

Abstract
Earlier we reported the discovery and design of NBD-556 and their analogs which demonstrated their potential as HIV-1 entry inhibitors. However, progress in developing these inhibitors has been stymied by their CD4-agonist properties, an unfavorable trait for use as drug. Here, we demonstrate the successful conversion of a full CD4-agonist (NBD-556) through a partial CD4-agonist (NBD-09027), to a full CD4-antagonist (NBD-11021) by structure-based modification of the critical oxalamide midregion, previously thought to be intolerant of modification. NBD-11021 showed unprecedented neutralization breath for this class of inhibitors, with pan-neutralization against a panel of 56 Env-pseudotyped HIV-1 representing diverse subtypes of clinical isolates (IC50 as low as 270 nM). The cocrystal structure of NBD-11021 complexed to a monomeric HIV-1 gp120 core revealed its detail binding characteristics. The study is expected to provide a framework for further development of NBD series as HIV-1 entry inhibitors for clinical application against AIDS.
Graphical abstract

INTRODUCTION
Entry of human immunodeficiency virus type 1 (HIV-1) to host cells consists of highly orchestrated events and is essential for maintaining the virus life-cycle.1 The HIV-1 entry process starts when its surface envelope glycoprotein gp120 binds to the host cell primary receptor CD4.2–4 CD4 binding triggers conformational changes in gp120, which facilitate its binding to the host cell co-receptor (secondary) CCR5 or CXCR4.5–7 Co-receptor binding initiates conformational changes in gp41, which trigger the insertion of its fusion protein and formation of a stable hexahelical bundle, which promotes fusion of the virus membrane with the cell membrane and virus cell entry. Each of these steps of the entry pathway has been recognized as targets for developing drugs for prevention and therapy of this deadly disease.8–11 Approval of two drugs by the FDA that target HIV-1 entry has validated entry inhibition to host cells as an effective strategy for developing drugs. One of the clinically applied drugs, maraviroc (Selzentry) works against CCR5, and the other drug enfuvirtide (Fuzeon) targets the HIV-1 envelope glycoprotein gp41. There are no licensed drugs currently available that target HIV-1 gp120.
In the quest to discover effective drugs that target gp120, we identified two inhibitors, NBD-556 and NBD-557, in 2005 by targeted screening of small molecules from commercial libraries.12 Subsequently, the report that these compounds, despite their small molecular weights (337 and 382 Da), mimic CD4 remarkably well13 sparked interest in multiple research groups to target this site to develop NBD-556 related inhibitors.13–26 In 2011, we reported the structure of NBD-556 bound to HIV-1 gp120 by X-ray crystallography.27 Several other crystal structures of NBD-556 analogs bound to HIV-1 gp120 have also been reported.15,16,28 These structures reveal that NBD-556 and its analogs bind to a cavity (termed the Phe43 cavity, because of the peripheral contact of the cavity by the side chain phenyl group of residue 43 of CD4); however, the aromatic chlorophenyl group of NBD-556 and its analogs penetrates deep into the Phe43 cavity. The structures confirm that NBD-556 does not retain the critical H-bond/salt bridge interaction with Asp368gp120 as was observed with Arg59CD4. Unfortunately, NBD-556 and its analogs behave as CD4-agonists and enhance HIV-1 infectivity in CD4−CCR5+ cells. We and others attempted to modify regions I, II, and III of NBD-556 (Figure 1a) and concluded that region I could be minimally modified and that region III is amenable for modification. However, it was generally concluded that modifications to region II were detrimental to binding and antiviral activity, because the oxalamide moiety contributes to the binding by forming two hydrogen bonds with gp120 residues. We therefore focused on making modifications to region I and especially to region III. To enhance binding affinity and antiviral potency, we reasoned that it might be essential to gain the critical H-bond/salt bridge interaction of the basic moieties in region III with the conserved Asp368gp120. Toward this goal, we explored different scaffolds with basic groups. We synthesized a series of compounds with a piperidine and a thiazole ring attached with a flexible linker, tested those molecules in a large set of Env-pseudotyped HIV-1, and observed measurable enhancement of antiviral activity.19 However, both functional and biophysical assays confirmed that this class of compounds retains agonist properties similar to NBD-556. Interestingly, one of these new compounds, NBD-09027, showed reduced agonist properties compared with NBD-556 both in functional and biophysical studies.19 These observations motivated us to determine the X-ray structure of NBD-09027 with HIV-1 gp120 in order to investigate the interactions.19 The structure revealed that although the 4-chlorophenyl oxalamide group was superimposable with the NBD-556 in the X-ray structure, the basic “nitrogen” of the piperidine ring of the new scaffold was within 4.4 Å of the Asp368gp120, though not close enough to form a H-bond or salt bridge indicating that additional modifications would be needed to obtain this critical interaction.
Figure 1.

Structures of NBD series compounds and their CD4-agonist/antagonist characterization by functional (cell-based assay) and biophysical (competitive-SPR) experiments. (a) The chemical structures of NBD series compounds indicating regions I, II, and III and the stereoisomers of NBD-11021. (b) Dose-dependent curve of CD4-positive Cf2Th/CD4–CCR5 cells infected with CD4-dependent HIV-1ADA (solid line) and CD4-negative Cf2Th–CCR5 cells infected with CD4-independent HIV-1ADAN197S (dashed line) in the presence of different concentrations of NBD-11021, NBD-556, and NBD-09027. Three independent experiments were performed in triplicate, and the graph is representative of one experiment; the values represent the mean ± standard deviation. (c) CD4-negative Cf2Th–CCR5 cells were infected with CD4-dependent HIV-1ADA in the presence of increasing concentrations of NBD-11021, NBD-556, NBD-09027, and BMS-378806. Relative virus infectivity specifies the amount of infection detected in the presence of the compounds relative to the infection detected in the absence of the compounds. Three independent experiments were performed in triplicate, and the graph is representative of one experiment; the values represent the mean ± standard deviation. (d) Impact of NBD series compounds on the binding between mAb 17b and YU2 core gp120 protein. The YU2 core gp120 was passed over the anti-human IgG-captured 17b in the absence or presence of saturated NBD series compounds or sCD4 (control). The injection time was 2 min, and the disassociation time was 5 min. The graph is representative of two independent experiments. (e, f) Competitive SPR to probe whether NBD-11021 mimics the binding site of sCD4 and VRC-PG04. The protein alone or in the presence of varied concentrations of NBD-11021 was passed over YU2 core-immobilized surface on the CM5 chip. The association time and the dissociation time were 2 and 5 min, respectively.
Here we report a systematic structure-based approach to modifying region II by preserving a key H-bond interaction of one of the most active agonists, NBD-09027, with gp120 but to alter positioning of the piperidine moiety used in NBD-09027 to form a second key H-bond with Asp368gp120. This report demonstrates that modifications in region II are feasible and may be critical for enhancing antiviral activity of the NBD series entry inhibitors. Most notably, we successfully convert a full CD-agonist (NBD-556) to a full CD4-antagonist (NBD-11021) and achieve the desired broad spectrum anti-HIV-1 activity, which has not been previously accomplished for this class of entry inhibitors.
RESULTS
Rational Design of NBD-11021
It has been recently reported that VRC01 and NIH45-46, two highly potent broadly neutralizing antibodies (bNAbs), induce conformational changes of monomeric gp120 in the bridging sheet similar to that induced by CD4; thereby showing enhanced binding to mAb 17b. Therefore, VRC01 and NIH45-46 induce exposure of the co-receptor binding site, suggesting that they are CD4 agonists.29 In contrast, VRC03 and VRC-PG04, two other highly potent bNAbs, induce monomeric gp120 conformation, especially within residues 428–431gp120 in strands β20/21, which is distinctly different, thus showing no enhancement in binding to mAb 17b. VRC03 and VRC-PG04 thus do not induce exposure of the co-receptor binding site in gp120, suggesting that they are CD4 antagonists, not agonists. In addition, they also observed that a critical residue, Met434 on β21 in gp120 in both VRC-PG04- and VRC03-bound structures, adopts a conformation that clashes with CDRH2 of mAb 17b, whereas this residue adopts a nonclashing conformation when bound to VRC01 and NIH45-46, explaining the differences in recognition by the mAb 17b.29 Interestingly, irrespective of their agonist or antagonist characteristics, they all have a positively charged arginine (Arg79) interacting with Asp368gp120. Mutation of Asp368gp120 abrogates the neutralizing capacity of these bNAbs.30 Therefore, this interaction is essential for binding of these highly potent bNAbs to gp120 with high affinity, as well as to elicit potent neutralization efficiency. We reasoned that in order to first enhance the potency of these new series of NBD inhibitors we also needed to introduce this interaction.
To enhance antiviral activity and to overcome the agonist properties of NBD-09027, we decided to modify region II by incorporating a pyrrole ring replacing part of the oxalamide ring toward the phenyl group. We kept the remaining structure identical. We reasoned that introduction of the pyrrole ring would help to preserve the H-bond interaction of the NH of pyrrole with Asn425, but its rigid geometry might alter the conformation of the scaffold in region III. This conformational change may facilitate movement of the piperidine ring toward Asp368gp120 for the desired H-bond/salt bridge interaction.
Synthesis of NBD-11021
As illustrated in Scheme-1, the target compound was envisioned to arise from coupling between pyrrole-2-carboxylic acid 2′ and amine 1′, which in turn could be prepared by the addition of the thiazolyl anion to a suitably protected aldehyde or imine 3′.
Scheme 1.

Retrosynthetic Analysis of NBD-11021 (15)
The thiazole core was prepared by the reaction between thioformamide and ethyl 2-chloroacetoacetate (Scheme-2). The reduction of the ester group with LiAlH4 and the subsequent protection of the alcohol group with TBSCl furnished the first of the three key building blocks 7. The synthesis of the second building block 11 commenced with the oxime preparation from 4′-chloroacetophenone followed by the Trofimov reaction.31 The Friedel–Crafts acylation of the resulting 2-arylpyrrole 9 with trifluoroacetic acid anhydride followed by the haloform reaction provided the required acid 11 in 78% yield over two steps.
Scheme 2.

Outline of the Synthesis of NBD-11021 (Diastereomeric Mixture), NBD-11021A (15a), and NBD-11021B (15b)
The synthesis of the third part of the target compound and an assembly of the fragments began with the protection of the pipecolic acid with allyl chloroformate. The formation of the Weinreb amide 2, followed by the reduction with LiAlH4, provided the aldehyde 3. The sulfinimide 4 was prepared from the racemic tert-butanesulfinamide32 and racemic aldehyde 3 as a mixture of stereoisomers. The addition of thiazolyl lithium derived from 7 to the imine 4 furnished a chromatographically separable mixture of isomers in a combined 85% yield. The first fraction contained one pure racemic diastereomer 12a (according to LCMS) and the second fraction contained presumably a mixture of two diastereomers (differing in the configuration of the sulfur atom) assigned together as 12b (Scheme-2). The isolated isomers 12a and 12b were used in the subsequent steps separately.
The synthetic intermediates derived from 12a have been labeled with an “a” letter in their numbering system (13a, 14a, 15a = NBD-11021A). The synthetic intermediates derived from 12b have been labeled with a “b” letter in the numbering system (13b, 14b, 15b = NBD-11021B).
After sulfinyl and TBS groups were cleaved with anhydrous methanolic HCl solution two pure racemic diastereomers 13a and 13b were produced separately from 12a and 12b. No stereochemical assignment was performed at this point, and both amines were transformed independently into the target compounds. Only inseparable mixtures of isomers were observed in the addition reaction when Boc or Cbz protecting groups were used. It is also important to note that separation could not be achieved in the next steps because of the low differences in mobility of the compounds on silica gel. Finally, amines 13a and 13b were coupled with acid 11, and the alloc group was removed according to the literature procedure33 to afford the title compounds 15a (NBD-11021A) and 15b (NBD-11021B) in racemic form.
The final compounds did show very similar 1H and 13C NMR spectra. To prove once again that these compounds are in fact isomers, we measured 1H and 13C NMR spectra from a mixture of 15a (NBD-11021A) and 15b (NBD-11021B). The spectral data showed a double set of signals clearly indicating that 15a (NBD-11021A) and 15b (NBD-11021B) are in fact stereoisomers.
Separation of NBD-11021 Diastereomers and Assignment of Absolute Configuration
Initial synthesis of NBD-11021 yielded a diastereomeric mixture (four isomers), which we designated “NBD-11021” and used in different assays. However, we were able to synthesize NDB-11021 as two separate isomeric mixtures (comprising of a mixture of two enantiomers of each single racemate) and designated these as NBD-11021A and NBD-11021B. We subsequently used chiral separation fluid chromatography (SFC) to separate these mixtures into individual enantiomers, which we named NBD-11021A1, NBD-11021A2, NBD-11021B1, and NBD-11021B2.
Vibrational circular dichroism (VCD) was used to delineate the absolute configuration of these four isomers. They were assigned (Figure 1a) as NBD-11021A1 (16S,25R), NBD-11021A2 (16R,25S), NBD-11021B1 (16S,25S), and NBD-11021B2 (16R,25R).
NBD-11021 Exhibits Anti-HIV-1 Activity in Multicycle and Single-Cycle Assay
Following purification of the two isomers A and B of NBD-11021 and subsequent separation of the fractions to NBD-11021A1, NBD-11021A2, NBD-11021B1, and NBD-11021B2, we evaluated the anti-HIV-1 activity and the cytotoxicity of these compounds in different cell lines using both multicycle and single cycle assays (Table 1) to select the best isomers for subsequent studies in larger panel of ENV pseudotyped reference viruses. NBD-11021A2 and NBD-11021B2 showed improvement in antiviral activity compared with the diastereomeric mixture of NBD-11021 exhibiting an IC50 as low as 0.8 μM in MT-2 cells against HIV-1IIIB. NBD-11021B1 showed a significant loss in antiviral activity with respect to the initial diastereomeric NBD-11021 and the isomers NBD-11021A and NBD-11021B in multicycle assays.
Table 1.
Anti-HIV-1 Activity (μM) and Cytotoxicity (μM) of NBD-11021 and Its Isomersa
| compound | cell line virus |
MT-2 HIV-1IIIB |
TZM-bl HIV-1HXB2 |
U87-CD4-CXCR4 HIV-1HXB2 |
U87-CD4-CCR5 HIV-1ADA |
|---|---|---|---|---|---|
| NBD-11021 (A+B) | CC50 | ~28 | ~28 | ~25 | ~30 |
| IC50 | 1.5 ± 0.06 | 2.5 ± 0.2 | 2.4 ± 0.1 | 1.7 ± 0.2 | |
| NBD-11021A (A1+A2) | CC50 | ~30 | ~30 | ~30 | ~30 |
| IC50 | 1.3 ± 0.2 | 2.8 ± 0.2 | 3 ± 0.4 | 2.5 ± 0.2* | |
| NBD-11021A1 | CC50 | ~30 | ~30 | ~30 | ~30 |
| IC50 | 1.7 ± 0.02 | 3.2 ± 0.3* | 2.4 ± 0.3 | 1.9 ± 0.1 | |
| NBD-11021A2 | CC50 | ~25 | ~24 | ~30 | ~30 |
| IC50 | 0.85 ± 0.06** | 2.2 ± 0.2 | 1.7 ± 0.1 | 1.3 ± 0.2 | |
| NBD-11021B (B1+B2) | CC50 | ~25 | ~28 | ~28 | ~24 |
| IC50 | 1.8 ± 0.05* | 2.4 ± 0.1 | 3.3 ± 0.3* | 3.1 ± 0.2** | |
| NBD-11021B1 | CC50 | ~20 | ~28 | ~28 | ~24 |
| IC50 | 7.9 ± 0.6** | 3 ± 0.1* | 4.7 ± 0.7** | 4.4 ± 0.2** | |
| NBD-11021B2 | CC50 | ~20 | ~24 | ~20 | ~24 |
| IC50 | 0.8 ± 0.07** | 0.99 ± 0.13** | 1.6 ± 0.15 | 1.6 ± 0.2 | |
| NBD-556 | CC50 | > 280 | ~60 | ~60 | ~60 |
| IC50 | 6.5 ± 0.1 | 6.9 ± 0.9 | 9.5 ± 0.6 | 7.4 ± 0.8 | |
| NBD-09027 | CC50 | >108 | 23.7 ± 1.1 | ~87 | >45 |
| IC50 | 4.7 ± 0.6* | 4.7 ± 1.1* | 9.1 ± 0.7 | 8.6 ± 0.7 |
Values, representing the mean ± standard deviation, were obtained from three independent experiments. The data was analyzed with the one-way ANOVA, implementing Dunnett’s test for adjustments of multiple comparisons, with a significance level set at 0.05.
P < 0.05,
P < 0.0001.
We detected a similar level of cytotoxicity in all the isomers with respect to the original diastereomeric mixture of NBD-11021 with minimal variations considering different sensitivity of the diverse cell lines we tested. While the antiviral neutralization appeared specific, we note that neutralization of A-MLV was observed (see below).
NBD-11021 Shows Similar Antiviral Activity against CXCR4- and CCR5-tropic HIV-1
Interaction of gp120 with the cellular receptor CD4 and co-receptors CCR5 or CXCR4 results in the formation of fusion pores and release of the HIV genome into target cells.34,35 CCR5-using viruses are responsible for both interindividual transmission and sustaining pandemics, whereas CXCR4-using viruses, usually dual-tropic R5X4, emerge in about 50% of individuals in the late, immunologically suppressed stage of the disease.36 Therefore, it was pertinent to verify whether NBD-11021 shows activity against both CCR5-tropic and CXCR4-tropic HIV viruses. To this end, U87-CD4-CCR5 and U87-CD4-CXCR4 cells were infected with pseudoviruses HIV-1ADA (CCR5-tropic) and HIV-1HXB2 (CXCR4-tropic) and treated with varying doses of NBD-11021 and its isomers. Our results indicate that this compound inhibited both CCR5- and CXCR4-tropic HIV-1 with similar efficiency with an IC50 of ~1.7–2.4 μM (Table 1), which was about 4–5-fold improvement in activity with respect to NBD-556 and NBD-09027.
