

Abstract
We performed genomewide gene expression analysis of 35 samples representing 6 common histologic subtypes of canine lymphoma and bioinformatics analyses to define their molecular characteristics. Three major groups were defined on the basis of gene expression profiles: (1) low-grade T-cell lymphoma, composed entirely by T-zone lymphoma; (2) high-grade T-cell lymphoma, consisting of lymphoblastic T-cell lymphoma and peripheral T-cell lymphoma not otherwise specified; and (3) B-cell lymphoma, consisting of marginal B-cell lymphoma, diffuse large B-cell lymphoma, and Burkitt lymphoma. Interspecies comparative analyses of gene expression profiles also showed that marginal B-cell lymphoma and diffuse large B-cell lymphoma in dogs and humans might represent a continuum of disease with similar drivers. The classification of these diverse tumors into 3 subgroups was prognostically significant, as the groups were directly correlated with event-free survival. Finally, we developed a benchtop diagnostic test based on expression of 4 genes that can robustly classify canine lymphomas into one of these 3 subgroups, enabling a direct clinical application for our results.
Keywords: canine, lymphoma subclassification, gene expression profiling, prognostication
Canine lymphoma is a heterogeneous group of diseases that share malignant transformation of lymphocytes as a common property. Several systems have been proposed to classify these diseases over the past 4 decades (see Valli et al29 for review), including the modified World Health Organization (WHO) classification system, which was initially proposed in 2002 but has not yet been universally adopted.28 The lack of documented prognostic significance has raised doubts about cost:benefit and risk:benefit ratios of the diagnostic procedures needed to assign a sample to its category in this classification. Furthermore, current therapeutic regimens are not tailored for lymphoma subtypes, highlighting opportunities for improvement and additional reasons for the observed resistance to the expense and effort of classification.
The most contemporary WHO classification system for B- and T-cell lymphomas includes approximately 30 subtypes.29 The system combines morphology, topography, immunophenotype, and clinical progression to define these disease entities. In our experience, 6 subtypes are commonly observed, including diffuse large B-cell lymphoma (DLBCL), marginal-zone lymphoma (MZL), Burkitt and Burkitt-like lymphoma (BL), lymphoblastic T-cell lymphoma (LBT), T-zone lymphoma (TZL), and peripheral T-cell lymphoma not otherwise specified (PTCL).4,13,29 Other recent studies support the frequency with which these tumor types are observed.16,29 However, a molecular foundation for the classification of these tumors remains to be established.
In human non-Hodgkin lymphomas, gene expression profiling has generated considerable insight into transcriptional differences between and within subtypes of this disease.3,12,15,24 For example, human DLBCL is subdivided into activated B-cell lymphoma, germinal center-like B-cell lymphoma, and peripheral mediastinal B-cell lymphoma, each corresponding to a distinct anatomical entity.2,24 The biological behavior and response to therapy in these human DLBCL subtypes also are reportedly different,7,18,21 indicating that molecular phenotyping can be prognostically significant. To date, molecular studies of gene expression in canine lymphoma are limited,23,25 and none has been large enough to address questions about the molecular basis of these diseases. Specifically, molecular evidence for the utility of the modified WHO classification in delineating biologically distinct disease entities has not been published. Here, we performed genomewide gene expression analysis in tumor samples from 35 dogs with lymphoma to provide insights into the molecular underpinnings of different subtypes of this disease.
Materials and Methods
Samples
Samples from dogs with naturally occurring lymphoma (N = 80) were collected from veterinary practices across the United States between 1999 and 2010 as described.1,4,8–10,27 Animal care and experimentation were carried out in accordance with all applicable institutional, local, and national guidelines; dogs were under the care of licensed veterinarians, and participation did not influence decisions of care. Sample collection protocols were approved and reviewed by the institutional review board of the University of Colorado and the institutional animal care and use committee of the University of Minnesota. Tumors from 35 dogs yielded high-quality RNA and passed quality control for gene expression profiling on microarrays. Samples were handled in separate batches by 2 individuals and consequently analyzed as independent cohorts of 29 samples (cohort 1) and 6 samples (cohort 2). Tumors were classified according to the modified WHO criteria based on morphology and immunophenotype.29 The sample population was purposefully biased to Golden Retrievers, although the dogs whose tumors we analyzed reflect the demographic distribution of lymphoma subtypes in the canine population as a whole.29 Table 1 shows the demographic characteristics, treatment, and breed distribution for the dogs. Demographics of dogs profiled using microarray were not significantly different from dogs not included on array (P > .4). For survival data, treatment protocols were summarized into 3 groups: none, palliative (prednisone only), or multiagent CHOP-based chemotherapy. For the last group, all CHOP-based protocols were considered equivalent based on existing outcome data.5,17,22
Table 1.
Demographic Characteristics of Complete Cohort of 80 Dogs With Lymphoma and Restricted Group of 35 Dogs Analyzed by Gene Expression Profiling.
| Recruited Cohort (n = 80) No. (%) | GEP Cohort (n = 35)a No. (%) | Dogs Not on Array (n = 45) No. (%) | |
|---|---|---|---|
| Sex | |||
| Male | 48 (60) | 15 (43) | 33 (73) |
| Female | 32 (40) | 20 (57) | 12 (27) |
| Age at diagnosis, y | |||
| Median | 8.5 | 8 | 8.8 |
| Mean ± SD | 8.5 + 3.1 | 7.9 + 3.3 | 8.1 + 3.0 |
| Breed | |||
| Golden Retrievers | 50 (62) | 23 (66) | 27 (60) |
| All other breedsb | 30 (38) | 12 (34) | 18 (40) |
| Classificationc | |||
| LBT | 9 | 5 | 4 |
| PTCL | 5 | 4 | 1 |
| TZL | 12 | 8 | 4 |
| T-ALCL | 1 | 0 | 1 |
| DLBCL | 29 | 10 | 19 |
| BL | 8 | 2 | 6 |
| MZL | 13 | 5 | 8 |
| B-ALCL | 2 | 0 | 2 |
| NTNBL | 1 | 1 | 0 |
| Median survival, mo | |||
| All dogs | 6.6 (n = 80) | 8 (n = 35) | 6.2 (n = 45) |
| Standard of care | 8.5 (n = 54) | 10.0 (n = 22) | 8.0 (n = 32) |
| Other treatment | 6.1 (n = 15) | 10.5 (n = 5) | 5.6 (n = 8) |
| No treatment | 0.25 (n = 5) | 3.1 (n = 2) | 0.03 (n = 3) |
Gene expression profiling (GEP) cohort was divided into 2 groups with 29 and 6 dogs, respectively. The demographic data for the 2 cohorts were not significantly different; the group of 6 dogs included 1 PTCL, 3 TZL, and 2 DLBCL.
