstrate superior efficacy to tenofovir 300 mg in a 10-day monotherapy study of HIV-1+ patients. In: 19th conference on retroviruses and opportunistic infections, 2012 March 5–8. Seattle, WA, USA; 2012. p. 103.
- 81.Babusis D., Phan T.K., Lee W.A., Watkins W.J., Ray A.S. Mechanism for effective lymphoid cell and tissue loading following oral administration of nucleotide prodrug GS-7340. Mol Pharm. 2013;10:459–466. doi: 10.1021/mp3002045. [DOI] [PubMed] [Google Scholar]
- 82.Holý A. Syntheses of enantiomeric N-(3-hydroxy-2-phosphonomethoxypropyl) derivatives of purine and pyrimidine bases. Collect Czech Chem Commun. 1993;58:649–674. [Google Scholar]
- 83.De Clercq E., Sakuma T., Baba M., Pauwels R., Balzarini J., Rosenberg I. Antiviral activity of phosphonylmethoxyalkyl derivatives of purine and pyrimidines. Antiviral Res. 1987;8:261–272. doi: 10.1016/s0166-3542(87)80004-9. [DOI] [PubMed] [Google Scholar]
- 84.Holý A., Votruba I., Masojídková M., Andrei G., Snoeck R., Naesens L. 6-[2-(Phosphonomethoxy)alkoxy]pyrimidines with antiviral activity. J Med Chem. 2002;45:1918–1929. doi: 10.1021/jm011095y. [DOI] [PubMed] [Google Scholar]
- 85.De Clercq E. The acyclic nucleoside phosphonates (ANPs): Antonín Holý's legacy. Med Res Rev. 2013;33:1278–1303. doi: 10.1002/med.21283. [DOI] [PubMed] [Google Scholar]
- 86.Krečmerová M., Holý A., Pískala A., Masojídková M., Andrei G., Naesens L. Antiviral activity of triazine analogues of 1-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]cytosine (cidofovir) and related compounds. J Med Chem. 2007;50:1069–1077. doi: 10.1021/jm061281+. [DOI] [PubMed] [Google Scholar]
- 87.Krečmerová M., Holý A., Pohl R., Masojídková M., Andrei G., Naesens L. Ester prodrugs of cyclic 1-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]-5-azacytosine: synthesis and antiviral activity. J Med Chem. 2007;50:5765–5772. doi: 10.1021/jm0707166. [DOI] [PubMed] [Google Scholar]
- 88.De Clercq E. Sofosbuvir in the current context of hepatitis C treatment. J Symptoms Signs. 2014;3:126–131. [Google Scholar]
Commentary

Libor Grubhoffer, Ph.D., Professor and Rector (Head), University of South Bohemia, Ceske Budejovice, Czech Republic.
First I ever met the name of Dr. Erik De Clercq was as a graduate student of virology long time ago. Since 70th of the last century he has been systematically working on research of antivirals in the Rega Institute for Biomedical Research at Catholic University in Louvain, Belgium. He introduced modern methods and approaches of testing as well as investigating of biochemical mechanisms of newly synthesized compounds with antiviral activity, especially those they could block genome replication of a broad array of animal/human viruses. As a former student and later on successor of Prof. P. De Somer has laid the foundations of rational design of drugs with antiviral activities. Despite of that Erik De Clercq does believe in serendipity which had played such important role in his enormously successful doing in antiviral research. One of the first result of such serendipity was when he proved suramin as an inhibitor of reverse transcriptase. It stimulated meaningfully his interest in reverse transcriptase, and thus when HIV as a causative agent of AIDS appeared in beginning of 80th of last century Erik De Clercq was experienced enough to make a milestone in research of anti-HIV compounds. The third great serendipity in the scientific career the friendship with Dr. Antonín Holý, a Czech organic chemist who linked his whole scientific career, with the Institute of Organic Chemistry and Biochemistry in Prague, and with the synthesis of nucleoside phosphonates as nucleic acid antimetabolites. Working together for nearly 30 years they achieved such great contribution for mankind eliminating or reducing fear from some of viral infectious diseases.

Fener Chen, Ph.D., Professor, Department of Chemistry, Fudan University, Shanghai, China.
Erik De Clercq was born in Belgium (1941). He received his Ph.D. from the Katholieke Universiteit Leuven in 1972. He served as the President of the Rega Foundation, and is a member of the Belgian (Flemish) Royal Academy of Medicine and fellow of the American Association for the Advancement of Science. He is an active Emeritus Professor of the Katholieke Universiteit Leuven (KU. Leuven), Belgium. He is honorary doctor of the Universities of Ghent, Belgium, Athens, Greece, Ferrara, Italy, Jinan (Shandong), China, Charles (Prague), Czech Republic, Jihoceska, Czech Republic, Tours, France and Hull,UK. For his pioneering efforts in antiviral research, in 1996 he received the Aventis award from the American Society for Microbiology, and in 2000 the Maisin Prize for Biomedical Sciences from the Belgian National Science Foundation. In 2008 he was elected Inventor of the Year by the European Union. Jointly with Dr. Anthony Fauci, he received the Dr. Paul Janssen Award for Biomedical Research in 2010. His scientific interests are in the antiviral chemotherapy field, and, in particular, the development of new antiviral agents for various viral infections, including herpes simplex virus (HSV), varicella-zoster virus (VZV), cytomegalovirus (CMV), human immunodeficiency virus (HIV), hepatitis B virus (HBV), human papilloma virus (HPV), and hepatitis C virus (HCV). He has (co)-discovered a number of antiviral drugs, currently used in the treatment of HSV infections (valaciclovir, Valtrex, Zelitrex), VZV infections (brivudin, Zostex, Brivirac, Zerpex), CMV infections (cidofovir, Vistide), HBV infections (adefovir dipivoxil, Hepsera), and HIV infections (AIDS) (tenofovir disoproxil fumarate, marketed as Viread, and, in combination with emtricitabine, as Truvada, and, in combination with both emtricitabine and efavirenz, as Atripla). Viread has also been approved for the treatment of HBV infections (chronic hepatitis B).

Denis De Keukeleire, Honorary Full Professor, Ghent University, Belgium.
At our first meeting years ago on a yearly bike outing with friendly research teams, Prof. De Clercq fascinated me by his sporting drive, as I had already learnt that he was a most outstanding medico-scientist not only in my country Belgium, but really on a global scale. Never before, I had met a person gifted in so many ways: from sharing numerous friends at his side taking part in social activities and being a reputed student educator, all the way to managing a top-quality laboratory that succeeded in combining fundamental science with real applications to the benefit of countless humans (“Chemistry at the Service of Medicine”). Such comprehensive accomplishments remain the priority of only very few! Joining forces with most prominent colleagues in academia and with visionary industry captains, Prof. De Clercq convincingly proved how chemistry, biology, and medicine could ultimately foster successful antiviral drug development. My greatest admiration to Prof. De Clercq concentrates on his virtues of dedication, creativity, perseverance, honesty, enthusiasm, and, finally, ambition to be the best. I am exceedingly proud to have been privileged to introduce Prof. De Clercq for a honorary doctorate in pharmacy at Ghent University, the first honor preceding an impressive list of recognitions and awards.