NBD-11021 Blocks gp120–CD4 Interaction
Previously we reported that several NBD-compounds, such as NBD-556, NBD-557, NBD-09027, and NBD-10007, bind to the Phe43 cavity and inhibit interaction between CD4 and gp120.12,20,27,28 Since, in this study, we made a major modification in region II, we investigated how this change might affect the interaction with both CD4-positive and CD4-negative cells. To accomplish this, we evaluated the effect of NBD-11021 on the infection of a CD4-dependent virus, HIV-1ADA, in Cf2Th/CD4-CCR5 target cells that express CD4 and CCR5 and a CD4-independent mutant, HIV-1ADAN197S, in Cf2Th-CCR5 target cells that express CCR5 but not CD4. As was observed with NBD-556 and NBD-09027,12,17 NBD-11021 inhibited the CD4-dependent virus in a dose-dependent manner with an IC50 of 2.4 ± 0.2 μM (Figure 1b) suggesting that this compound inhibits interaction between gp120 and CD4. However, while NBD-556 and NBD-09027 did not inhibit infection of the target cells by the CD4-independent virus HIV-1ADAN197S, NBD-11021 inhibited the CD4-independent virus with similar activity (IC50 = 2.1 ± 0.15 μM). A similar inhibition was reported for entry inhibitor BMS-378806.37 Moreover, we and others have recently shown that NBD-556 can also act as CD4-agonist, promoting CCR5 binding and enhancing HIV-1 entry into CD4-negative cells expressing CCR5,13,19 while the enhancement of infection by NBD-09027 was about 2-fold lower.19 To further evaluate the activity of NBD-11021, we infected CD4-negative Cf2Th-CCR5 cells with recombinant CD4-dependent HIV-1ADA in the presence of various concentrations of NBD-11021 (Figure 1c). NBD-556, NBD-09027, and BMS-378806 were used as controls. As reported previously,17,19 NBD-556 enhanced HIV-1 entry into CD4-negative cells and NBD-09027 showed moderate enhancement with respect to NBD-556; however, NBD-11021, similar to BMS-378806, did not enhance HIV-1 infectivity in CD4−CCR5+ cells suggesting that the structural modification of NBD-09027 successfully converted it to a CD4-antagonist.
To verify this finding biophysically, we used surface plasmon resonance (SPR) to determine whether NBD-11021 induced the antibody 17b-recognized conformation in gp120. sCD4, NBD-556, and NBD-09027 were used as controls. In the presence of a 15-fold excess of sCD4, the YU2 corem gp12027 demonstrated enhanced binding to antibody 17b compared with the YU2 corem gp120 alone. Among the NBD compounds tested, NBD-556 showed the highest enhancement of antibody 17b binding to YU2 corem gp120, whereas NBD-09027 showed much reduced enhancement. Remarkably, no enhancement in binding of mAb 17b to YU2 corem gp120 was observed in the presence of NBD-11021 (Figure 1d) suggesting that NBD-11021 functions as an antagonist.
NBD-11021 Binds to gp120
We used competitive SPR to validate and to characterize the target of NBD-11021 on gp120. As shown in Figure 1e, NBD-11021 competes with CD4 to bind to gp120 in a dose-dependent manner. The recent structure of antibody VRC-PG04 (also called antibody PGV04) with a trimeric form of HIV-1 Env (BG505 SOSIP.664)38 demonstrated that this antibody recognizes the prefusion mature closed conformation of HIV-1 Env. We used competitive-SPR to verify whether NBD-11021 shared the same binding site as VRC-PG04. The data in Figure 1f suggest that at higher ratios of NBD-11021 versus VRC-PG04, NBD-11021 effectively competed with VRC-PG04 to inhibit its binding to gp120 in a dose-dependent manner. The data suggest that the NBD-11021 binding site on gp120 overlapped with that of VRC-PG04, which binds to the Phe43 cavity in gp120.
NBD-11021 Shows Broad Spectrum Inhibition against a Large Diverse Panel of ENV Pseudotyped Reference Viruses
To further evaluate the breadth of anti-HIV-1 activity, we tested NBD-11021 against 56 HIV-1 Env-pseudotyped reference viruses of diverse clades including primary and transmitted/founder HIV-1 isolates.39 These ENV-pseudoviruses were derived from patient serum and exhibit a neutralization phenotype that is typical of most primary isolates. We also used the two purified isomers NBD-11021A2 and NBD-11021B2, which showed the best anti-HIV-1 activity in our preliminary assays, and we compared their activity with compounds NBD-556 and NBD-09027. Six representative dose-dependent inhibition plots of the neutralization assay of selected pseudoviruses (#11314, #11316, #11911, #11526, NIH #11888, and KNH1144) are shown in Figure S1. The results in Table 2 indicate that NBD-11021, even as mixture of isomers, showed improved antiviral activity against all viral isolates tested including subtype C with an overall range of activity of 0.6–5.3 μM of which 82% of the viruses showed sensitivity at ≤3 μM and 39% at ≤2 μM. By contrast, NBD-556 showed relatively much lower antiviral activity against most of the isolates tested with an overall range in activity of 1.9–27.8 μM of which only 7% of the viruses showed sensitivity at ≤3 μM and only one at ≤2 μM. Additionally, the two most active isomers NBD-11021A2 and NBD-11021B2 separated from the isomers A and B, respectively, exhibited improved activity over the diastereomeric mixtures of NBD-11021 (mean IC50 = 2.38 μM; Figure S2). For example, NBD-11021A2 isomer had an IC50 in the range of 0.32–3.7 μM (mean IC50 = 1.71 μM) with 68% of the viruses showing sensitivity ≤2 μM. Similarly, NBD-11021B2 isomer showed antiviral activity in the range of 0.27–2.7 μM (mean IC50 = 1.57 μM) with 71% of the viruses showing sensitivity ≤2 μM indicating that the separation of isomers resulted in improved anti-HIV activity. Surprisingly, NBD-11021 (isomer mixtures and pure isomers) also showed inhibition against HIV-1 pseudotyped with A-MLV envelope glycoprotein. Recently, A-MLV was shown to enter cells by macropinocytosis.40 HIV-1 entry has also been shown to occur by endocytic pathways41,42 including macropinocytosis in macrophages.43 By contrast, NBD-11021 and its two isomers showed poor activity against HIV-1 pseudotyped with VSV-G envelope. Furthermore, NBD-11021 had also poor activity against SHIVSF162P3, SIVmac186, and HIV-2 isolates (Table S2).
Table 2.
Neutralization Activity of NBD Compounds against a Panel of HIV-1 Env Pseudoviruses
| IC50(μM) ± S.D.a | |||||||
|---|---|---|---|---|---|---|---|
| Subtype | NIH # | ENVs | NBD-556 | NBD-09027 | NBD-11021 | NBD- 11021A2 |
NBD-11021B2 |
| A | 11887 | Q259env.w6 | 6.9±0.9c | 2±0.2c | 2.8±0.1 | 1.6±0.1 | 1.3±0.4 |
| 11888 | QB726.70M.ENV.C4 | 20.2±6 | 6.3±1 | 2.6±0.1 | 1.3±0.3 | 1.5±0.6 | |
| 11890 | QF495.23M.ENV.A1 | 13.6±2.3 | 2.3±0.3 | 1.8±0.5 | 1.3±0.4 | 0.86±0.4 | |
| 11889 | QB726.70M.ENV.B3 | 11.3±2.5 | 6.1±0.3 | 1.5±0.9 | 1.3±0.4 | 2.4±0.9 | |
| 11891 | QF495.23M.ENV.A3 | 27.8±9.3 | 5.7±2.2 | 3.2±0.4 | 0.88±0.4 | 1.3±0.3 | |
| 11892 | QF495.23M.ENV.B2 | 20.4±7 | 8.6±2 | 3.7±1 | 0.6±0.4 | 1.1±0.2 | |
| BG505-T332N | 11.5±6 | 2.5±0.9 | 2.4±1 | 1±0.1 | 1.3±0.6 | ||
| KNH1144 | 20±3.2 | 8.3±0.4 | 2.1±0.1 | 2.1±0.4 | 2.3±0.2 | ||
| A/D | 11901 | QA790.204I.ENV.A4 | 12.3±1.8c | 2.4±0.4c | 3.7±0.2 | 1.4±0.05 | 1±0.3 |
| 11903 | QA790.204I.ENV.C8 | 5±1c | 3±0.3c | 2±0.1 | 1.6±0.2 | 1.6±0.2 | |
| 11904 | QA790.204I.ENV.E2 | 7.1±0.5c | 1±0.1c | 3.4±0.1 | 1.2±0.3 | 2±0.05 | |
| A2/D | 11905 | QG393.60M.ENV.A1 | 43±2 | 1.2±0.2 | 1.8±0.6 | 0.41±0.2 | 1.1±0.3 |
| 11906 | QG393.60M.ENV.B7 | 3.5±0.2 | 1.7±0.4 | 1.7±0.2 | 0.8±0.3 | 1.4±0.5 | |
| 11907 | QG393.60M.ENV.B8 | 3.6±0.1 | 1.3±0.4 | 2.6±1.4 | 0.48±0.2 | 1.5±0.6 | |
| A/E | 11603 (potential) |
CRF01_AE clone 269 | 10.7±1.5 | 3.5±1.5 | 3±0.1 | 1.5±0.3 | 1.7±0.9 |
| AA058 | 4.1 ±0.8 | 1.9±0.8 | 4±0.8 | 3.3±1.2 | 3±0.6 | ||
| CM244 | 5.6±0.8 | 3±0.5 | 2.8±0.2 | 2.8±0.3 | 2.5±0.6 | ||
| A/G | 11601 | CRF02_AG clone 263 | 7±2.2 | 2.3±0.3 | 2.2±0.3 | 1.4±0.3 | 2.2±0.7 |
| 11602 | CRF02_AG clone 266 | 4.6±1.6 | 2±0.3 | 1.6±0.5 | 1±0.2 | 1.3±0.5 | |
| 11605 | CRF02_AG clone 278 | 6.5±1.1 | 2.3±0.3 | 2.2±0.4 | 1.3±0.5 | 1.8±0.2 | |
| B | B41 | 4.5±2 | 1±0.2 | 1.1±0.1 | 0.68±0.1 | 1.1±0.4 | |
| 11563 | p1058_11.B11.1550b | 1.9±1 | 0.8±0.1 | 5.3±0.3 | 1.1±0.8 | 2.1±0.7 | |
| 11578 | pWEAUd15.410.5017b | 4.9±0.8c | 1.9±0.2c | 3±0.1 | 3±0.1 | 1.7±0.03 | |
| 11018 | QH0692, clone 42 | 2±0.6 | 1.4±0.2 | 0.7±0.2 | 0.52±0.1 | 0.9±0.3 | |
| 11022 | PVO, clone 4 | 3.5±0.7 | 1.9±0.4 | 1.7±0.1 | 1.9±0.1 | 1.1±0.2 | |
| 11023 | TRO, clone 11 | 4.1±0.6 | 2.9±0.3 | 1.3±0.2 | 1.2±0.05 | 1±0.1 | |
| 11024 | AC10.0, clone 29 | 8.9±3c | 1.4±0.3c | 1±0.2 | 0.32±0.01 | 0.74±0.25 | |
| 11033 | pWIT04160 clone 33 | 4.8±0.8 | 2.6±0.2 | 2±0.05 | 1.7±0.1 | 1±0.2 | |
| 11035 | PREJ04541 clone 67 | 3.4±0.2 | 2.1±0.1 | 1.8±0.2 | 1.6±0.3 | 1.2±0.1 | |
| 11036 | pRHPA4259 clone 7 | 4.3±0.5 | 2.5±0.2 | 2.1±0.1 | 2±0.4 | 1.2±0.06 | |
| 11037 | PTHR04156 clone 18 | 8.4±2.4 | 2.3±0.2 | 3.1±1.6 | 1±0.2 | 2±0.2 | |
| 11038 | pCAAN5342 clone A2 | 2.4±0.3c | 2.3±0.3c | 1.7±0.1 | 0.6±0.07 | 0.64±0.01 | |
| 11058 | SC422661.8 | 4.8±0.4c | 2.3±0.2c | 0.6±0.1 | 0.33±0.01 | 0.27±0.02 | |
| C | 11306 | Du156, clone 12 | 5±0.9 | 3.4±0.3 | 2.910.6 | 2.2±0.5 | 1.2±0.2 |
| 11307 | Du172, clone 17 | 4.8±0.2 | 2.4±0.3 | 1.5±0.3 | 1.9±0.5 | 1.5±0.3 | |
| 11308 | Du422, clone 1 | 6.1 ±0.5 | 4.6±0.4 | 3±0.1 | 3.3±0.4 | 2±0.6 | |
| 11309 | ZM197M.PB7 | 3.8±0.3 | 3.7±0.8 | 2±0.2 | 2.2±0.5 | 0.96±0.2 | |
| 11310 | ZM214M.PL15 | 4.1±0.7 | 3.5±0.2 | 2.6±0.1 | 2.7±0.2 | 1.7±0.6 | |
| 11311 | ZM233M.PB6 | 6.6±0.8 | 2.8±0.2 | 1.4±0.2 | 1.3±0.1 | 1.2±0.1 | |
| 11312 | ZM249M.PL1 | 6.1±0.3 | 5.7±1.3 | 3.1±0.3 | 3.1±0.5 | 1.8±0.6 | |
| 11313 | ZM53M.PB12 | 14.6±1 | 2.5±0.1 | 1.3±0.2 | 1.5±0.2 | 1.3±0.3 | |
| 11314 | ZM109F.PB4 | 6.2±0.3 | 5±0.2 | 2.2±0.4 | 3.2±0.5 | 1.9±0.3 | |
| 11315 | ZM135M.PL10a | 2.2±0.6 | 2.2±0.8 | 2.5±0.3 | 2.7±1.2 | 2.2±0.5 | |
| 11316 | CAP45.2.00.G3 | 9.6±0.3 | 7.3±1.7 | 2.1±0.1 | 1.5±0.1 | 1.2±0.1 | |
| 11317 | CAP210.2.00.E8 | 5.3±0.6 | 4.1±0.4 | 2.7±0.1 | 2.8±0.7 | 1.3±0.3 | |
| 11908 | QB099.391 M.ENV.B1 | 5.7±0.9 | 3±0.5 | 2.2±0.4 | 1.7±0.4 | 1.3±0.2 | |
| 11909 | QB099.391 M.ENV.C8 | 3.6±0.9 | 3.9±1.3 | 1.8±0.4 | 1.8±0.5 | 2.5±0.5 | |
| 11910 | QC406.70M.ENV.F3 | 3.6±0.9 | 3.9±1.3 | 1.8±0.4 | 1.8±0.5 | 2.5±0.5 | |
| D | 11911 | QA013.70I.ENV.H1 | 9.4±2.6 | 4.6±0.6 | 2.7±0.2 | 2.6±0.2 | 2.1±0.4 |
| 11912 | QA013.70I.ENV.M12 | 5.4±0.3 | 3.3±0.3 | 2.4±0.2 | 2.2±0.3 | 1.7±0.2 | |
| 11913 | QA465.59M.ENV.A1 | 4.8±0.2c | 1.5±0.2c | 3±0.3 | 0.76±0.15 | 1±0.2 | |
| 11914 | QA465.59M.ENV.D1 | 6.1±0.5 | 5.3±0.8 | 3.8±0.2 | 3.1±0.5 | 2.5±0.2 | |
| 11916 | QD435.100M.ENV.B5 | 4.4±1 | 2.9±0.5 | 2±0.6 | 1.7±0.5 | 1.7±0.6 | |
| 11918 | QD435.100M.ENV.E1 | 6.2±1.7 | 3.5±0.4 | 4±0.8 | 3.7±0.1 | 2.7±0.5 | |
| D/A | 11526 | 2.MF535.W0M.ENV.C1 | 7.2±1.5 | 10.6±0.6 | 2.7±0.3 | 1.8±0.5 | 1.7±0.8 |
| (mother) | |||||||
| 11517 (infant) |
1.BF535.W6M.ENV.A1 | 9.4±0.7 | 11±1.8 | 2.8±0.1 | 2.8±0.6 | 2.1±0.4 | |
| Control | A-MLV | >60 | >30 | 7.5±0.2 | 2.5±0.5 | 2.6±1 | |
| Control | VSV-G | >60 | >30 | 10±0.5 | 8.1±1 | 6.4±0.5 | |
| IC50(μM) Color code |
< 1 | 1-3 | >3-10 | >10 | |||
The reported IC50 values are means ± standard deviations (n = 3).
R5X4-tropic viruses; all the rest were CCR5-tropic viruses.
Data previously published (ref 19).