Airedale Terrier (n = 1), Beagle (n = 2), Bichon Frise (n = 1), Boxer n = 4), Labrador Retriever (n = 6), Mastiff (n = 3), Rottweiler (n = 4), Scottish Terrier (n = 1), Shih Tzu (n = 1), Terrier (n = 1), Toy Poodle (n = 1), West Highland Terrier (n = 1), mix breed (n = 2), unknown (n = 2).
LBT = lymphoblastic T-cell lymphoma; PTCL = peripheral T-cell lymphoma not otherwise specified; TZL = T-zone lymphoma; ALCL = anaplastic large cell lymphoma; DLBCL = diffuse large B-cell lymphoma; BL = Burkitt lymphomas; MZL = marginal-zone lymphoma; NTNBL = non-T, non-B-cell lymphoma.
RNA Isolation and Array Hybridization
Biopsy samples were processed to single-cell suspensions as described previously.11 RNA was isolated from cells recovered from cryopreservation using the RNAeasy Mini Kit and QIAshredder (QIAGEN, Valencia, California). RNA concentration was determined using NanoDrop ND-1000 UV-Vis spectrophotometer (NanoDrop Technologies, Wilmington, Delaware), and quality was measured using a 2100 Bioanalyzer (Agilent, Santa Clara, California). All the samples included in the gene expression profiling experiment were suitable for microarray analysis based on RNA quality (RIN > 7.0). Samples were hybridized to Affymetrix Canine_2.0 gene chips (Affymetrix, Santa Clara, California) as described elsewhere.26 RNA was isolated twice from one DLBCL sample, and both isolates were arrayed separately to generate a technical replicate. The bioinformatics methods used for analysis can be found online as supplementary methods.
Quantitative Real-Time Reverse Transcriptase Polymerase Chain Reaction
Purified RNA was reverse transcribed to cDNA using the First Strand cDNA Synthesis Kit for RT-PCR (Roche Applied Science, Indianapolis, Indiana). Quantitative real-time reverse transcriptase polymerase chain reaction (qRT-PCR) was used to quantify the resulting cDNAs in an ABI7500 sequence detector with the Taqman PCR Master Mix Protocol (ABI, Foster City, California). Each reaction was performed by denaturing cDNAs at 95°C for 10 minutes, followed by 40 PCR cycles of denaturation (95°C) for 15seconds, annealing (57°C) for 30 seconds, and extension (68°C) for 30 seconds per cycle. Primers were designed using Primer3 software.19 The primers used for molecular stratification of lymphoma subtypes were as follows: glyceraldehyde 3-phosphate dehydrogenase gene, F: GCCAAGAGGGTCATCATCTC, R: CTTTGGCTAGAGG TGCCAAG; CD28 gene, F: GACCACCGTTAGCACAATGA, R: CCGGAACTCCTTTGAGAAGA; ATP-binding cassette transporter A5 (ABCA5) gene, F: TGGCCATTCATATCGTA GCA, R: CGCAGCTACTTTGAGGGAAT; SPARC-related modular calcium binding-2 (SMOC2) gene, F: GCTGGA GACCCAACCTCA, R: ATTGGTTTTGTTCTGCCGACT; and coiled-coiled domain containing-3 (CCDC3) gene, F: TGTTTTCCAGCCTTTTCCAG R: GCTGCTTGTTACGC TTCTCC. Additional validation of microarray data was performed by plotting log-transformed array values against log-transformed values of fold change in expression from 5 dogs using qRT-PCR for genes that were not necessarily differentially expressed between disease subtypes. These included inhibitor of NFκB Kinase E, F: CATCAAGCCTGGA AACATCA, R: TTACCCCGAATGTCTTCTGC, and inter leukin-17 receptor-A, F: TCCTTCATCCCCAAAAGATG, R: TTGGTGTTCAGTTGCAGGAC. The relationship between array and qRT-PCR values for the transcripts of interest was analyzed with the Pearson correlation.20
Immunophenotyping
Flow cytometry, immunohistochemistry (IHC), and PCR for antigen receptor rearrangement (PARR) were used to phenotype tumors and to identify subpopulations in the tumors. Flow cytometry,8,10 IHC,10 and PARR methods8 have been described. IHC was done by IHC Services (Smithville, Texas), and confirmatory staining was done as needed at the Masonic Cancer Center Shared Pathology Resource core facility. PARR was done by the Immunopathology Laboratory of Colorado State University (Ft Collins, Colorado), and confirmatory analyses were done in our laboratory at the University of Minnesota as needed.
Statistics
Descriptive statistics for variables of breed, sex, age at diagnosis, and treatment regimen were performed. Categorical data are expressed as percentages, while continuous data are expressed as means ± SD, medians, and/or ranges. Fisher exact test was performed to compare data from lymphoma subtypes and from the dogs used for gene expression profiling and the rest of the dogs in the recruitment cohort. Event-free survival was defined as the interval from diagnosis until recurrence (relapse). In most cases, dogs were euthanized at the time that recurrence was diagnosed. Censoring was done for dogs that received rescue therapy (on the date that recurrence was detected), for dogs that were lost to follow-up (on the last contact date), and for dogs that died from other causes (on the date of death). Kaplan-Meier analysis was done with a tool from the Walter and Elisa Hall Institute Department of Bioinformatics (http://bioinf.wehi.edu.au/software/russell/logrank/). Statistical significance was calculated using the log rank test, and a P < .05 was considered significant.