Antiviral Activity of NBD-11021 against a Diverse Set of Laboratory-Adapted and Primary HIV-1 Isolates
As a next step, we decided to test the inhibitory activity of NBD-11021 against a diverse set of seven laboratory-adapted HIV-1 strains and eight primary HIV-1 isolates representing the diverse subtypes (B, C, E, and G) and co-receptor usage (R5, X4, and R5X4). In addition, we have tested this compound against one RT inhibitor-resistant, one protease inhibitor-resistant, and one fusion inhibitor-resistant isolate. The inhibitory activity of NBD-11021 was compared with the activity of earlier compounds NBD-556 and NBD-09027. The data indicated substantial improvement of antiviral activity of NBD-11021 from the initially identified compounds (Table 3). In fact, NBD-556 inhibited HIV-1 lab-adapted isolates with an IC50 in the range of 4.8 to ≥118 μM, and the IC50 of NBD-09027 was in the range of 4–35.8 μM, while the IC50 of NBD-11021 was in the range of 1.2–7.3 μM. Similarly, for all the drug resistant and primary isolate HIV-1 strains, NBD-11021 was the only compound that showed a consistent inhibitory activity. Remarkably, NBD-11021 not only had a low micromolar activity against all the primary HIV-1 isolates we tested in peripheral blood mononuclear cells (PBMCs) but also was the only compound that exhibited activity against the HIV-1 isolates of subtype C that we tested in this study, HIV-193IN101 and HIV-193MW959, while NBD-556 and NBD-09027 showed poor to no activity against these HIV-1 strains. Also, the HIV-1BCF02 of group O showed practically no sensitivity to NBD-09027 but was sensitive to NBD-11021.
Table 3.
Antiviral Activity of the NBD Compounds in Laboratory-Adapted and Primary HIV-1 Isolates and Toxicity Values
| IC50 (μm) |
||||||
|---|---|---|---|---|---|---|
| HIV-1 virus | subtype | cell type | co-receptor | NBD-556 | NBD-09027 | NBD-11021 |
| Laboratory Strains | ||||||
| IIIB | B | MT-2 | X4 | 6.5 ± 0.1 | 4.7 ± 0.6 | 3.46 ± 0.2 |
| MN | B | MT-2 | X4 | 15.9 ± 1.6 | 4 ± 0.9 | 2.1 ± 0.1 |
| SF2 | B | MT-2 | R5X4 | ≥118 | 5.7 ± 0.9 | 2.6 ± 0.3 |
| RF | B | MT-2 | R5X4 | 18.7 ± 1.3 | 9.6 ± 0.8 | 7.3 ± 0.6 |
| BaL | B | PBMC | R5 | ≥118 | 35.8 ± 1.2 | 3.7 ± 0.4 |
| 89.6 | B | PBMC | R5X4 | 4.8 ± 1 | 6.7 ± 0.3 | 1.2 ± 0.1 |
| SF162 | B | PBMC | R5 | 48.9 ± 7.3 | 12.7 ± 0.7 | 2.6 ± 0.5 |
| RT Inhibitor-Resistant Isolate | ||||||
| AZT-R | B | MT-2 | X4 | 58 ± 14.3 | 4.4 ± 1.1 | 3 ± 0.1 |
| Protease Inhibitor-Resistant Isolate | ||||||
| HIV-1RF/L-323-12-3 | B | MT-2 | X4 | <59a | 14.7 ± 2.3 | 6.7 ± 0.3 |
| Fusion Inhibitor-Resistant Isolate | ||||||
| pNL4-3 gp41(36G)V38E,N42S | B | MT-2 | X4 | 11 ± 0.9 | 5.8 ± 0.3 | 2.2 ± 0.1 |
| Primary Isolates | ||||||
| 92US657 | B | PBMC | R5 | 48 ± 1.65 | 8.6 ± 0.9 | 3.3 ± 0.9 |
| 93IN101 | C | PBMC | R5 | >87a | 2.9 ± 0.1 | |
| 93MW959 | C | PBMC | R5 | 57.2 ± 8.7 | >43.5a | 2.3 ± 0.5 |
| 93TH060 | E | PBMC | R5 | >45a | 7.2 ± 0.6 | 5.5 ± 1.2 |
| RU570 | G | PBMC | R5 | 19.5 ± 2.3 | 8.5 ± 0.8 | 2.5 ± 0.6 |
| BCF02 | (Group 0) | PBMC | R5 | ~87 | 9.6 ± 1.1 | |
| NBD-556 | NBD-09027 | NBD-11021 | ||||
| MT-2 CC50 (μM) | >280a | >108a | ~28 | |||
| PBMC CC50 (μM) | >280a | >160a | ~36 | |||
50% toxicity or activity with respect to the untreated control at this dose was not reached.
NBD-11021 Prevents Cell-to-Cell Fusion
To investigate whether NBD-11021 and its two isomers NBD-11021A2 and NBD-11021B2 prevent cell-to-cell fusion, we cocultured MAGI-CCR5 cells and HL 2/3 cells in a virus-free culture in the presence of different concentrations of compounds. We used BMS-378806 and NBD-556 as controls because they were previously shown to inhibit cell–cell fusion.12,44 As expected, both controls BMS-378806 and NBD-556 inhibited fusion between MAGI-CCR5 and HL 2/3 with an IC50 of 14 nM and 6.5 μM, respectively (Table 4). Moreover, NBD-11021 and its two isomers NBD-11021A2 and NBD-11021B2 also inhibited cell-to-cell fusion with similar activity ranges as other NBD compounds.
Table 4.
Antiviral Activity of NBD Compounds in a HIV-1 Cell–Cell Fusion Assay and in Assays Using HIV-1 Reverse Transcriptase (RT) and Integrase Enzymes
| Cell–Cell Fusion Assay | |
|---|---|
| inhibitors | IC50 (μM) |
| NBD-11021 | 9.8 |
| NBD-11021A2 | 10.8 |
| NBD-11021B2 | 7.6 |
| NBD-556 (control) | 6.5 |
| BMS-378806 (control) | 0.014 |
| HIV-1 Reverse Transcriptase (RT) Assay | |
| inhibitors | IC50 (μM) |
| NBD-11021A2 | 43.4 |
| NBD-556 | >200 |
| nevirapine (control) | 0.20 |
| AZT-TP (control) | 0.008 |
| HIV-1 Integrase Assay | |
| inhibitors | IC50 (μM) |
| NBD-11021A2 | >100 |
| NBD-556 | >200 |
| raltegravir (control) | 0.21 |
NBD-11021 Also Targets HIV-1 Reverse Transcriptase (RT) but Shows No Activity against Integrase
The activity of NBD-11201 against both MLV and VSV-G clearly indicated that there may be an additional mechanism of action of this molecule. Since both MLV and VSV-G have different entry pathways, we hypothesized that the other target of this compound could be at a later step in the life cycle of the virus. This led us to test this molecule against HIV-1 RT and HIV-1 integrase. We did not select HIV protease (PR) because this series of compounds showed potent activity in a single cycle assay. For the RT assay, we tested NBD-11021A2 and NBD-556 and used nevirapine and AZT-TP as controls. NBD-11021 showed HIV-1 RT inhibition with an IC50 of 43.4 μM; however, NBD-556 did not show any activity even at 200 μM. Nevirapine and AZT-TP showed potent activity against HIVRT with IC50 of 0.20 and 0.008 μM, respectively (Table 4).
However, neither NBD-11021A2 nor NBD-556 showed any activity against HIV-1 integrase, whereas the control integrase inhibitor raltegravir showed inhibition with an IC50 of 0.21 μM (Table 4).
NBD-11021 Blocks Cell-to-Cell Transmission of HIV-1
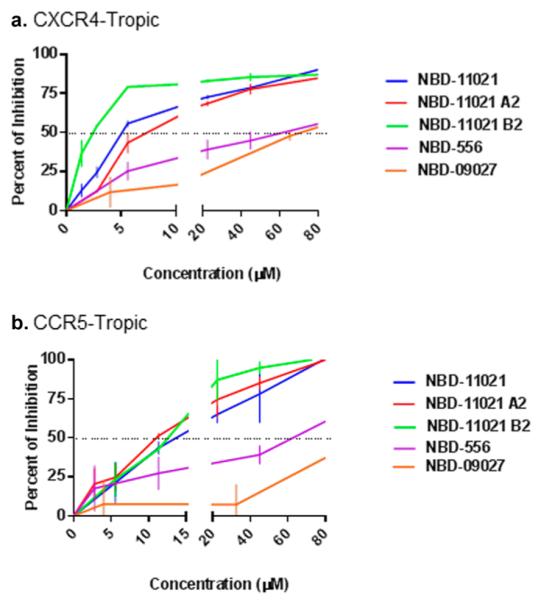
HIV-1 infection occurs via both cell-free and cell-associated transmission; however, the relative importance of the latter transmission pathway is not clear yet. Recently, Abela et al. elegantly demonstrated that some of the CD4BS directed antibodies and CD4M47, a peptide-based CD4-mimetic entry inhibitor, lost efficiency in inhibiting cell–cell transmission of HIV-1.45 However, in a more recent report by Agosto et al., it was shown that several antiretroviral drugs including some entry inhibitors effectively inhibited cell–cell transmission.46 In view of those findings, we investigated the ability of NBD-11021 to inhibit cell–cell transmission. To this end, we used Ghost X4/R5 as target cells and H9/HIV-1IIIB and MOLT-4/HIV-1ADA as effector cells. While NBD-556 and NBD-09027 had poor activity against the CXCR4-tropic HIV-1 cell–cell transmission (IC50 ≈ 60 μM) (Figure 2a), NBD-11021 and its two isomers NBD-11021A2 and NBD-11021B2 inhibited cell–cell transmission effectively. NBD-11021B2 was the most active inhibitor with an IC50 of 2 μM. With the CCR5-tropic cell–cell transmission assay, we found that NBD-556 had similar IC50 (~60 μM) as observed with CXCR4-tropic virus while the NBD-09027 had practically no inhibitory activity (Figure 2b). By contrast, NBD-11021 and the two isomers again showed inhibition though slightly higher than in the CXCR4-tropic cell–cell transmission system.
Figure 2.
Effect of NBD-11021 on cell-to-cell transmission of HIV-1. (a) Antiviral activity of NBD compounds in a CD4-dependent CXCR4-tropic HIV-1 cell–cell transmission assay. Ghost X4/R5 cells and H9/HIV-1IIIB cells were cocultured in the presence of escalating concentrations of test compounds. (b) Antiviral activity of NBD compounds in a CD4-dependent CCR5-tropic HIV-1 cell–cell transmission assay. Ghost X4/R5 cells and MOLT-4/HIV-1ADA cells were cocultured in the presence of escalating concentrations of test compounds. The plots represent the percentage of inhibition of p24 in the target cells Ghost X4/R5. Three independent experiments were performed in triplicate, and the graph is representative of one experiment; the values represent the mean ± standard deviation.
Crystal Structure of NBD-11021 in Complex with gp120 Core
To understand underlying mechanism(s) of improved antiviral potency of NBD-11021, we have determined the crystal structure of gp120 core in complex with NBD-11021 and compared it with the NBD-09027-bound gp120 structure. We used clade A/E 93TH057 H375S gp120 coree to determine the NBD-11021-bound gp120 structure. Among the four isomers of NBD-11021, NBD-11021A1 fits the best to the electron density map. NBD-11021 bound to the Phe43 cavity in a manner similar to that observed with other NBD analogs; the chlorophenyl ring and the linker region of the compound penetrate the cavity while region III of the compound resides in the vestibule of the cavity (Figure 3a,b,c). When the two structures were superimposed, the phenyl rings were on top of each other, but the two different linker regions (oxalamide of NBD-09027 and pyrrole of NBD-11021) prompted region III of the compounds to deviate substantially from each other in the cavity (Figure 3b,c). Val255 and Thr257 make hydrophobic interactions with the phenyl ring of region I, Trp427 interacts with the linker region (region II), and Gln428 and Gly429 make hydrophobic interactions with the five-membered thiazole ring of the region III of NBD-11021 (Figure 3d). Interestingly, the nitrogen atom (N13) in the pyrrole ring of the linker makes a hydrogen bond with Asn425 (Figure 3d). Most interestingly, the new linker of NBD-11021 allowed its piperdine ring to position near the Asp368gp120 and to make a hydrogen bond with the nitrogen atom of the piperidine ring (Figure 3d), which has not been observed in NBD-09027-bound gp120 structure. This interaction may contribute significantly to the improved affinity of NBD-11021 to gp120 and its antiviral properties over NBD-09027. Similar interactions between the guanidinium group of DMJ-I-228, an NBD analog, and Asp368gp120 was reported previously (PDB ID 4DKQ).15
Figure 3.

Crystal structure of NBD-11021-bound HIV-1 gp120. (a) NBD-11021 is shown in stick representation with its feature-enhanced map (FEM)85 (blue mesh) contoured at 1.5σ. The nitrogen atom in the piperidine ring makes a hydrogen bond with Asp368 of gp120. The average length of hydrogen bonds between the nitrogen atom and Asp368 is about 3.1 Å (There are four NBD-11021-bound gp120 molecules in the asymmetric unit). (b, c) Superposition of NBD-11021 (purple)-bound gp120 and NBD-09027 (yellow)-bound gp120 structures. The phenyl rings were superimposable (c), but the piperidine rings deviate substantially from each other (b, c), which results in the average distance between the nitrogen in the piperidine ring of NBD-11021 (NBD-09027) and Asp368 being about 3.1 Å (4.5 Å). (d) Detailed interactions between NBD-11021 and gp120.
DISCUSSION
The highly conserved Phe43 cavity has been validated as potential target for developing entry inhibitors as drugs for HIV-1 therapy and prophylaxis.12,19,20,47,48 The discovery of NBD-556 in 2005 by our group and the subsequent finding that this class of small molecules mimics CD4 and induces conformational changes in gp120 in a similar manner generated considerable interest to pursue designing NBD-556 analogs as entry inhibitors. However, these inhibitors, except one reported recently,48 induce gp120 conformation suitable for binding by CCR5 co-receptor; therefore, they function as CD4 agonists, an undesirable property for developing these inhibitors as drug candidates because they may promote HIV-1 entry and enhance infection in CD4−CCR5+ target cells.
Recently, we designed NBD-09027, a more active analog of NBD-556, by modifying the scaffold in region III. Interestingly, despite its higher potency in single-cycle HIV-1 infection assay, we demonstrated by both functional and biophysical studies that this molecule still possesses the undesired CD4-agonist property. However, we noted that the tendency of this molecule to function as agonist was about 50% less than that of NBD-556. Subsequently the X-ray structure of NBD-09027 bound to gp120 indicated the absence of a critical interaction between the piperidine ring and Asp368gp120 of gp120. We reasoned that to enhance binding affinity and antiviral activity, we needed further modification to achieve this interaction. In this study, we presented the successful modification of part of the oxalamide moiety with a pyrrole ring in region II, which was reported as intolerant to modification.48 The X-ray structure of NBD-11021-gp120 indicated that the introduction of a more rigid five-membered ring in region II forced the molecule to adopt a conformation different from NBD-09027 and bind to the cavity in a different orientation than NBD-09027. The structure confirmed that this modification helped in acquiring an H-bond interaction between the piperidine ring and Asp368gp120. However, this interaction alone cannot be attributed to the CD4-antagonist property of NBD-11021 because the X-ray structural information reported here is limited due to the deletion of the V3 loop in the gp120 structure. Therefore, the effect of this compound on the V3 loop conformation is not available. V3 loop has been implicated as the major binding site for the CCR5 co-receptor binding34,49,50 and mutations in the V3 loop affect CCR5 binding.51,52 Therefore, it is most likely that a cumulative effect of the binding of NBD-11021 in the Phe43 cavity in gp120, which is in the proximity of the V3 loop, may have altered the conformation of gp120 to be no longer suitable for CCR5 binding. Consequently, NBD-11021 behaves as CD4 antagonist.
Since NBD-11021A2 and NBD-11021B2 showed somewhat better antiviral activity, we made an attempt to fit these enantiomers to the electron density map of the gp120 structure in complex with NBD-11021 mixture. We found that both these isomers could be modeled in such a way that the thiazole ring is now facing to the Asp368 and the nitrogen atom in the thiazole ring is ~4 Å apart from Asp368 to form H-bond (Figure S10). Although the fit of these isomers is not as ideal as NBD-11021A1, these putative binding modes of isomers may provide additional guidance to optimize this lead compound.
The single-cycle infectivity assay with a large number of Env-pseudotyped HIV-1 demonstrated substantial improvement in antiviral potency of NBD-11021A2 and NBD-11021B2 compared with NBD-556. Considerable improvement in antiviral potency of NBD-11021 compared with NBD-556 and NBD-09027 was also observed against replicating laboratory strains and primary isolates in multicycle assay. Most significantly, the cell-based functional assay in CD4−CCR5+ cells demonstrated that NBD-11021 did not enhance HIV-1 infection in these cells. Furthermore, the SPR (BIAcore) based assessment of CCR5 antibody surrogate mAb 17b binding to the NBD-11021–gp120 core complex indicated that this inhibitor did not enhance mAb 17b binding. The data confirm that NBD-11021 is a CD4 antagonist. When we used A-MLV envelope pseudotyped with HIV-1 backbone to test the specificity of the NBD-11021, we surprisingly found that NBD-11021 also inhibits this virus, which belongs to the same retrovirus family. The A-MLV pseudovirus has HIV-1 backbone. Therefore, NBD-11021 may also inhibit a post entry step. A-MLV has been shown recently to enter cells by macropinicytosis.40 Furthermore, HIV-1 protease (PR) inhibitors have been shown to also work against MLV PR despite the fact that these two PR show substantial differences in specificity toward substrates and their molecular models of MLV PR suggested that the residues forming the subsites for substrate recognition in both PRs are different.53 Therefore, it is not unusual to find antiviral activity of the same compounds in different viruses with distinctly different structural features. In order to unravel other possible targets of NBD-11021, we tested one of its isomers, NBD-11021A2, against HIV-1 RT and integrase, and indeed we found that it inhibits HIV-1 RT but at relatively higher dose (IC50 of 43.4 μM). NBD-11021A2 did not show any activity against HIV-1 integrase. Recently, a pyrazolo-piperidine based HIV-1 entry inhibitor that targets coreceptors CCR5/CXCR4 has been shown to also target HIV-1 RT with an IC50 of 9.0 μM. This compound was ~5-fold more potent against HIV-1 RT than NBD-11021.54
CONCLUSION
In this study, we report successful conversion of a CD4-agonist NBD-556 to a CD4-antagonist NBD-11021 by systematic structure-based design, synthesis, and functional and biophysical assays. The antagonist showed the level of breadth in antiviral activity against a panel of diverse HIV-1 Env-pseudotyped viruses in single-cycle assay never reported before for small molecule inhibitors that target the Phe43 cavity. Furthermore, our study indicates that NBD-11021 also efficiently inhibits cell-to-cell fusion and cell-associated virus infection. Besides inhibiting viral entry, we also uncovered that NBD-11021 inhibits HIV-1 RT but at much higher concentration. This discovery is expected to pave the way to further optimize NBD-11021 to a clinical candidate for HIV-1 prevention and therapy.
EXPERIMENTAL SECTION
Cells, Virus, and Plasmids
MT-2 cells (human T-cell leukemia cells obtained through the NIH AIDS Reagent Program (ARP) from Dr. D. Richman), H9/HTLV-/HIV-1IIIB cells (obtained through the NIH ARP from Dr. R. Gallo), and human PBMC (isolated from buffy coats of healthy HIV-1 negative donor obtained from the New York Blood Center, NY) were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), penicillin, and streptomycin (100 U/μL each).
TZM-bl cells (a HeLa cell line that expresses CD4, CXCR4, and CCR5 and expresses luciferase and β-galactosidase under control of the HIV-1 promoter, obtained from Dr. J. C. Kappes, Dr. X. Wu, and Tranzyme Inc. through the NIH ARP),55,56 HEK 293T cells (ATCC), Cf2Th/CD4-CCR5, and CD4-Cf2Th-CCR5 cells (kindly provided by Dr. J. G. Sodroski57) were grown in Dubecco’s modified Eagle medium (DMEM) supplemented with 10% FBS, penicillin, and streptomycin.
U87-CD4-CCR5 and U87-CD4-CXCR4 cells (obtained through the ARP from Drs. H. Deng and D. Littman)58 were grown in DMEM supplemented with 15% FBS, 1 μg/mL puromycin, 300 μg/mL G418, penicillin, and streptomycin.
GHOST (3) X4/R5 cells (obtained through the NIH ARP from Drs. V. N. Kewalramani and D. Littman)59 were grown in DMEM supplemented with 10% FBS, 500 μg/mL G418, 100 μg/mL hygromycin, 1 μg/mL puromycin, penicillin, and streptomycin.
MOLT-4/CCR5 cells (obtained through the NIH ARP from Drs. M. Baba, H. Miyake, and Y. Iizawa) were grown in RPMI 1640 medium supplemented with 10% FBS, 1 mg/mL G418 penicillin, and streptomycin.
MAGI-CCR5 cells (obtained from Dr. J. Overbaugh through the NIH ARP) and HL 2/3 cells (obtained from Drs. B. K. Felber and G. N. Pavlakis through the NIH ARP) were grown in DMEM medium supplemented with 10% FBS, penicillin, and streptomycin.
HIV-1 lab-adapted and primary isolates, SIV, SHIV, and HIV-2 strains were obtained through the NIH ARP.
Env expression vector pSVIIIenv-ADA and mutant pSVIIIenv-ADA-N197S DNA were kindly provided by Dr. J. G. Sodroski.57 HIV-1 Env molecular clone expression vector pHXB2-env (X4) DNA was obtained through the ARP from Drs. K. Page and D. Littman.60 HIV-1 Env molecular clones of gp160 genes for HIV-1 Env pseudovirus production were obtained as follows: clones representing the standard panel A, A/D, A2/D, D, and C (QB099.391M.ENV.B1, QB099.391M.ENV.C8, and QC406.70M.ENV.F3) were obtained through the NIH ARP from Dr. J. Overbaugh.61,62 The HIV-1 Env molecular clones panel of subtype A/G and CRF01_AE clone 269 were obtained through the NIH ARP from Drs. D. Ellenberger, B. Li, M. Callahan, and S. Butera.63 The AE clones AA058 and CM244 were kindly provided by Drs. Robert J. McLinden and Agnés-Laurence Chenine from US Military HIV Program, Henry M. Jackson Foundation (Silver Spring, MD). The HIV-1 Env panel of standard reference subtype B Env clones were obtained through the NIH ARP from Drs. D. Montefiori, F. Gao and M. Li (PVO, clone 4 (SVPB11); TRO, Clone 11 (SVPB12); AC10.0, clone 29 (SVPB13); QH0692, clone 42 (SVPB6); SC422661, clone B (SVPB8)), from Drs. B. H. Hahn and J. F. Salazar-Gonzalez (pREJO4541, clone 67 (SVPB16); pRHPA4259, clone 7 (SVPB14); pWITO4160 clone 33 (SVPB18)), and from Drs. B. H. Hahn and D. L. Kothe (pTHRO4156 clone 18 (SVPB15), pCAAN5342 clone A2 (SVPB19)).56,64,65 The subtype B pWEAUd15.410.5017 and p1058_11.B11.1550 were obtained through the NIH ARP from Drs. B. H. Hahn, B. F. Keele, and G. M. Shaw.39 The subtype C HIV-1 reference panel of Env clones were also obtained through the NIH ARP from Drs. D. Montefiori, F. Gao, S. A. Karim, and G. Ramjee (Du 156.12; Du172.17), from Drs. D. Montefiori, F. Gao, C. Williamson, and S. A. Karim (Du422.1), from Drs. B. H. Hahn, Y. Li, and J.F. Salazar-Gonzalez (ZM197M.PB7; ZM233M.PB6; ZM249M.PL1; ZM214M.PL15), from Drs. E. Hunter and C. Derdeyn (ZM53M.PB12; ZM135M.PL10a; ZM109F.PB4), and from Drs. L. Morris, K. Mlisana, and D. Montefiori, (CAP45.2.00.G3; CAP210.2.00.E8).66–68 The paired envelope variants subtype D/A Maternal (MF535.W0M.ENV.C1) and Infant (BF535.W6M.ENV.A1) molecular clones were obtained through the NIH ARP from Dr. J. Overbaugh.69
The ENV pseudotyped genes of BG505.T332N, KNH1144, and B41 were kindly provided by Dr. J. P. Moore of the Weil Cornell Medical College, NY. SV-A-MLV-env from Drs. N. Landau and D. Littman and pHEF-VSVG from Dr. L. Chang were obtained through the NIH ARP.
The ENV-deleted pro-viral backbone plasmids, pNL4-3.Luc.R-.E-DNA (Dr. N. Landau)70,71 and pSG3Δenv DNA (Drs. J. C. Kappes and X. Wu)56,65 were obtained through the NIH ARP. The Env-deleted proviral backbone plasmid pNL4-3KFS DNA was kindly provided by Dr. E. Freed from NIH/NCI. The DNA fragment encoding rev-gp160 was subcloned from the YU2 strain. The expression vector was constructed as previously described.72
Soluble CD4 (sCD4) was purchased (ImmunoDiagnostics, Woburn, MA), and 17b hybridoma was kindly provided by Dr. James Robinson.
Chemistry
General
Commercial reagents and solvents were used without further purification. All reactions were performed in air atmosphere unless otherwise stated. Reactions were monitored by thin layer chromatography (TLC) carried out on Merck TLC silica gel plates (60 F254), using UV light for visualization and basic aqueous potassium permanganate or iodine fumes as developing agent. NMR 1H and 13C spectra were recorded on Bruker Avance 400 instrument with operating frequency of 400 and 100 MHz, respectively, and calibrated using residual undeuterated chloroform (δH = 7.28 ppm) and CDCl3 (δC = 77.16 ppm) or undeuterated DMSO (δH = 2.50 ppm) and DMSO-d6 (δC = 39.51 ppm) as internal references. The following abbreviations are used to set multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad.
1-((Allyloxy)carbonyl)piperidine-2-carboxylic Acid (1)
dl-Pipecolic acid (18.20 g, 0.141 mol, 1.5 equiv) was dissolved in water (70 mL), containing NaOH (5.60 g, 0.140 mol). To this solution, THF was added (70 mL), and the reaction mixture was cooled to 0 °C. At this temperature from two separate funnels were added allyl chloroformate (10 mL, 94.1 mmol in 20 mL of THF) and NaOH solutions (5.60 g, 0.140 mol in 70 mL of water). The reaction mixture was allowed to warm to an ambient temperature, acidified with 24 mL of concentrated aqueous hydrochloric acid, and extracted with dichloromethane (3 × 100 mL). The combined organic phases were dried over anhydrous Na2SO4 and evaporated to give a white solid, which was used without further purification. M = 18.00 g; yield = 90%.
1H NMR (CDCl3, 400 MHz): δ = 1.26–1.54 (m, 2 H), 1.63–1.78 (m, 3 H), 2.21–2.34 (m, 1 H), 2.92–3.14 (m, 1 H), 4.09 (dd, J = 1.0 Hz, 1 H), 4.63 (s, 2 H), 4.96 (d, J = 27.5, 13.0 Hz, 1 H), 5.16–5.29 (m, 1 H), 5.33 (d, J = 16.6 Hz, 1 H), 5.86–6.02 (m, 1 H), 9.94 (br s, 1 H).
13C NMR (CDCl3, 100 MHz): δ = (20.75, 20.82), (24.6, 24.8), (26.7, 26.8), (41.8, 41.9), (54.2, 54.4), (66.5, 66.6), 117.6, 132.9, (155.9, 156.6), 177.4.
Allyl 2-(Methoxy(methyl)carbamoyl)piperidine-1-carboxylate (2)
CDI (15.20 g, 93.8 mmol, 1.1 equiv,) was added portionwise to a solution of 1 (18.00 g, 84.5 mmol) in MeCN (90 mL) (Caution! CO2 evolution!). The resulting solution was stirred for 5 min, and N,O-dimethylhydroxylamine hydrochloride (10.00 g, 109.3 mmol, 1.2 equiv) was added in one portion. The reaction mixture was stirred overnight and evaporated to half of a volume. The residue was dissolved in 500 mL of CH2Cl2 and washed with 5–10% aqueous hydrochloric acid and then with 5% K2CO3 solution. The organic phase was dried over anhydrous Na2SO4 and evaporated to give the pure compound 2 as a nonviscous liquid. M = 18.91 g; yield = 87%.
1H NMR (CDCl3, 400 MHz): δ = 1.35–1.58 (m, 2 H), 1.59–1.67 (m, 1 H), 1.76 (m, 2 H), 1.93–2.07 (m, 1 H), 3.18 (s, 3 H), 3.43–3.65 (m, 1 H), 3.71 (s, 1 H), 3.81 (s, 2 H), 4.03 (m, 1 H), 4.53–4.68 (m, 2 H), 5.02–5.14 (m, 1 H), 5.16–5.37 (m, 2 H), 5.94 (m, 1 H).
13C NMR (CDCl3, 100 MHz): δ = 19.5, (24.75, 24.83), 26.5, (31.9, 32.1) (br), (42.1, 42.2), (51.3, 51.5), 61.34, 66.10, (117.2, 117.7), 133.1, 156.9, 173.2.
Allyl 2-Formylpiperidine-1-carboxylate (3)
LiAlH4 (2.65 g, 69.7 mmol) was added in small portions to the solution of 2 (17.91 g, 70.0 mmol) in THF (70 mL) at 0 °C. The resulting reaction mixture was stirred at 0 °C until the complete consumption of the starting material (~1 h, TLC control, 1:1 hexanes/EtOAc). After cooling to −20 °C, the excess of LiAlH4 was quenched by a dropwise addition of water (~5 mL). After that 10% aqueous HCl (100 mL) was added dropwise, and the resulting cold solution was immediately extracted with CH2Cl2 (3 × 100 mL). The combined organic phases were dried over anhydrous Na2SO4 and evaporated to give an oil, which was used without further purification. M = 9.16 g; yield = 66%.
1H NMR (CDCl3, 400 MHz): δ = 1.16–1.36 (m, 1 H), 1.46 (q, J = 12.7 Hz, 1 H), 1.55–1.78 (m, 3 H), 2.16–2.32 (m, 1 H), 2.80–3.08 (m, 1 H), 3.96–4.26 (m, 1 H), 4.57–4.79 (m, 3 H), 5.13–5.40 (m, 2 H), 5.85–6.08 (m, 1 H), 9.62 (s, 1 H).
13C NMR (CDCl3, 100 MHz): δ = 20.9, (23.5, 23.7), (24.6, 24.8), (42.4, 42.8), 61.1, 66.5, 117.7, 132.8, 156.4, (201.0, 201.2).
Allyl 2-(((tert-Butylsulfinyl)imino)methyl)piperidine-1-carboxylate (4)
To a solution of 3 (9.16 g, 46.5 mmol) in THF (46 mL), tert-butylsulfonamide (7.62 g, 63.0 mmol, 1.35 equiv) was added in one portion, followed by the addition of Ti(OiPr)4 (28 mL, 94.6 mmol, 2 equiv). The resulting solution was stirred overnight and loaded directly onto silica. Chromatography with 1:1 hexanes/EtOAc and then sole EtOAc as eluent gave fractions that contained the title compound and some Ti(OiPr)4 as admixture (cloudy fractions). Ti(OiPr)4 was quenched by the addition of 1 mL of water, TiO2·nH2O was filtered through paper, and the filtrate was evaporated and dried in vacuo. M = 10.61 g; yield = 76%. The title compound exists as a mixture of two diastereomers. Diastereomeric ratio was not determined. The configuration of the double bond is also unknown; however it should probably be E-configuration.
1H NMR (CDCl3, 400 MHz): δ = 1.18 (s, 4 H), 1.22 (s, 5H), 1.25–1.57 (m, 3 H), 1.59–1.91 (m, 3 H), 2.17 (d, J = 11.5 Hz, 1 H), 2.84–3.10 (m, 1 H), 3.97–4.18 (m, 1 H), 4.43–4.67 (m, 1 H), 4.99–5.35 (m, 3 H), 5.83–5.99 (m, 1 H), 7.93 (dd, J = 5.8, 1.7 Hz, 1 H).
13C NMR (CDCl3, 100 MHz): δ = (20.2, 20.4), (22.5, 22.7) (3C), (24.9, 25.0) (br), 26.5 (br), 41.6 (br), 55.9 (br), 57.0, 66.3, (117.5, 117.9 (br)), 133.0, 155.9, (168.0, 168.5 (br)).
Ethyl 4-Methylthiazole-5-carboxylate (5)
To a solution of formamide (14.5 mL, 0.364 mol, 1.5 equiv) in 1,4-dioxane (100 mL), P2S5 (17.30 g, 77.7 mmol, 0.3 equiv) was added in small portions in the course of 5 h under vigorous stirring. The reaction mixture was stirred for 2 h, and ethyl 2-chloroacetoacetate (35.4 mL, 0.243 mol) was added dropwise. The reaction mixture was heated at ~60 °C for 8 h, poured into cold water (300 mL), and extracted with CH2Cl2 (3 × 100 mL). Combined organic phases were washed with 5% K2CO3, dried over anhydrous Na2SO4, evaporated, and distilled at reduced pressure. M = 25.0 g; yield = 60%.
1H NMR (CDCl3, 400 MHz): δ = 1.39 (t, J = 7.2 Hz, 3 H), 2.79 (s, 3 H), 4.36 (q, J = 7.1 Hz, 2 H), 8.84 (s, 1 H).
13C NMR (CDCl3, 100 MHz): δ = 14.2, 17.2, 61.2, 122.2, 155.2, 160.4, 161.9.
(4-Methylthiazol-5-yl)methanol (6)
A solution of 5 (25.0 g, 0.146 mol) in THF (150 mL) was added dropwise to a suspension of LiAlH4 (5.55 g, 0.146 mmol) in THF (150 mL). The reaction mixture was stirred for 1 h and quenched by successive addition of water (6 mL), 10% NaOH (6 mL) solution, and water (12 mL). The precipitate was filtered and washed several times with THF. The filtrate was evaporated to give 6, which was used without purification. M = 17.90 g; yield = 95%.
1H NMR (CDCl3, 400 MHz): δ = 2.44 (s, 3 H), 4.83 (s+br s, 2 + 1 H), 8.65 (s, 1 H).
13C NMR (CDCl3, 100 MHz): δ = 14.5, 55.9, 132.2, 148.7, 151.1.
5-(((tert-Butyldimethylsilyl)oxy)methyl)-4-methylthiazole (7)
Compound 6 (36.0 g, 0.279 mol) was dissolved in DMF (280 mL), and imidazole (28.5 g, 0.419 mol, 1.5 equiv) was added in one portion, followed by portionwise addition of TBSCl (50.2 g, 0.335 mol, 1.2 equiv). The reaction mixture was stirred overnight at 50–60 °C, cooled to room temperature, diluted with water (0.5 L), and extracted with hexane (3 × 100 mL). The combined organic phases were dried over anhydrous Na2SO4 and evaporated to give an oil, which was purified by distillation at reduced pressure. M = 61.20 g; yield = 90%.
1H NMR (CDCl3, 400 MHz): δ = 0.12 (s, 6 H), 0.94 (s, 9 H), 2.42 (s, 3 H), 4.85 (s, 2 H), 8.64 (s, 1 H).