Diagnostic Algorithm
Pairs of genes with potential diagnostic utility were chosen per the following 3 criteria: (1) highly significant P values between groups, (2) large and opposite fold changes between groups, and (3) low intragroup variance within each group that was to be separated. Messenger RNA for 17 independent samples (not on the arrays) was quantified with qRT-PCR. The diagnostic algorithm consisted of applying the ratio of gene 1:gene 2 to define a separation between B-cell and T-cell tumors and for the T-cell tumors, a second level of separation between high- and low-grade lymphomas. A patent application protecting intellectual property with regard to use of gene expression data and the resulting algorithms from this study as they apply to diagnosis, subclassification, and prognostication of lymphomas has been filed by the University of Minnesota Office for Technology Commercialization.
Results
Gene Expression Profiling Stratifies Canine Lymphoma Into 3 Major Subgroups
Recently, an international consensus was reached to subclassify canine lymphomas into multiple subtypes according to the modified WHO criteria.29 We sought to determine if these subtypes also could be distinguished at the molecular level. Initially, we profiled gene expression in samples from cohort 1, which included 4 LBT, 3 PTCL, and 6 TZL, totaling 13 T-cell lymphomas; 2 BLs, 8 DLBCL, and 5 MZL, totaling 15 B-cell lymphomas; and 1 non-T, non-B-cell lymphoma (NTNBL). Figure S1a shows unsupervised principal component analysis (PCA) of expression data from these samples. Three groups (a, b, c) were discernible, separated along the 3 principal components. Group a represented dogs with LBT and PTCL (n = 7). Group b represented dogs with TZL (n = 6). Group c represented dogs with B-cell malignancies (n = 15). The NTNBL sample appeared to be most closely related to T-cell lymphomas but did not segregate into any of the 3 groups (data not shown). The major differences in gene expression underlying this stratification were observable using unsupervised hierarchical clustering with a heat map showing 859 genes with variance > 1 and with > eightfold difference in expression across 3 or more samples (Figure S1b).
The possibility that breed was a significant driver for gene expression profiling was examined using supervised methods (Golden Retriever vs non–Golden Retriever) and the false discovery rate. No genes showed statistically significant differential expression between these groups.
The data show that LBT and PTCL composed a single molecular group (henceforth, high-grade T-cell lymphoma, or “T-high”) and TZL composed a distinct molecular group (henceforth, low-grade T-cell lymphoma, or “T-low”). Segregation among B-cell tumors was more subtle. It was challenging to separate out BL and DLBCL samples (henceforth, high-grade B-cell lymphoma, or “B-high”) from MZL samples (henceforth, low-grade B-cell lymphoma, or “B-low”), although MZL formed a subcluster in the PCA. Figure 1a recapitulates the heat map shown in Figure S1b, except that using a 2-group t test (BH q ≤.001), we found that 624 differentially expressed genes separated T-cell lymphomas from B-cell lymphomas. Figure 1b shows 389 differentially expressed genes that separated the T-high group from the T-low group; this gene cluster also stratified samples of high-grade T-cell lymphoma from all other lymphomas tested. Figure 1c shows 25 differentially expressed genes that could help to distinguish between the B-high and B-low groups. These genes also served to separate the T-high from the T-low group and included a subset of the gene cluster in Figure 1b, as illustrated by the Venn diagram in Figure 1d, which shows shared genes identified in each 2-group analysis.
Figure 1.
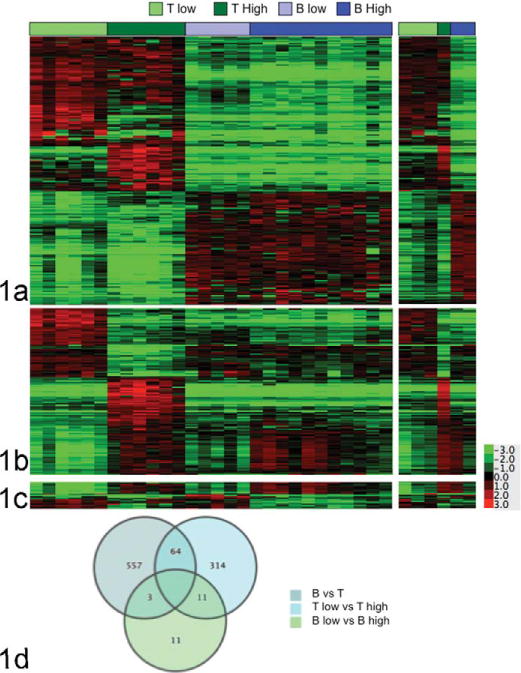
Statistically significant genes define molecular subtypes of canine lymphoma. Genes differentially expressed with > threefold-average change and P values < .001 were identified for the comparison of groups composed of (a) B-cell and T-cell lymphomas (n = 624), (b) high-grade and low-grade T-cell lymphomas (n = 389), and (c) high-grade and low-grade B-cell lymphomas (n = 25) using t test statistics. The second panel of (a–c) is an independent “validation” set (6 samples, right inset) of the results obtained in the initial set (29 samples, left panel). (d) Venn diagram showing the number of unique and overlapping genes for each 2-group test.
We next examined the statistically significant genes from cohort 1 in a second independent cohort (cohort 2), which consisted of 6 samples, including 3 TZL, 1 PTCL, and 2 DLBCL. The analysis for these samples is shown on the right inset for Figure 1a–1c. The molecular signatures statistically characterized in cohort 1 were clearly observable in cohort 2.
To more carefully assess the potential for heterogeneity of canine B-cell malignancies, we compared the molecular properties of DLBCLs and MZLs in our sample set, and we extended this comparison to a sample set that included human DLBCLs and MZLs,6 using an approach that we previously applied successfully to define gene expression signatures across different platforms and species.20 There is significant overlap in the morphological features of canine MZL and DLBCL,31 and these 2 tumor types were challenging to stratify according to their molecular signatures (Figure 1c). We saw comparable results in the human tumors: Figure 2a shows PCA for human DLBCLs (n = 22) and nodal MZLs (n = 13). However, as was true for the canine sample set, MZLs formed an identifiable, relatively homogeneous subgroup. Figure 2b shows hierarchical clustering of human samples using the 71 genes with highly statistically significant (P ≤ 10−6) differential expression between human MZL and DLBCL subtypes. To determine if this signature was retained in the canine samples, we name mapped 37 genes overexpressed in human MZL and 34 genes overexpressed in human DLBCL to their canine homologues, identifying 27 and 29 orthologous genes, respectively. To track the patterns of expression, we used a “gene vector–based” strategy. Each gene vector was assigned a yellow or blue tag within a toe-bar, where the color denoted its cluster assignment and the intensity reflected the relative variance in expression. The composite gene vectors (toe-bars) thus illustrate the relative conservation of the gene clusters between datasets. The corollary to the hypothesis that the molecular characteristics of DLBCL and MZL in humans and dogs are conserved is that a comparable arrangement of gene vectors would be apparent in datasets from both species.