13C NMR (CDCl3, 100 MHz): δ = −5.22, 15.2, 18.4, 25.9, 58.0, 132.5, 148.0, 150.6.
1-(4-Chlorophenyl)-N-hydroxyethanimine (8)
Hydroxylamine hydrochloride (50.11 g, 0.72 mol) was added to a solution of 4′-chloroacetophenone (85.74 g 0.55 mol) in 140 mL of ethanol. Then with stirring a solution of NaOH (28.84 g, 0.72 mol) in 140 mL of water (Caution! exothermic reaction!) was added in one portion. The biphasic reaction mixture was refluxed for 3 h and poured into 1000 mL of ice water. The precipitate was filtered off and dried thoroughly on a rotary evaporator to afford 91.00 g (97%) of an off-white to light-yellow solid (the color depends on a quality of the starting acetophenone) with mp = 98–99 °C.
1H NMR (DMSO, 400 MHz): δ = 2.11 (s, 3H), 7.25 (d, J = 8.6 Hz, 2H), 7.54 (d, J = 8.6 Hz, 2H), 10.93 (br s, 1H).
2-(4-Chlorophenyl)-1H-pyrrole (9)
KOH (74.02 g, 1.32 mol) was added to a solution of 1-(4-chlorophenyl)-N-hydroxyethanimine (80.00 g, 0,47 mol) in DMSO (230 mL). Further scale up led to a significant decrease in yield. The heating was turned on, and KOH was fused with a heatgun (T > 100 °C). The reaction mixture was stirred at 130–140 °C, and the solution of dibromoethane (81.2 mL, 0.94 mol) in DMSO (80 mL) was slowly added dropwise during the course of 6–7 h. Every hour a 30 g portion of KOH (totally five portions) was added and fused with a heatgun to achieve a fine suspension (otherwise the reaction mixture could not be stirred properly, especially at the last 2–3 h of the dibromoethane addition).
After the addition was finished, the reaction mixture was cooled to RT and poured into ~1500 g of ice water. The resulting dark solution was extracted with benzene (5 × 300 mL). The first two extractions were difficult to execute because the phase separation level was hardly noticeable. The combined organic phases were dried over anhydrous Na2SO4, and the solvent was evaporated on a rotary evaporator. The residue was distilled under high vacuum, and the high-boiling fractions were collected. Fractions with bp > 130 °C were collected. The product was crystallized in a wide condenser. The product was melted with a heat gun and washed off in a beaker with light petroleum ether. The pyrrole was filtered off, washed with petroleum ether, and dried in air to afford 10.60 g (13%) of off-yellow solid with a strong odor (mp = 140 °C). The product is readily oxidized in air. Thus, it should be used in the next step promptly.
1H NMR (CDCl3, 400 MHz): δ = 6.30 (s, 1H), 6.51 (s, 1H), 6.86 (s, 1H); 7.33 (d, J = 8.4 Hz, 2H), 7.39 (d, J = 8.6 Hz, 2H), 8.38 (br s, 1H).
13C NMR (CDCl3, 100 MHz): δ = 106.6, 110.5, 119.4, 125.1 (2C), 129.2 (2C), 131.1, 131.4, 131.8.
1-(5-(4-Chlorophenyl)-1H-pyrrol-2-yl)-2,2,2-trifluoroethanone (10)
To the solution containing 2-(4-chlorophenyl)-1H-pyrrole, 9 (18.20 g, 0.102 mol), and pyridine (11.0 mL, 0.13 mol) in 140 mL of diethyl ether, trifluoroacetic anhydride (18.9 mL, 0.14 mol) was added dropwise with an external cold water bath cooling. It should be noted that shortly after the beginning of the addition a precipitate starts to form. After the addition was complete, the mixture was stirred for 3 h and poured into 500 mL of cold water. The resulting precipitate was filtered off, washed with water, and dried on a rotary evaporator to afford 25.23 g (90%) of an off-white solid with mp = 189–191 °C.
1H NMR (DMSO, 400 MHz): δ = 6.92–6.96 (m, 1H), 7.25–7.29 (m, 1H), 7.51 (d, J = 6.9 Hz, 2H), 8.00 (d, J = 8.8 Hz, 2H), 12.93 (br s, 1H).
13C NMR (DMSO, 100 MHz): δ = 111.1, 117.1 (q, J = 289.8 Hz), 123.3 (q, J = 2.9 Hz), 126.2, 128.1 (2C), 128.7, 128.9 (2C), 133.7, 142.3, 167.8 (q, J = 35.0 Hz).
5-(4-Chlorophenyl)-1H-pyrrole-2-carboxylic Acid (11)
A solution of NaOH (2.86 g, 72 mmol) in 25 mL of water was added to the solution of 1-(5-(4-chlorophenyl)-1Hpyrrol-2-yl)-2,2,2-trifluoroethanone, 10 (9.79 g, 36 mmol), in 25 mL of ethanol. The resulting reaction mixture was refluxed for 6 h and cooled to RT. Ethanol was removed on a rotary evaporator. The resulting precooled solution was carefully acidified to pH 3 with an equivalent quantity of concentrated HCl solution (~3 mL). The resulting precipitate was filtered off, washed successively with cold water and diethyl ether, and dried on a rotary evaporator to afford 6.90 g (87%) of gray solid with mp = 171–172 °C.
1H NMR (DMSO, 400 MHz): δ = 6.60 (d, J = 3.5 Hz, 1H), 6.71 (d, J = 3.7 Hz, 1H), 7.39 (d, J = 8.6 Hz, 2H), 7.86 (d, J = 8.6 Hz, 2H), 11.87 (br s, 1H). CO2H missing due to broadening.
13C NMR (DMSO, 100 MHz): δ = 107.8, 115.3, 126.5 (2C), 126.9, 128.6 (2C), 130.8, 131.1, 134.0, 162.8.
Allyl 2-((5-(((tert-Butyldimethylsilyl)oxy)methyl)-4-methylthiazol-2-yl)(1,1-dimethylethylsulfinamido)methyl)piperidine-1-carboxylate (12)
Compound 7 (12.90 g 53.1 mmol, 1.5 equiv) was dissolved in THF (33 mL) and cooled to −78 °C. Please note that large excess of 7 is not necessary. It was used in excess because of the limited quantity of 4. At this temperature, n-BuLi (2.5 M, 22 mL, 55 mmol, 1.5 equiv) was added dropwise under the nitrogen atmosphere. The reaction mixture was stirred for 30 min at −78 °C, and 4 (10.64 g, 35.5 mmol) was added dropwise as a solution in THF (35 mL). The reaction mixture was slowly (~1 h) warmed to 0 °C, acidified with acetic acid (3.18 mL, 53 mmol), and quenched with water (200 mL). The biphasic mixture was extracted with CH2Cl2 (3 × 100 mL). The combined organic phases were dried over anhydrous Na2SO4 and evaporated to give a mixture of compounds 7, 12a, and 12b (brown oil), which was separated by column chromatography. Eluent, hexanes/EtOAc (1:1) then pure EtOAc.
First fraction (starting material 7): Rf ≈ 0.6–0.7 (EtOAc)
Second fraction (12a): Rf = 0.56 (EtOAc)
Third fraction (12b): Rf = 0.39 (EtOAc)
Collected: pure 12a 1.98 g, mixed fraction (12a + 12b) 8.52 g, pure 12b 5.98 g (85% overall).
Isomers 12a and 12b were used in subsequent steps separately.
N1 (12a)
1H NMR (CDCl3, 400 MHz): δ = 0.10 (s, 6 H), 0.92 (s, 9 H), 1.19 (s, 8 H), 1.25 (s, 1 H), 1.41–1.55 (m, 2 H), 1.58–1.87 (m, 5 H), 2.33 (s, 3 H), 3.01–3.22 (m, 1 H), 4.14 (m, 1 H), 4.42–4.53 (m, 1 H), 4.58–4.70 (m, 2 H), 4.79 (s, 2 H), 5.07 (d, J = 10.0 Hz, 1 H), 5.24 (d, J = 10.4 Hz, 1 H), 5.35 (d, J = 16.6 Hz, 1 H), 5.98 (ddt, J = 16.9, 11.0, 5.4, 5.4 Hz, 1 H).
13C NMR (CDCl3, 100 MHz): δ = −5.2, 14.3, 15.3, 18.3, 19.2, 22.6 (3C), 24.9 (br), 25.4, 25.9 (3C), 40.2 (br), 55.9, 58.1, 60.4, 66.7, 117.7, 132.9, 133.5, 147.1. (3 signals at the downfield regions are missing).
N2 (12b)
1H NMR (CDCl3, 400 MHz): δ = 0.08 (s, 6 H), 0.91 (s, 9 H), 1.23 (s, 6 H), 1.27 (s, 3 H), 1.38–1.84 (m, 6 H), 2.09–2.27 (m, 1 H), 2.30 (s, 3H), 2.62–3.05 (m, 1 H), 3.84–4.07 (m, 1 H), 4.45 (br s, 3 H), 4.73–4.79 (m, 2 H), 4.82–5.03 (m, 1 H), 5.12–5.25 (m, 2 H), 5.63–5.90 (m, 1 H).
13C NMR (CDCl3, 100 MHz): δ = −5.3, 15.0, 18.6, 22.5 (3C), 22.8, 24.1, 24.7 (br), 25.7, 25.75 (3C), 25.8, 40.0, (56.4, 56.6), 56.5, 57.8, 66.1, 117.1, 117.7, 133.0, 147.0, 166.0, 168.2.
Allyl 2-(Amino(5-(hydroxymethyl)-4-methylthiazol-2-yl)methyl)-piperidine-1-carboxylate (13a and 13b)
The 1 M HCl–MeOH solution was prepared by dropwise addition of AcCl (2.10 mL, 29.5 mmol) to methanol (30 mL). The resulting solution was cooled to an ambient temperature and added to the flask containing 12a (1.9752 g, 3.64 mmol). After the dissolution of 12a, the reaction mixture was stirred for 1 h, evaporated (without heating), and dissolved in MeOH (50 mL), and solid KOH (~1g) was added to the solution. The mixture was stirred for 10 min and loaded on silica gel. Eluting with CH2Cl2/MeOH (50:1 → 20:1 → 5:1) provided the title amine 13a. M = 0.9052 g; yield = 76%. The second isomer (13b) was prepared according to the same procedure from 12b. M = 0.8544 g; yield = 73%. Rf = 0.42 CHCl3/MeOH (for both amines).
N1 (13a)
1H NMR (CDCl3, 400 MHz): δ = 1.35–1.54 (m, 3 H), 1.54–1.74 (m, 3 H), 2.33 (s, 3 H), 2.4–3.4 (br s, 3 H) 2.77–2.98 (br s, 1 H), 4.05–4.31 (br s, 1 H), 4.31–4.50 (br s, 1 H), 4.48–4.53 (m, 1 H), 4.62 (d, J = 4.4 Hz, 2 H), 4.73 (s, 2 H), 5.22 (dd, J = 10.5, 1.2 Hz, 1 H), 5.32 (br d, J = 18.2 Hz, 1 H), 5.96 (ddd, J = 22.5, 10.7, 5.4 Hz, 1 H).
13C NMR (CDCl3, 100 MHz): δ = 15.0, 19.4, 23.2, (25.2, 25.7) (br), (39.8, 40.0) (br), (52.4, 52.8) (br), 56.5, (56.7, 57.0) (br), 66.3, 117.5, 131.7 (br), 133.1, 148.5 (br), 156.4, (171.7, 172.2) (br).
N2 (13b)
1H NMR (CDCl3, 400 MHz): δ = 1.40–1.54 (m, 1 H), 1.55–1.71 (m, 4 H), 2.11–2.20 (m, 1 H), 2.30 (s, 3 H), 2.34 (br s, 3 H), 3.03 (t, J = 12.8 Hz, 1 H), 3.98–4.07 (m, 1 H), 4.21–4.31 (m, 2 H), 4.32–4.40 (m, 1 H), 4.49 (d, J = 10.0 Hz, 1 H), 4.67 (s, 2 H), 5.12 (dd, J = 10.5, 1.3 Hz, 1 H), 5.16 (dd, J = 17.3, 1.5 Hz, 1 H), 5.77 (ddd, J = 22.6, 10.8, 5.5 Hz 1 H).
13C NMR (CDCl3, 100 MHz): δ = 15.0, 19.1, 25.1, 25.4, 40.4, 52.4, 56.4, 56.8, 66.0, 117.1, 131.2, 133.2, 148.3, 155.4, 171.9.
Allyl 2-((5-(4-Chlorophenyl)-1H-pyrrole-2-carboxamido)(5-(hydroxymethyl)-4-methylthiazol-2-yl)methyl)piperidine-1-carboxylate (14a and 14b)
To a suspension of 5-(4-chlorophenyl)-1H-pyrrole-2-carboxylic acid (556 mg, 2.51 mmol) in DMF (4.2 mL), N,N-diisopropylethylamine (DIPEA; 0.43 mL, 2.47 mmol) was added followed by 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU; 951 mg, 2.50 mmol). The resulting solution was stirred for 10 min, and a solution of 13a (905.2 mg, 2.78 mmol) in DMF (4.2 mL) was added in one portion. The reaction mixture was stirred for 2 h and loaded on silica gel. Eluting with CH2Cl2/MeOH (50:1) gave 14a. M = 1.4751 g; yield = 100%. The title compounds contained DMF, DIPEA, (Me2N)2CO, CH2Cl2, and MeOH as admixtures. The second isomer (14b) was prepared according to the same procedure from 13b. M = 1.1172 g; yield = 80%.
14a
1H NMR (CDCl3, 400 MHz): δ = 1.20–1.60 (m, 7 H), 1.65 (d, J = 12.3 Hz, 1 H), 2.29 (s, 3 H), 3.0–4.0 (br s 1H), 3.97–4.10 (m, 1 H), 4.59 (br s, 2 H), 4.75 (s+m, 2 + 1 H), 4.99–5.23 (m, 2 H), 5.63–5.86 (m, 2 H), 6.47 (t, J = 2.8 Hz, 1 H), 6.68–6.77 (m, 1 H), 7.33 (d, J = 8.6 Hz, 2 H), 7.50 (d, J = 8.4 Hz, 2 H), 10.22 (br s, 1 H).
14b
1H NMR (CDCl3, 400 MHz): δ = 1.34–1.72 (m, 5 H), 1.71–1.89 (m, 1 H), 1.88–2.04 (m, 1 H), 2.30 (s, 3 H), 2.30–2.41 (m, 1 H), 3.38–4.49 (m, 4 H), 4.49–4.60 (m, 1 H), 4.70 (s, 2 H), 5.10–5.24 (m, 2 H), 5.66–5.91 (m, 2 H), 6.49 (s, 1 H), 6.76–6.91 (m, 1 H), 7.31 (d, J = 8.1 Hz, 2 H), 7.49 (d, J = 8.6 Hz, 2 H), 10.01–10.41 (br d, 1 H).
5-(4-Chlorophenyl)-N-((5-(hydroxymethyl)-4-methylthiazol-2-yl)-(piperidin-2-yl)methyl)-1H-pyrrole-2-carboxamide (15a = NBD-11021A and 15b = NBD-11021B)
To the solution containing 14a (1.4751 g, 2.79 mmol) and N,N’-dimethylbarbituric acid (NDMBA; 2.17 g, 13.9 mmol, 5 equiv) in 1,2-dichloroethane (28 mL), PPh3 (160 mg, 20 mol %) was added under a nitrogen atmosphere followed by Pd(dba)2 (146 mg, 10 mol %). The mixture was stirred for 1–2 h at ~60 °C (TLC control). After cooling, 100 mL of CH2Cl2 was added, and the organic phase was extracted twice with 5% aqueous K2CO3 to remove the unreacted NDMBA. The organic phase was dried over anhydrous Na2SO4 and concentrated. Purification by flash chromatography (20:1 CH2Cl2/MeOH) afforded amine 15a as a slightly brown solid. M = 572.1 mg; yield = 46%. The second isomer (15b) was prepared according to the same procedure from 14b. M = 648.2 mg; yield = 69%.
15a
Mp = 195 °C.
1H NMR (CDCl3, 400 MHz): δ = 1.17–1.52 (m, 4 H), 1.54–1.68 (m, 2 H), 1.76–1.85 (m, 1 H), 2.29 (s, 3 H), 2.63 (t, J = 10.6 Hz, 1 H), 3.03 (d, J = 10.9 Hz, 1 H), 3.39 (d, J = 11.2 Hz, 1 H), 4.69 (s, 2 H), 5.25–5.35 (m, 1 H), 6.47 (d, J = 3.8 Hz, 1 H), 6.90 (d, J = 3.8 Hz, 1 H), 7.28 (d, J = 8.6 Hz, 2 H), 7.49 (d, J = 8.6 Hz, 2 H), 7.77 (br s, 1 H), 10.50 (br s, 1 H). Both 1H spectra contain broad signal (1H) in the aliphatic region (1.0–4.0 ppm).
13C NMR (CDCl3, 100 MHz): δ = 15.1, 24.2, 25.4, 29.0, 46.8, 54.9, 56.2, 59.0, 107.8, 113.0, 126.1 (2C), 126.4, 129.1 (2C), 130.3, 132.0, 133.0, 135.1, 149.5, 161.5, 169.2.
HRMS (ESI) calcd for C22H26ClN4O2S [M + H]+ 445.1460, found 445.1453.
15b
Mp = 215 °C.
1H NMR (CDCl3, 400 MHz): δ = 1.18–1.34 (m, 3 H), 1.35–1.50 (m, 1 H), 1.54–1.63 (m, 1 H), 1.67–1.74 (m, 1 H), 1.78–1.86 (m, 1 H), 2.33 (s, 3 H), 2.60 (t, J = 13.3 Hz, 1 H), 2.98–3.05 (m, 1 H), 3.05–3.14 (m, 1 H), 4.73 (s, 2 H), 5.29 (dd, J = 8.2, 5.0 Hz, 1 H), 6.47 (d, J = 3.8 Hz, 1 H), 6.80 (d, J = 3.9 Hz, 1 H), 7.31 (d, J = 8.6 Hz, 2 H), 7.48 (d, J = 8.6 Hz, 2 H), 7.59 (br s, 1 H), 10.25 (br s, 1 H).
13C NMR (CDCl3, 100 MHz): δ = 15.0, 24.2, 26.1, 29.0, 46.4, 53.9, 56.0, 60.3, 107.8, 113.4, 126.0 (2C), 126.4, 129.0 (2C), 130.3, 132.4, 132.8, 135.2, 148.7, 160.9, 166.6.
HRMS (ESI) calcd for C22H26ClN4O2S [M + H]+ 445.1460, found 445.1453.
15 (15a + 15b)
Compound 15 was initially achieved in very low yields (1–3%) in three steps and according to Scheme S2; however, by the use of the Boc derivative of 12. This synthetic route was abandoned due to the reproducibility problem, time of execution, low yield, and the difficulties in separating the title compound from the reaction mixture. Mp = 200–210 °C.
1H NMR (CDCl3, 400 MHz): δ = 1.23–1.50 (m, 8 H), 1.54–1.74 (m, 5 H), 1.76–1.85 (m, 2 H), 2.30 (s, 6 H), 2.54–2.67 (m, 2 H), 2.98–3.05 (m, 2 H), 3.05–3.14 (m, 1 H), 3.39 (d, J = 11.2 Hz, 1 H), 4.70 (s, 2 H), 4.71 (s, 2 H), 5.25–5.35 (m, 2H), 6.45 (d, J = 3.8 Hz, 1 H), 6.48 (d, J = 3.8 Hz, 1 H), 6.80 (d, J = 3.9 Hz, 1 H), 6.90 (d, J = 3.8 Hz, 1 H), 7.28 (d, J = 8.6 Hz, 2 H), 7.31 (d, J = 8.6 Hz, 2 H), 7.48 (d, J = 8.6 Hz, 2 H), 7.49 (d, J = 8.6 Hz, 2 H), 7.59 (br s, 1 H), 7.77 (br s, 1 H), 0.43 (br s, 2H).
13C NMR (CDCl3, 100 MHz): δ = 15.1, 24.3, 24.4, 25.5, 26.5, 29.0, 29.2, 46.6, 46.8, 54.0, 54.9, 56.3, 59.0, 60.3, 107.8, 107.9, 112.7, 113.0, 126.00 (2C), 126.03 (2C), 126.44, 126.48, 129.2 (2C), 130.3, 130.4, 132.0, 132.1, 133.0, 133.1, 135.0, 135.1, 149.1, 149.5, 160.8, 161.4, 166.7, 169.2.
HRMS (ESI) calcd for C22H26ClN4O2S [M + H]+ 445.1460, found 445.1453.
Chiral Separation of NBD-11021 by High Pressure Liquid Chromatography (HPLC)
The diastereomer, NBD-11021 (220 mg), was resolved to individual isomers using chiral supercritical fluid chromatography (SFC). Two chromatographic methods were required to isolate the four isomers to diasteromeric excess (de) > 97%. Method 1 resolved fractions 3 and 4 at high purity from NBD-11021A and NBD-11021B, which eluted as a single peak. A second method, Method 2, was used to resolve the mixture of fractions 1 and 2.
Method 1 used a 3.0 cm × 25.0 cm ChiralPak AD-H (Chiral Technologies, West Chester PA) column with a mobile phase consisting of 50% CO2 and 50% (isopropanol with 1.0% v/v isopropylamine) at a combined flow rate of 80 mL/min, 100 bar of system backpressure, and 25 °C. Method 2 used a 3.0 cm × 25.0 cm (S,S) Whelk-O1 column (Regis Technologies, Morton Grove IL) with a mobile phase consisting of 60% CO2 and 40% (isopropanol with 1.0% v/v isopropylamine) at a combined flow rate of 80 mL/min, 100 bar of system backpressure, and 25 °C. Fractions were collected as solutions in the respective liquid cosolvents, evaporated to dryness at ambient temperature on a rotary evaporator, and redissolved and dried from acetonitrile three times to remove residual isopropylamine.
Fractions recovered were weighed and analyzed for diastereomeric excess. Recoveries and de values were as follows: fraction 1 (34 mg, 98.4% de), fraction 2 (81 mg, 98.0% de), fraction 3 (76 mg, 97.1% de), and fraction 4 (52 mg, 99.3% de).
Assignment of Absolute Configuration of NBD-11021 Diastereomers
Absolute configuration of NBD-11021A1, NBD-11021A2, NBD-11021B1, and NBD-11021B2 fractions were determined by vibrational circular dichroism (VCD) by BioTools, Inc.
Experimental Procedure for Assignment of Absolute Configuration
NBD-11021A1 and NBD-11021A2 were dissolved in CDCl3 (10 mg/0.2 mL) and placed in a 100 μm path length cell with BaF2 windows. IR and VCD spectra were recorded on a ChiralIR VCD spectrometer (BioTools, Inc.) equipped with DualPEM accessory, with 4 cm−1 resolution, 6 h collection for both sample and solvent, and instrument optimized at 1400 cm−1. The solvent-subtracted IR and enantiomer subtracted VCD spectra are shown in Figure S3.
NBD-11021B1 and NBD-11021B2 were dissolved in CDCl3 (10 mg/0.2 mL) and placed in a 100 μm path length cell with BaF2 windows. IR and VCD spectra were recorded on a ChiralIR VCD spectrometer (BioTools, Inc. Jupiter, FL) equipped with DualPEM accessory, with 4 cm−1 resolution, 6-h collection for both sample and solvent, and instrument optimized at 1400 cm−1. The solvent-subtracted IR and enantiomer subtracted VCD spectra are shown in Figure S4.
Theoretical Calculations for Assignment of Absolute Configuration
The (16R,25S) and (16S,25S) configurations were built with ComputeVOA program (BioTools, Inc., Jupiter, FL). A conformational search was carried out with ComputeVOA for the entire structure at the molecular mechanics level. Geometry, frequency, and IR and VCD intensity calculations were carried out at the DFT level (B3LYP functional/6-31G(d) basis set) with Gaussian 09 (Gaussian Inc., Wallingford, CT). The calculated frequencies were scaled by 0.96, and the IR and VCD intensities were converted to Lorentzian bands with 6 cm−1 half-width for comparison to experiment.
Gaussian calculations of the conformers resulted in eight (16R,25S) and eight (16S,25S) conformers that have energies within 1.5 kcal/mol from the lowest-energy conformer. The comparison of the observed VCD and IR spectra with those of the calculated lowest-energy conformers are shown in Figures S5–8. Based on the overall agreement between VCD pattern (especially between 1300 and 1400 cm−1) for the observed and the Botlzmann sum of the calculated spectra of both (16R,25S) and (16S,25S) configurations, the absolute configuration of NBD-11021A1 is assigned as (16S,25R), that of NBD-11021A2 as (16R,25S), that of NBD-11021B1 as (16S,25S) and that of NBD-11021B2 as (16R,25R) (Figure 1a and Figure S9). The assignment was evaluated by CompareVOA program, and the confidence level of the assignment is 100% based on current database that includes 106 previous correct assignments for different chiral structures.
Surface Plasmon Resonance (SPR) by BIAcore
To probe whether NBD-11021 induces a mAb 17b-recongnized conformation, we designed mAb 17b-captured BIAcore experiments. NBD-556, NBD-09027, and sCD4 were used as positive controls. The final concentration of YU2 core protein was fixed at 62.5 nM in HBS buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA, and 0.005% surfactant P20) containing 3% DMSO (v/v). The buffer, sCD4, or compounds were added into the core proteins and incubated at room temperature for 1 h before passing over the chip. sCD4 or compounds were used at 15-fold or 50-fold excess, respectively, to the core protein. Anti-human IgG was immobilized on the CM5 chip to capture IgG 17b to 300 response units (RU). Then, the core protein alone or the core protein in the presence of sCD4 or compounds was passed over the 17b-captured surface. The association time was 2 min and the dissociation time was 5 min. MgCl2 (3 M) was used to regenerate the surface via two pulse injections (25 s per injection) at 60 μL/min. We analyzed the data by using BiaEvaluation software (BIAcore).
In addition, we designed a competitive BIAcore to probe whether NBD-11021 mimics one of the broadly neutralizing antibodies (bNAbs), VRC-PG04, that binds to the CD4-binding site (CD4BS) in gp120 and functions as antagonist. HIV-1 YU2 core gp120 was immobilized on the CM5 chip to 6000 RU. Then, 60 nM of VRC-PG04 or sCD4 protein alone or in the presence of NBD-11021 at varied concentrations (200, 100, 50, 25, and 12.5 μM) was passed over gp120-immobilized surface. The association time was 2 min, and the dissociation time was 5 min. We used 6.25 mM NaOH to regenerate the surface via two pulse injections (25 s per injection) at 60 μL/min. We used the BiaEvaluation software (BIAcore) to analyze the data.
Pseudovirus Preparation
Pseudoviruses capable of single cycle infection were prepared as previously described.19,20 Briefly, 5 × 106 HEK 293T cells were plated in a T75 flask 24 h before transfection. Cells were transfected in 15 mL of medium with a mixture of 10 μg of an env-deleted pro-viral backbone plasmid pNL4-3.Luc.R-.E- DNA or pSG3Δenv DNA and 10 μg of an Env expression vector using FuGENE 6 (Roche). Amphotropic MLV (A-MLV) and VSV-G pseudoviruses were prepared by transfecting 293T cells with a mixture of 20.25 μg of the Env-deleted proviral backbone plasmid pNL4-3KFS DNA and 2.25 μg of the respective Env expression vectors by using Lipofectamin 2000 reagent (Life Technologies). Pseudovirus-containing supernatants were collected 2 days after transfection, filtered, and stored in aliquots at −80 °C. Pseudoviruses were titered to calculate the 50% tissue culture infectious dose (TCID50) by infecting different cell types. Cells were plated in 96 well plates 24 h before infection. On the day of the infection, 100 μL of serial 2-fold dilutions of pseudoviruses was added to the cells. After 3 days incubation, the cells were washed 2 times with PBS and lysed with 50 μL of cell culture lysis reagent (Promega). Lysates were transferred to a white 96 well plate (Costar) and mixed with 100 μL of luciferase assay reagent (Luciferase Assay System, Promega). The luciferase activity was immediately measured with a Tecan infinite M1000 reader (Tecan US). Wells producing relative luminescence units (RLU) 4 times the background were scored as positive, and the TCID50 was calculated by the Spearman–Karber statistical method.
Infectivity Assay against CD4-Dependent and CD4-Independent Viruses
CD4-expressing cells Cf2Th/CD4-CCR5 and CD4-negative cells Cf2Th-CCR5 were seeded at 6 × 103 cells/well in a 96 well tissue culture plate and cultured at 37 °C overnight. Cf2Th/CD4-CCR5 cells were infected with the HIV-1 expressing luciferase and the ENV glycoprotein from HIV-1ADA (CCR5-tropic). Cf2Th-CCR5 cells were infected with CD4-independent HIV-1ADAN197S expressing luciferase and HIV-1ADA as previously reported.18 Briefly, 50 μL of a test compound at graded concentrations was mixed with an equal volume of the respective recombinant virus. After incubation at 37 °C for 30 min, the mixtures were added to the cells and incubated at 37 °C for 48 h. Cells were washed 2 times with PBS and lysed with 40 μL of cell culture lysis reagent. Lysates were transferred to a white 96 well plate and mixed with 100 μL of luciferase assay reagent. The luciferase activity was immediately measured as described above to obtain the percent inhibition with respect to the control. The IC50 values were calculated using the GraphPad Prism software (GraphPad).
Measurement of Antiviral Activity
Multicycle Infection Assay in MT-2 Cells
The inhibitory activity of test compounds on infection by laboratory-adapted HIV-1 strains was determined as previously described.73 Briefly, 1 × 104 MT-2 cells were infected with HIV-1IIIB and other lab-adapted HIV-1 at 100 TCID50 (0.01 MOI) in the presence or absence of test compounds at graded concentrations overnight. The culture supernatants were then removed, and fresh media were added. On the fourth day postinfection, 100 μL of culture supernatants was collected from each well, mixed with equal volume of 5% Triton X-100, and tested for p24 antigen by “sandwich” enzyme-linked immunosorbent assay (ELISA).
Multicycle Infection Assay in PBMC
The inhibitory activity of test compounds on infection by primary HIV-1 isolates was determined as previously described.73 PBMCs were isolated from the blood of healthy donors at the New York Blood Center by standard density gradient centrifugation using Histopaque-1077 (Sigma-Aldrich). The cells were cultured at 37 °C for 2 h. Nonadherent cells were collected and cultured at 5 × 106 cells/mL in RPMI-1640 medium containing 10% FBS, 5 μg/mL phytohemagglutinin (PHA), and 100 U/mL interleukine 2 (IL-2; Sigma-Aldrich), followed by incubation at 37 °C for 3 days. The PHA-stimulated cells (5 × 104 cells/well) were infected with lab-adapted and primary HIV-1 isolates at 500 TCID50 (0.01 MOI) in the absence or presence of inhibitors at graded concentrations. Culture media were replaced every 3 days with fresh media. The supernatants were collected 7 days postinfection and tested for p24 antigen by ELISA. The percent inhibition of p24 production and IC50 values were calculated by the GraphPad Prism software.
Single-Cycle Infection Assay in TZM-bl Cells
The inhibitory activity of test compounds was measured on HIV-1 pseudotyped viruses expressing HIV-1HXB-2 ENV or ENV from the panel of standard reference subtypes A, A/D, A2/D, A/E, A/G, B, C, D, and D/A. Pseudoviruses were obtained by transfecting HEK 293T cells with a mixture of an Env-deleted backbone proviral plasmid pSG3Δenv and an Env expression vector DNA. Briefly, 100 μL of TZM-bl cells at 1 × 105 cells/mL was added to the wells of a 96 well tissue culture plate and cultured at 37 °C overnight. Test compound (50 μL) at graded concentrations was mixed with 50 μL of the HIV-1 pseudovirus. After incubation at 37 °C for 30 min, the mixture was added to the cells and incubated at 37 °C for 3 days. Cells were washed 2 times with PBS and lysed with 50 μL of cell culture lysis reagent. Lysates (20 μL) were transferred to a white 96 well plate and mixed with 100 μL of luciferase assay reagent. The luciferase activity was immediately measured with a Tecan infinite M1000 reader, and the percent inhibition by the compounds and IC50 values were calculated using the GraphPad Prism software.
Single-Cycle Infection Assay in U87-CD4-CXCR4 and U87-CD4-CCR5 cells
U87-CD4-CCR5 and U87-CD4-CXCR4 cells were seeded in 96 well plates at 104 cells/well and incubated overnight at 37 °C. Pseudoviruses expressing luciferase HIV-1ADA (CCR5-tropic) and HIV-1HXB2 (CXCR4-tropic) were obtained by transfecting the HEK 293T cells with a mixture of pNL4-3.Luc.R-.E- DNA and Env expression vector pSVIIIenv-ADA or Env expression vector pHXB2-env (X4) DNA. Briefly, pseudoviruses were preincubated with varying doses of test compounds for 30 min. The mixtures were then added to the respective cells and incubated for 3 days. Cells were washed 2 times with PBS and lysed with 40 μL of cell culture lysis reagent. Lysates were transferred to a white 96 well plate and mixed with 100 μL of luciferase assay reagent. The luciferase activity was immediately measured as described above to calculate IC50.
Cell-Associated CD4-Dependent Transmission of CCR5- and CXCR4-tropic HIV-1
The CD4-dependent cell-associated HIV-1 transmission inhibition assays were performed as previously described74,75 with some minor modifications. Briefly, target GHOST (3) X4/R5 cells were plated at 104/well in a 96 well plate 24 h before the experiment. For the CXCR4-tropic assay, we used chronically infected H9/HIV-1IIIB at 2 × 103 cells/well, and for the CCR5-tropic assay, we used MOLT-4/CCR5 cells chronically infected with HIV-1ADA at 5 × 104 cells/well as transmitting cells. The cells were treated with 200 μg/mL mitomycin C (Sigma-Aldrich) for 1 h at 37 °C, washed with PBS, and incubated with the target cells and drugs for 4 h. Medium was removed, and the target cells were washed three times with PBS; 100 μL/well of GHOST cells medium was added to the cells to continue the incubation for 16 h. Medium was removed, and cells were washed with PBS and lysed with 1% Triton X-100. Intracellular p24 contents were determined by sandwich ELISA.
Cell-to-Cell Fusion
To assess the ability of NBD compounds to block cell-to-cell fusion mediated by HIV-1, we performed cell fusion assay as previously described74,76,77 with some modifications. We used MAGI-CCR5 cells, a HeLa cell clone expressing human CD4, both coreceptors CXCR4 and CCR5, and HIV-LTR-β-gal,77,78 as target cells and HL 2/3 cells, a HeLa-derived cell line that expresses HIV-1HXB2 Env on the surface and Tat, Gag, Rev, and Nef proteins in the cytoplasm and does not produce detectable amounts of mature virions,79 as effector cells. Following fusion of the two cell lines, Tat induces the expression of the β-gal enzyme. Briefly, following preincubation of 1.5 × 104/well MAGI-CCR5 cells for 1 h with escalating concentrations of NBD compounds, 7.5 × 103/well HL 2/3 cells were added to the culture and incubated for 24 h at 37 °C. β-gal expression was quantified with the Beta-Glo assay system (Promega) following the manufacturer’s instructions. The percent inhibition and the IC50 values were calculated using the GraphPad Prism software.