Figure 2.
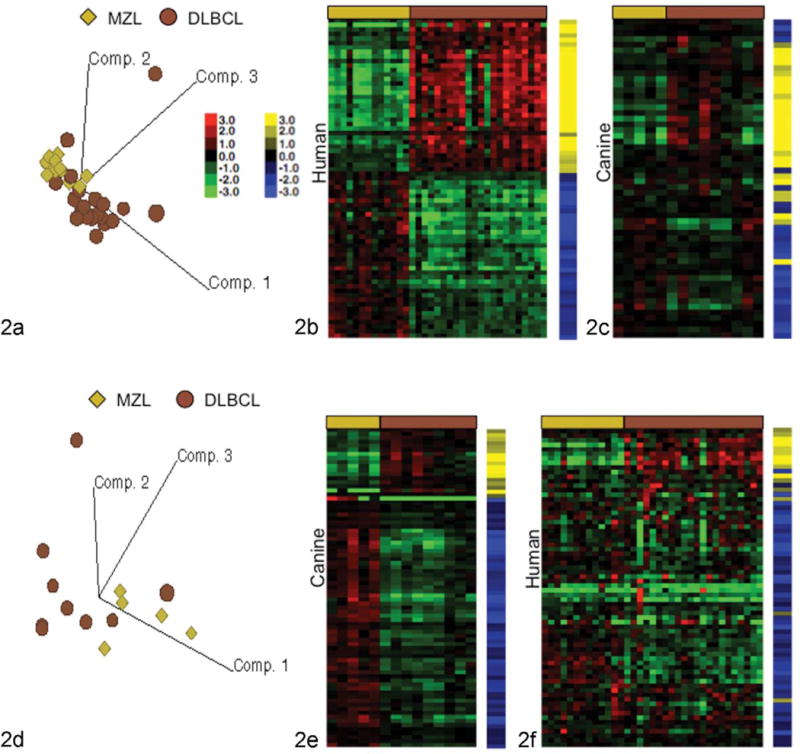
Orthologous gene expression signatures indicate canine and human B-cell lymphomas are molecularly homologous diseases. (a) Principal component analysis of human marginal-zone lymphoma (MZL) and diffuse large B-cell lymphoma (DLBCL) cases. (b) Heat map showing differential expression for 71 genes in human nodal MZL and DLBCL. (c) 71 Human gene vectors from panel b mapped to 56 canine genes and applied to canine MZL and DLBCL samples. (d) Principal component analysis of canine MZL and DLBCL cases. (e) Heat map showing differential expression for 79 genes between canine MZL and DLBCL. (f) Seventy-nine canine gene vectors from panel e mapped to 70 human genes and applied to human MZL and DLBCL samples. Yellow and blue toe-bars identify specific gene vectors in the signature for tracking from the data set of origin (human in b, canine in e) onto the comparison dataset (canine in c and human in f). Color intensity shows intersample variance.
Unsupervised hierarchical clustering of the dog cohort with the human gene vectors was consistent with the prediction of molecular homology, resulting in similar clustering for MZL and DLBCL to that observed in the human cohort (Figure 2c). The same analysis was used to examine stratification between canine MZL and DLBCL. Figure 2d shows PCA for canine DLBCL (n = 9) and nodal MZL (n = 5) samples, where the latter formed a subgroup that resembled the one seen in the human samples (compare with Figure 2a). Figure 2e shows hierarchical clustering of the canine samples using 79 genes with statistically significant (P ≤ 103) differential expression between the canine MZL and DLBCL subtypes (as shown in Figure 2c). To determine if this signature was retained in the human data set, we name mapped 62 genes overexpressed in canine MZL and 17 genes overexpressed in canine DLBCL to their human homologues; 55 and 15 orthologous genes were identifiable in the human data set, respectively. Unsupervised hierarchical clustering of the human cohort using these canine vectors was also was consistent with the prediction of molecular homology, resulting in similar clustering patterns for MZL and DLBCL to that observed in the canine cohort (Figure 2f).
Molecular Drivers for Stratification of Canine Lymphomas
Assessment of gene expression differences using Ingenuity Pathway Analysis (IPA) and the 624 genes shown in Figure 1a revealed segregation of T- and B-cell lymphoma according to predictable functions, including activation, proliferation, and development of T and B cells, respectively (Table S1a, S1b). Similarly, segregation of high-grade from low-grade T-cell tumors was based on enrichment of cell cycle–related genes (principally chromosomal segregation and mitosis; Table S1c). The low-grade T-cell tumors, in contrast, showed enrichment for functions associated with T-cell activation and survival (Table S1d). As noted above, the molecular signatures separating B-cell lymphomas were less robust. A subset of genes associated with cell division and chromosome segregation that were enriched in the high-grade T-cell tumors also were enriched in high-grade B-cell tumors. Intriguingly, IPA also showed enrichment of functions associated with T-cell signaling in the low-grade B-cell tumors.
We confirmed the IPA results using gene set enrichment analysis. We first examined genes that were differentially expressed between B-cell and T-cell lymphoma subtypes and obtained similar results as we did with IPA. The same was true when we analyzed differences between T-high and T-low tumors, and assessment of the B-high versus the B-low group provided further evidence of gene enrichment in T-cell signaling pathways.
Similar enrichment of T-cell signatures in B-cell disease has been reported for subsets of human DLBCL.2,14 Thus, we investigated the possibility that the IPA and gene set enrichment analysis data highlighted signatures of residual or infiltrating T cells associated with our low-grade B-cell lymphomas. We performed IHC on the tumors from this cohort, as well as IHC and flow cytometry on an independent series of B-high and B-low tumors to quantify T cells in B-cell lymphomas. Figure 3 shows examples of canine DLBCL and MZL samples stained for CD3 and CD20 that illustrate the relative T-cell enrichment that was seen recurrently in MZLs. Figure S2 shows the presence of T cells in B-cell lymphoma quantitatively: the box and whisker plots describe the relative abundance of T cells in high-grade B-cell lymphomas (n = 28) versus low-grade B-cell lymphomas (n = 20). There was a significant difference (P = .0079) between these 2 lymphoma subgroups with regard to the abundance of T cells present in the tumors.