In Vitro Biochemical HIV RT and Integrase Assay
Purified recombinant HIV (pNL4-3) heterodimeric (p66/p51) reverse transcriptase (RT) was purchased from a commercially available source. The assay was performed in 96-well filter plate, where RT activity was determined by the incorporation of radiolabeled deoxyribonucleotides into the newly synthesized DNA strand. The standard RT reaction mixture contains in vitro transcribed viral RNA derived from the HIV-1NL4-3 5′-LTR region (position 454 to 652) and primer that is complementary to the primer binding site (PBS, nucleotide residues nucleotides 636 to 652), radiolabeled deoxyribonucleotide, dNTPs, and reverse transcriptase. Briefly, the reaction was carried out in a volume of 50 μL containing 50 mM Tris HCl, pH 7.8, 50 mM KCl, 5 mM MgCl2, 1 mM DTT, 50 μM each of dATP, dCTP, and dGTP, 50 nM dTTP, 1 μCi of [3H] dTTP (70–90 Ci/mM), and 5 nM template/primer. The reaction was initiated by the addition of 10 nM RT.
Compounds were diluted in 100% DMSO to 40 mM. Each compound was diluted in the appropriate reaction buffer for the biochemical assay per protocol. Serially diluted test compounds were added to the reaction followed by the addition of RT. The reaction mixture was incubated at 37 °C for 1 h and then quenched by the addition of ice-cold trichloroacetic acid (TCA) to a final concentration of 10%. The plate was incubated at 4 °C for 1 h to precipitate the synthesized DNA, then rinsed 3-times with 10% TCA and 1 time with 70% ethanol. After addition of 25 μL of scintillation fluid to completely dried wells, radioactivity was counted by MicroBeta scintillation counter (PerkinElmer). The reduction of radioactivity represents the potency of compound inhibition.
HIV-1 integrase assay was performed as per the protocol in the HIV-1 Integrase assay kit from ExpressBio (Thurnmont, MD).
Determination of Cytotoxicity
MT-2 Cells
Cytotoxicity of test compounds in MT-2 cells was measured by a colorimetric method using XTT [(sodium 3′-(1-(phenylamino)-carbonyl)-3,4-tetrazoliumbis(4-methoxy-6-nitro) bezenesulfonic acid hydrate)] (PolySciences) as previously described.80 Briefly, 100 μL of a test compound at graded concentrations was added to an equal volume of cells (1 × 105 cells/mL) in 96 well plates followed by incubation at 37 °C for 4 days, which ran parallel to the neutralization assay in MT-2. Following the addition of XTT, the soluble intracellular formazan was quantitated colorimetrically at 450 nm 4 h later. The percent of cytotoxicity and the CC50 (the concentration for 50% cytotoxicity) values were calculated by the GraphPad Prism software.
TZM-bl and U87 Cells
The cytotoxicity of test compounds in these cells was also measured by the XTT method described previously.80 Briefly, 100 μL of a compound at graded concentrations was added to equal volume of cells (105/mL) in wells of 96 well plates followed by incubation at 37 °C for 3 days and addition of XTT. The soluble intracellular formazan was quantitated colorimetrically at 450 nm 4 h later. The percent of cytotoxicity and the CC50 values were calculated as above.
PBMCs
For the PBMC toxicity assay, we used 5 × 105 cells/m,L and the cytotoxicity of the compounds was measured after 7 days of incubation as previously reported.81 Following the addition of XTT, the soluble intracellular formazan was quantitated colorimetrically at 450 nm 4 h later. The percent of cytotoxicity and the CC50 values were calculated as above.
Crystallization, Structure Determination, And Refinement
Clade A/E93TH057 HIV-1 gp120 H375S core was expressed and purified as described.27 The protein was then concentrated to ~10 mg/mL in buffer containing 2.5 mM Tris-HCl, pH 7.5, 350 mM NaCl, and 0.02% NaN3. The gp120 core crystals were grown by mixing 0.5 μL of protein and 0.5 μL of reservoir solution containing 10% PEG 8000, 5% isopropanol, and 0.1 M HEPES, pH 7.5, by the hanging-drop vapor-diffusion method. Crystals were soaked in a stabilizing solution containing 1 mM NBD-11021, 10% DMSO, 10% PEG 8000, 5% isopropanol, and 0.1 M HEPES, pH 7.5, for 1 h, transferred to cryoprotectant solution containing 30% ethylene glycol, 12% PEG 8000, 5% isopropanol, and 0.1 M HEPES, pH 7.5, and flash-frozen in liquid nitrogen. X-ray diffraction data were collected from a single crystal of NBD-11021-bound HIV-1 gp120 core H375S on beamline 22-ID of SER-CAT at the APS. Data were integrated and scaled with HKL2000.82 The structure was solved by molecular replacement using unliganded clade A/E93TH057 HIV-1 gp120 core structure (PDB ID 3TGT) as a search model with AutoMR in the PHENIX software suite.83 We used COOT84 to manually fit NBD-11021 into the Phe43 cavity of gp120 and refined the structure with PHENIX. Data collection and refinement statistics were summarized in Table S1. Figures were generated with PyMOL (The PyMOL Molecular Graphics System, Version 2.0, Schrödinger, LLC).
Data Analysis
We performed statistical analysis to compare the NBD compounds with the control NBD-556. For adjustment of multiple comparisons, we implemented one-way ANOVA with Dunnett’s test, with significance level set at 0.05. We conducted all analyses using GraphPad Prism 6.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by funds from NIH Grant RO1 AI104416 (A.K.D.) and the New York Blood Center (A.K.D.), by Intramural funding to the Vaccine Research Center, and by the Intramural AIDS–Targeted Antiretroviral Program (IATAP) of the NIH. Use of sector 22 (Southeast Region Collaborative Access Team) at the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science Contract W-31-109-Eng-38. We thank Nicole Doria-Rose and John Mascola of Vaccine Research Center, NIH, for guidance and consultation. We thank Vijay Nandi of the New York Blood Center for her assistance in statistical analyses. We thank Carol Lackman-Smith, Junzhong Peng, and Jiayi Wei of Southern Research Institute (SR) for performing the HIV RT and integrase assays.
ABBREVIATIONS USED
- HIV-1
human immunodeficiency virus type-1
- AIDS
acquire immune deficiency syndrome
- CD4BS
CD4 binding site
- ENV
envelope
- A-MLV
amphotropic murine leukemia virus
- LCMS
liquid chromatography mass spectrometry
- NMR
nuclear magnetic resonance
- BOC
tert-butyloxycarbonyl
- Cbz
carboxybenzyl
- Alloc
allyloxycarbonyl
- HRMS
high resolution mass spectrometry
- SPR
surface plasmon resonance
- SFC
supercritical fluid chromatography
- VCD
vibrational circular dichroism
Footnotes
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b00709.
Dose–response plots of selected inhibition of NBD compounds against ENV-pseudotyped HIV-1, comparison of IC50 of fractions of NBD-11021A2 and NBD-11021B2 versus NBD-11021 tested on a large panel of ENV-pseudotyped HIV-1; IR and VCD spectra of NBD-11021 diastereomers, assignment of absolute configuration, putative binding mode of NBD-11021A2 and NBD-11021B2, crystallographic data collection and refinement statistics, and antiviral activities of NBD-compounds against SIV, SHIV and HIV-2 (PDF) Molecular formula strings for compounds NBD-556, NBD-09027, NBD11021, NBD-11021A2, and NBD-11021B2 (CSV)
Accession Codes
X-ray crystallographic structure of NBD-11021 with HIV-1 clade A/E 93TH057 H375S gp120 coree (PDB ID 4RZ8) has been deposited at RCSB PDB (http://www.pdb.org).
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Stevenson M. HIV-1 pathogenesis. Nat. Med. 2003;9:853–860. doi: 10.1038/nm0703-853. [DOI] [PubMed] [Google Scholar]
- (2).Bour S, Geleziunas R, Wainberg MA. The human immunodeficiency virus type 1 (HIV-1) CD4 receptor and its central role in promotion of HIV-1 infection. Microbiol. Rev. 1995;59:63–93. doi: 10.1128/mr.59.1.63-93.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Dalgleish AG, Beverly PCL, Clapham PR, Crawford DH, Greaves MF, Weiss RA. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature. 1984;312:763–767. doi: 10.1038/312763a0. [DOI] [PubMed] [Google Scholar]
- (4).Sattentau QJ, Weiss RA. The CD4 antigen: physiological ligand and HIV receptor. Cell. 1988;52:631–633. doi: 10.1016/0092-8674(88)90397-2. [DOI] [PubMed] [Google Scholar]
- (5).Ray N, Doms RW. HIV-1 coreceptors and their inhibitors. Curr. Top. Microbiol. Immunol. 2006;303:97–120. doi: 10.1007/978-3-540-33397-5_5. [DOI] [PubMed] [Google Scholar]
- (6).Moore JP, Trkola A, Dragic T. Co-receptors for HIV-1 entry. Curr. Opin. Immunol. 1997;9:551–562. doi: 10.1016/s0952-7915(97)80110-0. [DOI] [PubMed] [Google Scholar]
- (7).Sattentau QJ, Moore JP. The role of CD4 in HIV binding and entry. Philos. Trans. R. Soc., B. 1993;342:59–66. doi: 10.1098/rstb.1993.0136. [DOI] [PubMed] [Google Scholar]
- (8).Este JA, Telenti A. HIV entry inhibitors. Lancet. 2007;370:81–88. doi: 10.1016/S0140-6736(07)61052-6. [DOI] [PubMed] [Google Scholar]
- (9).Sodroski JG. HIV-1 entry inhibitors in the side pocket. Cell. 1999;99:243–246. doi: 10.1016/s0092-8674(00)81655-4. [DOI] [PubMed] [Google Scholar]
- (10).Chan DC, Kim PS. HIV entry and its inhibition. Cell. 1998;93:681–684. doi: 10.1016/s0092-8674(00)81430-0. [DOI] [PubMed] [Google Scholar]
- (11).Debnath AK. Rational design of HIV-1 entry inhibitors. Methods Mol. Biol. 2013;993:185–204. doi: 10.1007/978-1-62703-342-8_13. [DOI] [PubMed] [Google Scholar]
- (12).Zhao Q, Ma L, Jiang S, Lu H, Liu S, He Y, Strick N, Neamati N, Debnath AK. Identification of N-phenyl-N’-(2,2,6,6-tetramethyl-piperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology. 2005;339:213–225. doi: 10.1016/j.virol.2005.06.008. [DOI] [PubMed] [Google Scholar]
- (13).Schon A, Madani N, Klein JC, Hubicki A, Ng D, Yang X, Smith AB, III, Sodroski J, Freire E. Thermodynamics of binding of a low-molecular-weight CD4 mimetic to HIV-1 gp120. Biochemistry. 2006;45:10973–10980. doi: 10.1021/bi061193r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lalonde JM, Elban MA, Courter JR, Sugawara A, Soeta T, Madani N, Princiotto AM, Kwon YD, Kwong PD, Schon A, Freire E, Sodroski J, Smith AB., III Design, synthesis and biological evaluation of small molecule inhibitors of CD4-gp120 binding based on virtual screening. Bioorg. Med. Chem. 2011;19:91–101. doi: 10.1016/j.bmc.2010.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Lalonde JM, Kwon YD, Jones DM, Sun AW, Courter JR, Soeta T, Kobayashi T, Princiotto AM, Wu X, Schon A, Freire E, Kwong PD, Mascola JR, Sodroski J, Madani N, Smith AB., III Structure-Based Design, Synthesis, and Characterization of Dual Hotspot Small-Molecule HIV-1 Entry Inhibitors. J. Med. Chem. 2012;55:4382–4396. doi: 10.1021/jm300265j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lalonde JM, Le-Khac M, Jones DM, Courter JR, Park J, Schon A, Princiotto AM, Wu X, Mascola JR, Freire E, Sodroski J, Madani N, Hendrickson WA, Smith AB., III Structure-Based Design and Synthesis of an HIV-1 Entry Inhibitor Exploiting X-Ray and Thermodynamic Characterization. ACS Med. Chem. Lett. 2013;4:338–343. doi: 10.1021/ml300407y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Madani N, Schon A, Princiotto AM, Lalonde JM, Courter JR, Soeta T, Ng D, Wang L, Brower ET, Xiang SH, Kwon YD, Huang CC, Wyatt R, Kwong PD, Freire E, Smith AB, III, Sodroski J. Small-molecule CD4 mimics interact with a highly conserved pocket on HIV-1 gp120. Structure. 2008;16:1689–1701. doi: 10.1016/j.str.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Si Z, Madani N, Cox JM, Chruma JJ, Klein JC, Schon A, Phan N, Wang L, Biorn AC, Cocklin S, Chaiken I, Freire E, Smith AB, III, Sodroski JG. Small-molecule inhibitors of HIV-1 entry block receptor-induced conformational changes in the viral envelope glycoproteins. Proc. Natl. Acad. Sci. U. S. A. 2004;101:5036–5041. doi: 10.1073/pnas.0307953101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Curreli F, Kwon YD, Zhang H, Yang Y, Scacalossi D, Kwong PD, Debnath AK. Binding mode characterization of NBD series CD4-mimetic HIV-1 entry inhibitors by X-ray structure and resistance study. Antimicrob. Agents Chemother. 2014;58:5478–5491. doi: 10.1128/AAC.03339-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Curreli F, Choudhury S, Pyatkin I, Zagorodnikov VP, Bulay AK, Altieri A, Kwon YD, Kwong PD, Debnath AK. Design, synthesis and antiviral activity of entry inhibitors that target the CD4-binding site of HIV-1. J. Med. Chem. 2012;55:4764–4775. doi: 10.1021/jm3002247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Narumi T, Ochiai C, Yoshimura K, Harada S, Tanaka T, Nomura W, Arai H, Ozaki T, Ohashi N, Matsushita S, Tamamura H. CD4 mimics targeting the HIV entry mechanism and their hybrid molecules with a CXCR4 antagonist. Bioorg. Med. Chem. Lett. 2010;20:5853–5858. doi: 10.1016/j.bmcl.2010.07.106. [DOI] [PubMed] [Google Scholar]
- (22).Narumi T, Arai H, Yoshimura K, Harada S, Nomura W, Matsushita S, Tamamura H. Small molecular CD4 mimics as HIV entry inhibitors. Bioorg. Med. Chem. 2011;19:6735–6742. doi: 10.1016/j.bmc.2011.09.045. [DOI] [PubMed] [Google Scholar]
- (23).Narumi T, Arai H, Yoshimura K, Harada S, Hirota Y, Ohashi N, Hashimoto C, Nomura W, Matsushita S, Tamamura H. CD4 mimics as HIV entry inhibitors: lead optimization studies of the aromatic substituents. Bioorg. Med. Chem. 2013;21:2518–2526. doi: 10.1016/j.bmc.2013.02.041. [DOI] [PubMed] [Google Scholar]
- (24).Yamada Y, Ochiai C, Yoshimura K, Tanaka T, Ohashi N, Narumi T, Nomura W, Harada S, Matsushita S, Tamamura H. CD4 mimics targeting the mechanism of HIV entry. Bioorg. Med. Chem. Lett. 2010;20:354–358. doi: 10.1016/j.bmcl.2009.10.098. [DOI] [PubMed] [Google Scholar]
- (25).Hashimoto C, Narumi T, Otsuki H, Hirota Y, Arai H, Yoshimura K, Harada S, Ohashi N, Nomura W, Miura T, Igarashi T, Matsushita S, Tamamura H. A CD4 mimic as an HIV entry inhibitor: Pharmacokinetics. Bioorg. Med. Chem. 2013;21:7884–7889. doi: 10.1016/j.bmc.2013.10.005. [DOI] [PubMed] [Google Scholar]
- (26).Yoshimura K, Harada S, Shibata J, Hatada M, Yamada Y, Ochiai C, Tamamura H, Matsushita S. Enhanced exposure of human immunodeficiency virus type 1 primary isolate neutralization epitopes through binding of CD4 mimetic compounds. J. Virol. 2010;84:7558–7568. doi: 10.1128/JVI.00227-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kwon YD, Finzi A, Wu X, Dogo-Isonagie C, Lee LK, Moore LR, Schmidt SD, Stuckey J, Yang Y, Zhou T, Zhu J, Vicic DA, Debnath AK, Shapiro L, Bewley CA, Mascola JR, Sodroski JG, Kwong PD. Unliganded HIV-1 gp120 Core Structures Assume the CD4-Bound Conformation with Regulation by Quaternary Interactions and Variable Loops. Proc. Natl. Acad. Sci. U. S. A. 2012;109:5663–5668. doi: 10.1073/pnas.1112391109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kwon YD, Lalonde JM, Yang Y, Elban MA, Sugawara A, Courter JR, Jones DM, Smith AB, III, Debnath AK, Kwong PD. Crystal structures of HIV-1 gp120 envelope glycoprotein in complex with NBD analogues that target the CD4-binding site. PLoS One. 2014;9:e85940. doi: 10.1371/journal.pone.0085940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Falkowska E, Ramos A, Feng Y, Zhou T, Moquin S, Walker LM, Wu X, Seaman MS, Wrin T, Kwong PD, Wyatt RT, Mascola JR, Poignard P, Burton DR. PGV04, an HIV-1 gp120 CD4 Binding Site Antibody, Is Broad and Potent in Neutralization but Does Not Induce Conformational Changes Characteristic of CD4. J. Virol. 2012;86:4394–4403. doi: 10.1128/JVI.06973-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Gach JS, Quendler H, Tong T, Narayan KM, Du SX, Whalen RG, Binley JM, Forthal DN, Poignard P, Zwick MB. A human antibody to the CD4 binding site of gp120 capable of highly potent but sporadic cross clade neutralization of primary HIV-1. PLoS One. 2013;8:e72054. doi: 10.1371/journal.pone.0072054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Trofimov BA, Atavib AS, Mikhaleva AI, Kalabin GA, Chebotreva EG. Cyclohexanone oxime condensation with acetylene. Zh. Org. Khim. 1973;10:2205–2206. [Google Scholar]