Figure 3.
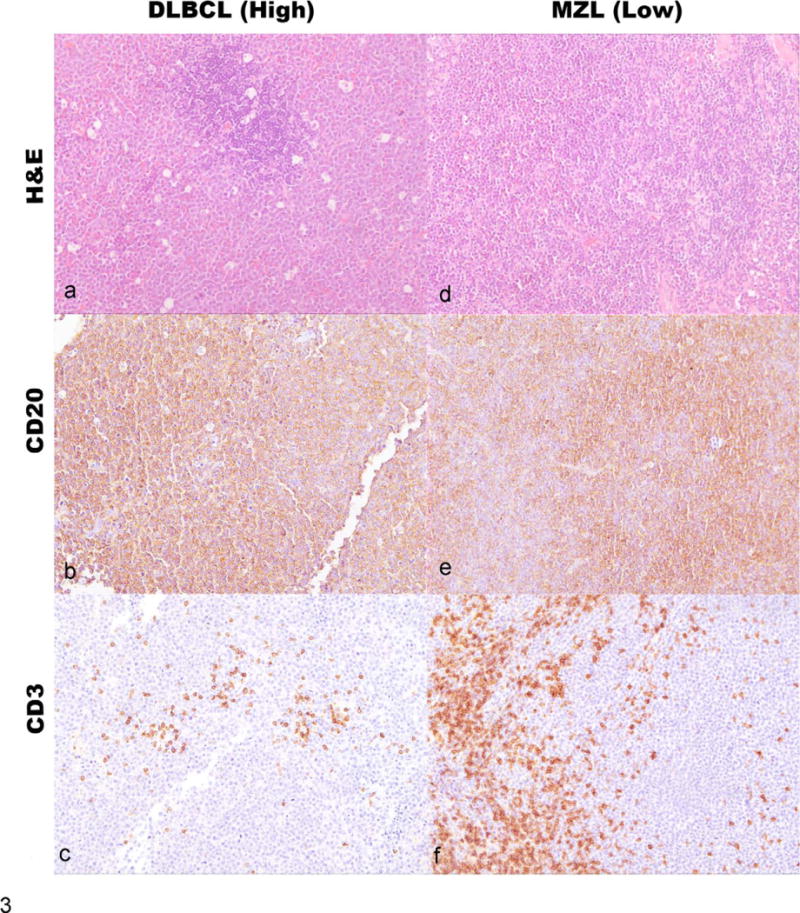
(Left column; panels a, b, c) Dog lymph node, case ID AMC 26, DLBCL. (a) Effacement of nodal architecture by a monomorphic population of large lymphoid cells (~2 red cell diameters) with many tingible body macrophages. HE. (b) Immunolabeling with anti-CD20, hematoxylin counterstain. Neoplastic cells are positive for CD20. (c) Immunolabeling with anti-CD3, hematoxylin counterstain. Neoplastic cells are negative for CD3. Few residual or infiltrating nonneoplastic T cells (CD3+) are visible in the section. (Right column; panels d, e, f) Dog lymph node, case ID AMC 63, MZL. (d) Effacement of nodal architecture by a monomorphic population of intermediate lymphoid cells (~1.5 red cell diameters) with few tingible body macrophages. HE. (e) Immunolabeling with anti-CD20, hematoxylin counterstain. Neoplastic cells are positive for CD20. (f) Immunolabeling with anti-CD3, hematoxylin counterstain. Neoplastic cells are negative for CD3. Clusters of residual or infiltrating nonneoplastic T cells (CD3+) are visible in the section.
Molecular Stratification Defines Subtypes of Disease That Are Predictive for Event-Free Survival for Dogs With Lymphoma
We reexamined the predictive value of this classification using event-free survival data (i.e., time from diagnosis to relapse), a reliable indicator for outcome. Seventy-four dogs were evaluable, including the 35 dogs used for the gene expression profiling experiment, which increased the statistical power to detect differences in event-free survival among the WHO groups. Kaplan-Meier survival probability curves for dogs that received any treatment or that received standard of care CHOP-based chemotherapy treated are shown in Figure 4a and 4b, respectively. Consistent with previous data,13 survival times were longest for dogs with low-grade T-cell lymphomas (TZL), shortest for dogs with high-grade T-cell lymphomas (LBT and PTCL), and intermediate for dogs with B-cell lymphomas. The survival benefit for dogs with low-grade T-cell lymphomas treated with CHOP-based protocols (8 of 11 dogs) appeared to be modest. In contrast to what we observed with the T-cell subtypes, there was no significant difference in event-free (or overall) survival between high- and low-grade B-cell malignancies.
Figure 4.
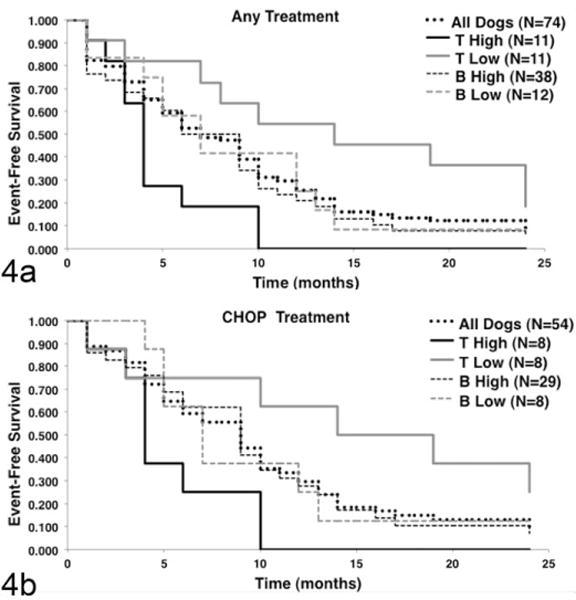
Event-free survival is different for the 3 major molecular subtypes of canine lymphoma. Event-free survival data were available for 74 of 80 dogs recruited. (a) Kaplan-Meier event-free survival curves for dogs that received any treatment, classified according to molecular subgroups. (b) Kaplan-Meier event-free survival curves for dogs that received multiagent chemotherapy treatment consisting of cyclophosphamide (Cytoxan), doxorubicin (also called Hydroxydaunorubicin), vincristine (Oncovin), and prednisone, generally referred to as CHOP, were classified according to molecular subgroups. Median survival for all treated dogs was 6.6 months, and for CHOP-treated dogs it was 8.5 months. Event-free survival of dogs with low-grade T-cell lymphoma was significantly longer (P < .05) than that of dogs with high-grade T-cell lymphoma or dogs with B-cell lymphoma regardless of treatment, as determined using the log-rank test. The event-free survival of dogs with high-grade T-cell lymphoma was significantly shorter (P < .05) than that of dogs with high-grade B-cell lymphoma. Two dogs classified as intermediate grade were censored from the subgroup analysis in panel a, and 1 dog classified as intermediate grade was censored from the analysis in panel b.