- (32).Robak MT, Herbage MA. Synthesis and Applications of tert-Butanesulfinamide. Chem. Rev. 2010;110:3600–3740. doi: 10.1021/cr900382t. [DOI] [PubMed] [Google Scholar]
- (33).Garro-Helion F, Merzouk A, Guibe F. Mild and selective palladium(0)-catalyzed deallylation of allylic amines. Allylamine and diallylamine as very convenient ammonia equivalents for the synthesis of primary amines. J. Org. Chem. 1993;58:6109–6113. [Google Scholar]
- (34).Trkola A, Dragic T, Arthos J, Binley JM, Olson WC, Allaway GP, Cheng-Mayer C, Robinson J, Maddon PJ, Moore JP. CD4-dependent, antibody-sensitive interactions between HIV-1 and its co-receptor CCR-5. Nature. 1996;384:184–187. doi: 10.1038/384184a0. [DOI] [PubMed] [Google Scholar]
- (35).Doms RW, Moore JP. HIV-1 membrane fusion: targets of opportunity. J. Cell Biol. 2000;151:F9–14. doi: 10.1083/jcb.151.2.f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Mariani SA, Vicenzi E, Poli G. Asymmetric HIV-1 co-receptor use and replication in CD4(+) T lymphocytes. J. Transl. Med. 2011;9(Suppl1):S8. doi: 10.1186/1479-5876-9-S1-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Madani N, Perdigoto AL, Srinivasan K, Cox JM, Chruma JJ, LaLonde J, Head M, Smith AB, III, Sodroski JG. Localized changes in the gp120 envelope glycoprotein confer resistance to human immunodeficiency virus entry inhibitors BMS-806 and #155. J. Virol. 2004;78:3742–3752. doi: 10.1128/JVI.78.7.3742-3752.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Lyumkis D, Julien JP, de VN, Cupo A, Potter CS, Klasse PJ, Burton DR, Sanders RW, Moore JP, Carragher B, Wilson IA, Ward AB. Cryo-EM structure of a fully glycosylated soluble cleaved HIV-1 envelope trimer. Science. 2013;342:1484–1490. doi: 10.1126/science.1245627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, Sun C, Grayson T, Wang S, Li H, Wei X, Jiang C, Kirchherr JL, Gao F, Anderson JA, Ping LH, Swanstrom R, Tomaras GD, Blattner WA, Goepfert PA, Kilby JM, Saag MS, Delwart EL, Busch MP, Cohen MS, Montefiori DC, Haynes BF, Gaschen B, Athreya GS, Lee HY, Wood N, Seoighe C, Perelson AS, Bhattacharya T, Korber BT, Hahn BH, Shaw GM. Proc. Natl. Acad. Sci. U. S. A. 2008;105:7552–7557. doi: 10.1073/pnas.0802203105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Rasmussen I, Vilhardt F. Macropinocytosis is the entry mechanism of amphotropic murine leukemia virus. J. Virol. 2015;89:1851–1866. doi: 10.1128/JVI.02343-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Daecke J, Fackler OT, Dittmar MT, Krausslich HG. Involvement of clathrin-mediated endocytosis in human immunodeficiency virus type 1 entry. J. Virol. 2005;79:1581–1594. doi: 10.1128/JVI.79.3.1581-1594.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Miyauchi K, Kim Y, Latinovic O, Morozov V, Melikyan GB. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell. 2009;137:433–444. doi: 10.1016/j.cell.2009.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Marechal V, Prevost MC, Petit C, Perret E, Heard JM, Schwartz O. Human immunodeficiency virus type 1 entry into macrophages mediated by macropinocytosis. J. Virol. 2001;75:11166–11177. doi: 10.1128/JVI.75.22.11166-11177.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Herschhorn A, Finzi A, Jones DM, Courter JR, Sugawara A, Smith AB, III, Sodroski JG. An inducible cell-cell fusion system with integrated ability to measure the efficiency and specificity of HIV-1 entry inhibitors. PLoS One. 2011;6:e26731. doi: 10.1371/journal.pone.0026731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Abela IA, Berlinger L, Schanz M, Reynell L, Gunthard HF, Rusert P, Trkola A. Cell-Cell Transmission Enables HIV-1 to Evade Inhibition by Potent CD4bs Directed Antibodies. PLoS Pathog. 2012;8:e1002634. doi: 10.1371/journal.ppat.1002634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Agosto LM, Zhong P, Munro J, Mothes W. Highly active antiretroviral therapies are effective against HIV-1 cell-to-cell transmission. PLoS Pathog. 2014;10:e1003982. doi: 10.1371/journal.ppat.1003982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Courter JR, Madani N, Sodroski J, Schon A, Freire E, Kwong PD, Hendrickson WA, Chaiken IM, Lalonde JM, Smith AB., III Structure-based design, synthesis and validation of CD4-mimetic small molecule inhibitors of HIV-1 entry: conversion of a viral entry agonist to an antagonist. Acc. Chem. Res. 2014;47:1228–1237. doi: 10.1021/ar4002735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Speck RF, Wehrly K, Platt EJ, Atchison RE, Charo IF, Kabat D, Chesebro B, Goldsmith MA. Selective employment of chemokine receptors as human immunodeficiency virus type 1 coreceptors determined by individual amino acids within the envelope V3 loop. J. Virol. 1997;71:7136–7139. doi: 10.1128/jvi.71.9.7136-7139.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, Wu L, Mackay CR, LaRosa G, Newman W, Gerard N, Gerard C, Sodroski J. The β-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell. 1996;85:1135–1148. doi: 10.1016/s0092-8674(00)81313-6. [DOI] [PubMed] [Google Scholar]
- (51).Suphaphiphat P, Thitithanyanont A, Paca-Uccaralertkun S, Essex M, Lee TH. Effect of amino acid substitution of the V3 and bridging sheet residues in human immunodeficiency virus type 1 subtype C gp120 on CCR5 utilization. J. Virol. 2003;77:3832–3837. doi: 10.1128/JVI.77.6.3832-3837.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Suphaphiphat P, Essex M, Lee TH. Mutations in the V3 stem versus the V3 crown and C4 region have different effects on the binding and fusion steps of human immunodeficiency virus type 1 gp120 interaction with the CCR5 coreceptor. Virology. 2007;360:182–190. doi: 10.1016/j.virol.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Fehér A, Boross P, Sperka T, Milkóssy G, Kádas J, Bagossi P, Oroszlan S, Weber IT, Tózsár J. Characterization of the murine leukemia virus protease and its comparison with the human immunodeficiency virus type 1 protease. J. Gen. Virol. 2006;87:1321–1330. doi: 10.1099/vir.0.81382-0. [DOI] [PubMed] [Google Scholar]
- (54).Cox BD, Prosser AR, Sun Y, Li Z, Lee S, Huang MB, Bond VC, Snyder JP, Krystal M, Wilson LJ, Liotta DC. Pyrazolo-Piperidines Exhibit Dual Inhibition of CCR5/CXCR4 HIV Entry and Reverse Transcriptase. ACS Med. Chem. Lett. 2015;6:753–757. doi: 10.1021/acsmedchemlett.5b00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol. 1998;72:2855–2864. doi: 10.1128/jvi.72.4.2855-2864.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Wei X, Decker JM, Wang S, Hui H, Kappes JC, Wu X, Salazar-Gonzalez JF, Salazar MG, Kilby JM, Saag MS, Komarova NL, Nowak MA, Hahn BH, Kwong PD, Shaw GM. Antibody neutralization and escape by HIV-1. Nature. 2003;422:307–312. doi: 10.1038/nature01470. [DOI] [PubMed] [Google Scholar]
- (57).Kolchinsky P, Mirzabekov T, Farzan M, Kiprilov E, Cayabyab M, Mooney LJ, Choe H, Sodroski J. Adaptation of a CCR5-using, primary human immunodeficiency virus type 1 isolate for CD4-independent replication. J. Virol. 1999;73:8120–8126. doi: 10.1128/jvi.73.10.8120-8126.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Bjorndal A, Deng H, Jansson M, Fiore JR, Colognesi C, Karlsson A, Albert J, Scarlatti G, Littman DR, Fenyo EM. Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. J. Virol. 1997;71:7478–7487. doi: 10.1128/jvi.71.10.7478-7487.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Morner A, Bjorndal A, Albert J, Kewalramani VN, Littman DR, Inoue R, Thorstensson R, Fenyo EM, Bjorling E. Primary human immunodeficiency virus type 2 (HIV-2) isolates, like HIV-1 isolates, frequently use CCR5 but show promiscuity in coreceptor usage. J. Virol. 1999;73:2343–2349. doi: 10.1128/jvi.73.3.2343-2349.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Page KA, Landau NR, Littman DR. Construction and use of a human immunodeficiency virus vector for analysis of virus infectivity. J. Virol. 1990;64:5270–5276. doi: 10.1128/jvi.64.11.5270-5276.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Blish CA, Jalalian-Lechak Z, Rainwater S, Nguyen MA, Dogan OC, Overbaugh J. Cross-subtype neutralization sensitivity despite monoclonal antibody resistance among early subtype A, C, and D envelope variants of human immunodeficiency virus type 1. J. Virol. 2009;83:7783–7788. doi: 10.1128/JVI.00673-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Long EM, Rainwater SM, Lavreys L, Mandaliya K, Overbaugh J. HIV type 1 variants transmitted to women in Kenya require the CCR5 coreceptor for entry, regardless of the genetic complexity of the infecting virus. AIDS Res. Hum. Retroviruses. 2002;18:567–576. doi: 10.1089/088922202753747914. [DOI] [PubMed] [Google Scholar]
- (63).Kulkarni SS, Lapedes A, Tang H, Gnanakaran S, Daniels MG, Zhang M, Bhattacharya T, Li M, Polonis VR, McCutchan FE, Morris L, Ellenberger D, Butera ST, Bollinger RC, Korber BT, Paranjape RS, Montefiori DC. Highly complex neutralization determinants on a monophyletic lineage of newly transmitted subtype C HIV-1 Env clones from India. Virology. 2009;385:505–520. doi: 10.1016/j.virol.2008.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Li M, Gao F, Mascola JR, Stamatatos L, Polonis VR, Koutsoukos M, Voss G, Goepfert P, Gilbert P, Greene KM, Bilska M, Kothe DL, Salazar-Gonzalez JF, Wei X, Decker JM, Hahn BH, Montefiori DC. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J. Virol. 2005;79:10108–10125. doi: 10.1128/JVI.79.16.10108-10125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 2002;46:1896–1905. doi: 10.1128/AAC.46.6.1896-1905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Derdeyn CA, Decker JM, Bibollet-Ruche F, Mokili JL, Muldoon M, Denham SA, Heil ML, Kasolo F, Musonda R, Hahn BH, Shaw GM, Korber BT, Allen S, Hunter E. Envelope-constrained neutralization-sensitive HIV-1 after heterosexual transmission. Science. 2004;303:2019–2022. doi: 10.1126/science.1093137. [DOI] [PubMed] [Google Scholar]
- (67).Li M, Salazar-Gonzalez JF, Derdeyn CA, Morris L, Williamson C, Robinson JE, Decker JM, Li Y, Salazar MG, Polonis VR, Mlisana K, Karim SA, Hong K, Greene KM, Bilska M, Zhou J, Allen S, Chomba E, Mulenga J, Vwalika C, Gao F, Zhang M, Korber BT, Hunter E, Hahn BH, Montefiori DC. Genetic and neutralization properties of subtype C human immunodeficiency virus type 1 molecular env clones from acute and early heterosexually acquired infections in Southern Africa. J. Virol. 2006;80:11776–11790. doi: 10.1128/JVI.01730-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Williamson C, Morris L, Maughan MF, Ping LH, Dryga SA, Thomas R, Reap EA, Cilliers T, van HJ, Pascual A, Ramjee G, Gray G, Johnston R, Karim SA, Swanstrom R. Characterization and selection of HIV-1 subtype C isolates for use in vaccine development. AIDS Res. Hum. Retroviruses. 2003;19:133–144. doi: 10.1089/088922203762688649. [DOI] [PubMed] [Google Scholar]
- (69).Wu X, Parast AB, Richardson BA, Nduati R, John-Stewart G, Mbori-Ngacha D, Rainwater SMJ, Overbaugh J. Neutralization Escape Variants of Human Immunodeficiency Virus Type 1 Are Transmitted from Mother to Infant. J. Virol. 2006;80:835–844. doi: 10.1128/JVI.80.2.835-844.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Connor RI, Chen BK, Choe S, Landau NR. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology. 1995;206:935–944. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- (71).He J, Choe S, Walker R, di MP, Morgan DO, Landau NR. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 1995;69:6705–6711. doi: 10.1128/jvi.69.11.6705-6711.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Si Z, Phan N, Kiprilov E, Sodroski J. Effects of HIV type 1 envelope glycoprotein proteolytic processing on antigenicity. AIDS Res. Hum. Retroviruses. 2003;19:217–226. doi: 10.1089/088922203763315722. [DOI] [PubMed] [Google Scholar]
- (73).Zhang H, Zhao Q, Bhattacharya S, Waheed AA, Tong X, Hong A, Heck S, Curreli F, Goger M, Cowburn D, Freed EO, Debnath AK. A cell-penetrating helical peptide as a potential HIV-1 inhibitor. J. Mol. Biol. 2008;378:565–580. doi: 10.1016/j.jmb.2008.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Lackman-Smith C, Osterling C, Luckenbaugh K, Mankowski M, Snyder B, Lewis G, Paull J, Profy A, Ptak RG, Buckheit RW, Jr., Watson KM, Cummins JE, Jr., Sanders-Beer BE. Development of a comprehensive human immunodeficiency virus type 1 screening algorithm for discovery and preclinical testing of topical microbicides. Antimicrob. Agents Chemother. 2008;52:1768–1781. doi: 10.1128/AAC.01328-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Gupta P, Lackman-Smith C, Snyder B, Ratner D, Rohan LC, Patton D, Ramratnam B, Cole AM. Antiviral activity of retrocyclin RC-101, a candidate microbicide against cell-associated HIV-1. AIDS Res Hum Retroviruses. 2013;29:391–396. doi: 10.1089/aid.2012.0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).McMahon JB, Currens MJ, Gulakowski RJ, Buckheit RW, Jr., Lackman-Smith C, Hallock YF, Boyd MR. Michellamine B, a novel plant alkaloid, inhibits human immunodeficiency virus-induced cell killing by at least two distinct mechanisms. Antimicrob. Agents Chemother. 1995;39:484–488. doi: 10.1128/aac.39.2.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Yang QE, Stephen AG, Adelsberger JW, Roberts PE, Zhu W, Currens MJ, Feng Y, Crise BJ, Gorelick RJ, Rein AR, Fisher RJ, Shoemaker RH, Sei S. Discovery of small-molecule human immunodeficiency virus type 1 entry inhibitors that target the gp120-binding domain of CD4. J. Virol. 2005;79:6122–6133. doi: 10.1128/JVI.79.10.6122-6133.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Chackerian B, Long EM, Luciw PA, Overbaugh J. Human immunodeficiency virus type 1 coreceptors participate in postentry stages in the virus replication cycle and function in simian immunodeficiency virus infection. J. Virol. 1997;71:3932–3939. doi: 10.1128/jvi.71.5.3932-3939.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Ciminale V, Felber BK, Campbell M, Pavlakis GN. A bioassay for HIV-1 based on Env-CD4 interaction. AIDS Res. Hum. Retroviruses. 1990;6:1281–1287. doi: 10.1089/aid.1990.6.1281. [DOI] [PubMed] [Google Scholar]
- (80).Jiang S, Lu H, Liu S, Zhao Q, He Y, Debnath AK. N-substituted pyrrole derivatives as novel human immunodeficiency virus type 1 entry inhibitors that interfere with the gp41 six-helix bundle formation and block virus fusion. Antimicrob. Agents Chemother. 2004;48:4349–4359. doi: 10.1128/AAC.48.11.4349-4359.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Debnath AK, Radigan L, Jiang S. Structure-based identification of small molecule antiviral compounds targeted to the gp41 core structure of the human immunodeficiency virus type 1. J. Med. Chem. 1999;42:3203–3209. doi: 10.1021/jm990154t. [DOI] [PubMed] [Google Scholar]
- (82).Otwinowski Z, Minor W. In Macromolecular Crystallography, part A. In: Carter CW Jr., Sweet RM, editors. Methods in Enzymology. Vol. 276. Academic Press; San Diego, CA: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- (83).Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- (85).Afonine PV, Moriarty NW, Mustyakimov M, Sobolev OV, Terwilliger TC, Turk D, Urzhumtsev A, Adams PD. FEM: feature-enhanced map. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2015;71:646–666. doi: 10.1107/S1399004714028132. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.