A Simplified 4-gene Signature Is Robust to Classify Molecular Subtypes of Lymphoma
We next evaluated the potential to use a simplified profile of gene expression that could be translated into a diagnostic platform to rapidly and accurately distinguish among the 3 defined molecular groups. The array data showed that CD28 and ABCA5 consistently showed differential expression in T-cell lymphomas and B-cell lymphomas, and the ratio of gene expression for these 2 genes was sufficient to establish whether any tumor originated from the T-cell or B-cell lineage (Figure 5a, arrays 1 and 2). Similarly, the ratio of CCDC3 and SMOC2 expression was sufficient to classify T-cell tumors into T-low (TZL) or T-high (LBT or PTCL) categories (Figure 5b, arrays 1 and 2). To validate this molecular approach of classification for canine lymphoma, we prospectively evaluated expression of these genes using qRT-PCR in an independent cohort of 17 cases to verify their utility to provide a definitive molecular classification. Samples were immunophenotyped and classified according to their morphologic appearance by a pathologist (TDO) without knowledge of the molecular results. For each sample, we first divided the value for CD28 expression over the value for ABCA5 expression. If the ratio was > 1, the tumor was determined to originate from a T cell; conversely, if the ratio was < 1 the tumor was determined to originate from a B cell (Figure 5a, qRT-PCR). For each T-cell tumor, we then divided the value for CCDC3 expression over the value for SMOC2 expression. If the ratio was > 1, the tumor was classified as a low-grade T-cell lymphoma, and if the ratio was < 1, the tumor was classified as a high-grade T-cell lymphoma (Figure 5b, qRT-PCR). Using this test, we correctly classified 17 of 17 samples into the correct phenotype (B or T cell), suggesting the probability of obtaining an incorrect classification using this algorithm is < 1 in 1 000 000 000 (Fisher exact test P = 8 × 10−10). We similarly classified 9 of 9 samples into the correct T-high or T-low subgroup, suggesting the probability of obtaining an incorrect classification using this algorithm is < 1 in 10 000 (Fisher exact test P = 4 × 10−5).
Figure 5.
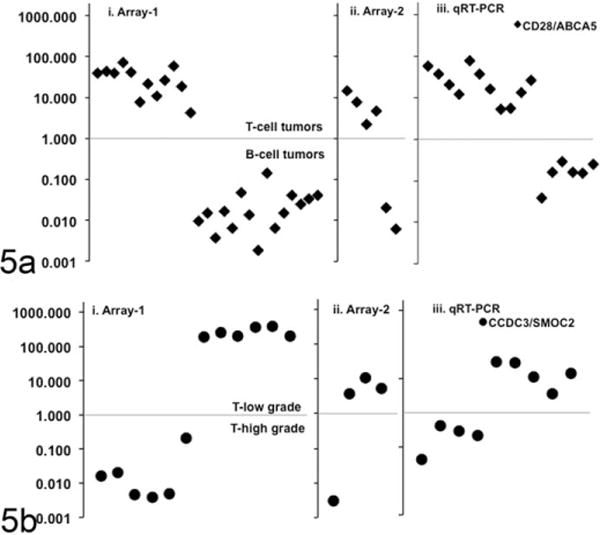
A 4-gene signature accurately classifies canine lymphomas into 3 molecular subgroups. Array data were surveyed to identify genes that showed robust and significantly different expression between groups and within group variance < 1.0. For each sample in the array set i (n = 29) and set ii (n = 6), the ratio of expression levels for the chosen genes was calculated from normalized array values; an additional validation set iii (n = 17) included samples that were not part of either gene expression array set, with expression ratios calculated from qRT-PCR values using the formula 1/2(Ct[gene-1]−Ct[gene-2]). For each sample, qRT-PCR also was also done using the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) to test confirm RNA integrity. (a) The graph shows the calculated value for the ratio of CD28/ABCA5 in each of the 3 independent sample sets. This gene expression ratio was > 1.0 for T-cell lymphoma samples, whereas it was < 1.0 for B-cell lymphoma samples in each group. Seventeen of seventeen samples were correctly identified according to their histologic phenotype (B cell or T cell). (b) The graph shows the calculated value for the ratio of CCDC3/SMOC2 for T-cell lymphomas in each of the 3 independent sample sets. This gene expression ratio was > 1.0 for low-grade T-cell lymphomas, whereas it was < 1.0 for high-grade T-cell lymphomas. Nine of nine samples are correctly identified according to their histologic classification as low-or high-grade T-cell lymphoma.
Discussion
A molecular classification for subtypes of canine lymphoma remains to be defined. This is a significant gap in our understanding of the disease, and it contributes to the uncertainty about the relevance and utility of morphological and topographical classification systems. It also precludes precise comparisons between some types of canine lymphoma and human non-Hodgkin lymphoma, as the extent of disease homology is unclear.
Here, we sought to identify molecular correlates for common morphological subtypes of canine lymphoma classified using the modified WHO classification. Our data show that the most common subtypes of canine lymphoma (DLBCL, BL, MZL, LBT, PTCL, and TZL)29 can be generally subdivided into 3 molecular subgroups consisting of high-grade T-cell lymphomas (LBT, PTCL), low-grade T-cell lymphomas (TZL), and B-cell lymphomas (DLBCL, BL, and MZL). The BL samples were generally indistinguishable from DLBCL using gene expression profiles; one NTNBL sample was most similar to TZL but nevertheless appeared to be definable as its own entity by PCA.
The classification of samples into 3 major groups was prognostically significant. TZLs have been reported to show indolent progression,30 and our results here and elsewhere13 support that observation. These tumors might initially benefit from conservative management (watchful waiting) or low-intensity chemotherapy that would reduce the likelihood of treatment-related toxicity with low risk for accelerated tumor progression. In contrast, high-grade LBTs and PTCLs are aggressive, rapidly progressive tumors that respond poorly to conventional chemotherapy. This information undoubtedly can help dog owners and veterinarians make more educated treatment decisions, but its intrinsic benefit has yet to outweigh common resistance to biopsy procedures. Genomewide gene expression profiling is cost-prohibitive and impractical; thus, we developed a simple and reliable qRT-PCR-based test to provide preliminary stratification into low-grade or high-grade T-cell lymphomas (or B-cell lymphomas). This test has the potential to be performed in fine-needle aspirate samples preserved at the point of care, potentially improving our diagnostic capability with acceptable risk and with favorable assessments of cost and benefit. It is noteworthy that one of the genes expressed at higher levels in TZL than in LBT and PTCL was CR2 (complement receptor-2 or CD21). This provides an explanation for the observed reactivity of a widely used anti-CD21 antibody, which is conventionally used as a B-cell marker, in canine lymphomas that also express CD3 and CD5 by flow cytometry.32 The expression of CD21 in TZLs also suggests that this represents a bona fide subset of T-cell malignancies in dogs and not an unusually high proportion of tumors with bilineage differentiation.
In the case of B-cell lymphomas, the molecular similarity between DLBCL and MZL was not surprising. Among all the canine lymphoma subtypes defined by the WHO classification, DLBCL and nodal MZL are the most challenging to distinguish,31 and our data suggest that these conditions might represent a continuum of the same disease. We have observed similar patterns using DNA copy number abnormalities to classify canine B-cell lymphomas (R Thomas et al, manuscript in preparation). In contrast, BL has distinguishable morphological characteristics16,28,29,31 and can be defined by the peculiar translocation t(8;13) involving the IGH locus in canine chromosome 8 (CFA 8) and the MYC locus in CFA 13.1 The different survival estimates observed in this study and reported by Ponce et al16 suggest that there may be molecular heterogeneity in BL and that additional diagnostics to confirm the presence of a t(11;13) translocation may be advisable to confirm this diagnosis.
It is possible that the cells that give rise to canine DLBCL and MZL are functionally and topographically related, originating from the germinal or follicle center, and thus share immunophenotypic and molecular activation markers despite different degrees of anatomical lymph node effacement and slightly to moderately different morphologic appearance. Human and canine MZL and DLBCL showed similar groupings and conserved driver genes, suggesting that these 2 conditions could represent a continuum of one disease in both species. Preliminary analyses suggest that the DLBCL samples in our study did not conform to the gene expression profiles that are characteristic of human activated B-cell and germinal centerlike B-cell lymphoma–DLBCL (not shown). However, analysis of larger data sets might help to establish if these subtypes can be defined in canine DLBCL.
The enrichment of cell cycle–related pathways in high-grade B-cell lymphomas was intriguing, and it suggests that analysis of a larger sample set of tumors might provide a robust basis to refine segregation between DLBCL and MZL even if they represent distinct stages of the same disease. We showed that a related cell cycle signature was useful to stratify canine and human osteosarcomas according to their biological behavior in vivo,20 and preliminary data indicate that similar transcription factors may be dysregulated in both diseases. It is nonetheless unclear whether such stratification would have prognostic value for canine lymphoma. A previous report suggested that progression of MZL was indolent and that the relatively poor outcome of this disease might be related to the absence of clinical signs in the early stages of disease, leading to diagnosis at an advanced stage when the probability to undergo conversion to DLBCL was greater.30 We cannot exclude that possibility, but it is apparent that the response to therapy and survival of dogs with DLBCL and MZL are heterogeneous and unpredictable. Indeed, we were unable to identify differentially expressed genes that passed robust tests of significance using 2-group supervised analysis based on outcome (ie, event-free survival or overall time below and above the median or even limited to the extremes).
We observed enrichment of gene sets associated with activated T cells in MZL samples. This result was consistent with the quantifiable excess of T cells seen in MZL samples by flow cytometry, and the frequent identification of T-cell aggregates by IHC. This could be explained by the presence of residual T cells in lymph nodes that have not undergone complete effacement (indolent disease) or by recruitment of infiltrating T cells. The latter could be effector (inflammatory) T cells responding to anatomical disruption or immunological cues, or they could be regulatory T cells co-opted by the tumor to mitigate inflammation and antitumor immune responses. Signals associated with the tumor “microenvironment,” especially T cells, were defined in the earliest genomewide gene expression studies of human DLBCL2 and are now becoming a routine part of prediction in human patients diagnosed with DLBCL.11,14 Unlike these predictive signatures, however, our data did not show enrichment for genes associated with histiocytic infiltrates. We also did not observe enrichment of genes associated with increased blood vessel density (angiogenesis), but this could be due to sample processing. Our analysis was done using samples that were disaggregated into single-cell suspensions with removal of connective tissue stroma, whereas some studies analyzing human DLBCLs have used frozen whole lymph node sections that would retain connective tissue stromal components. The enriched T-cell-associated gene sets we found were most consistent with regulatory T cells, which might provide independent explanations both for the presence of T cells in MZL samples and for the rather “poor” survival seen in some dogs diagnosed with this otherwise “indolent” disease.
In summary, we have shown that canine lymphoma can be stratified into at least 3 molecular subgroups that are prognostically significant, and we have developed a robust test to establish this classification measuring the standardized expression of 4 genes. Additional work will be necessary to establish the sensitivity and specificity for this test, as well as its positive and negative predictive values for outcome. Thus far, our classification also was limited to the most commonly observed morphological subtypes of canine lymphoma. A broad collaboration among veterinary pathologists and veterinary oncologists will be needed to accrue sufficient, suitable material to define molecular signatures and their prognostic significance in other less common subtypes of canine lymphoma.
Supplementary Material
Acknowledgments
We thank Drs James Cerhan and Vivian Bardwell for helpful discussions and review of the manuscript; Susan P. Fosmire, Katherine J. Gavin, and Okyong Cho for technical assistance preparing and hybridizing RNA samples; John Wojcieszyn, Josh Parker, and Paula Overn for histology services and immunohistochemistry; dog owners who allowed their pets to participate in this study; veterinarians who assisted in collection of biopsy samples and provided follow-up information; and Mitzi Lewellen for coordinating sample collection, maintaining the sample biobanks, and editorial assistance. We wish to acknowledge infrastructure support and staff assistance from the Minnesota Supercomputing Institute.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This work was supported by grants 2254 (JFM and MB), 615 (JFM, MB, and LEH), and 1113 (TDO and JFM) from the AKC Canine Health Foundation; grant D10-CA-501 (JFM, MB, KLT) from the Golden Retriever Foundation and Morris Animal Foundation; grants P30CA046934 (UCCC Core) and P30CA077598 (MCC Core) from the National Institutes of Health, by the Starlight Fund, the Land of PureGold Foundation, the WillPower Fund, and other philanthropic donations to the University of Minnesota Animal Cancer Care and Research Program and by the Companion Animal Health Foundation and a Tufts University internal seed grants (KEB). AMF was supported by the DVM/PhD combined degree program of the College of Veterinary Medicine, University of Minnesota, by a predoctoral fellowship from Morris Animal Foundation (D09CA-405) and by a doctoral dissertation fellowship from the Graduate School, University of Minnesota. DI is the recipient of a Morris Animal Foundation FIRST Award (D12CA-302). KLT is the recipient of a EURYI award from the ESF.
Footnotes
Supplemental material may be retrieved from http://vet.sagepub.com/supplemental.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Abramson JS, Shipp MA. Advances in the biology and therapy of diffuse large B-cell lymphoma: moving toward a molecularly targeted approach. Blood. 2005;106(4):1164–1174. doi: 10.1182/blood-2005-02-0687. [DOI] [PubMed] [Google Scholar]
- 2.Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403(6769):503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 3.Breen M, Modiano JF. Evolutionarily conserved cytogenetic changes in hematological malignancies of dogs and humans: man and his best friend share more than companionship. Chromosome Res. 2008;16(1):145–154. doi: 10.1007/s10577-007-1212-4. [DOI] [PubMed] [Google Scholar]
- 4.Fosmire SP, Thomas R, Jubala CM, et al. Inactivation of the p16 cyclin-dependent kinase inhibitor in high-grade canine non-Hodgkin’s T-cell lymphoma. Vet Pathol. 2007;44(4):467–478. doi: 10.1354/vp.44-4-467. [DOI] [PubMed] [Google Scholar]
- 5.Garrett LD, Thamm DH, Chun R, et al. Evaluation of a 6–month chemotherapy protocol with no maintenance therapy for dogs with lymphoma. J Vet Intern Med. 2002;16(6):704–709. doi: 10.1892/0891-6640(2002)016<0704:eoacpw>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 6.Gomez-Abad C, Pisonero H, Blanco-Aparicio C, et al. PIM2 inhibition as a rational therapeutic approach in B-cell lymphoma. Blood. 2011;118(20):5517–5527. doi: 10.1182/blood-2011-03-344374. [DOI] [PubMed] [Google Scholar]
- 7.Gutierrez-Garcia G, Cardesa-Salzmann T, Climent F, et al. Gene-expression profiling and not immunophenotypic algorithms predicts prognosis in patients with diffuse large B-cell lymphoma treated with immunochemotherapy. Blood. 2011;117(18):4836–4843. doi: 10.1182/blood-2010-12-322362. [DOI] [PubMed] [Google Scholar]
- 8.Ito D, Endicott MM, Jubala CM, et al. A tumor-related lymphoid progenitor population supports hierarchical tumor organization in canine B-cell lymphoma. J Vet Intern Med. 2011;25(4):890–896. doi: 10.1111/j.1939-1676.2011.0756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ito D, Frantz AM, Williams C, et al. CD40 ligand is necessary and sufficient to support primary diffuse large B-cell lymphoma cells in culture: a tool for in vitro preclinical studies with primary B-cell malignancies. Leuk Lymphoma. 2012;53(7):1390–1398. doi: 10.3109/10428194.2011.654337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jubala CM, Wojcieszyn JW, Valli VEO, et al. CD20 expression in normal canine B cells and in canine non-Hodgkin’s lymphoma. Vet Pathol. 2005;42(4):468–476. doi: 10.1354/vp.42-4-468. [DOI] [PubMed] [Google Scholar]
- 11.Lenz G, Wright G, Dave SS, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med. 2008;359(22):2313–2323. doi: 10.1056/NEJMoa0802885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lenz G, Wright GW, Emre NC, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci U S A. 2008;105(36):13520–13525. doi: 10.1073/pnas.0804295105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Modiano JF, Breen M, Valli VE, et al. Predictive value of p16 or Rb inactivation in a model of naturally occurring canine non-Hodgkin’s lymphoma. Leukemia. 2007;21(1):184–187. doi: 10.1038/sj.leu.2404392. [DOI] [PubMed] [Google Scholar]
- 14.Monti S, Savage KJ, Kutok JL, et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood. 2005;105(5):1851–1861. doi: 10.1182/blood-2004-07-2947. [DOI] [PubMed] [Google Scholar]
- 15.Piccaluga PP, Agostinelli C, Tripodo C, et al. Peripheral T-cell lymphoma classification: the matter of cellular derivation. Expert Rev Hematol. 2011;4(4):415–425. doi: 10.1586/ehm.11.37. [DOI] [PubMed] [Google Scholar]
- 16.Ponce F, Marchal T, Magnol JP, et al. A morphological study of 608 cases of canine malignant lymphoma in France with a focus on comparative similarities between canine and human lymphoma morphology. Vet Pathol. 2010;47(3):414–433. doi: 10.1177/0300985810363902. [DOI] [PubMed] [Google Scholar]
- 17.Rassnick KM, McEntee MC, Erb HN, et al. Comparison of 3 protocols for treatment after induction of remission in dogs with lymphoma. J Vet Intern Med. 2007;21(6):1364–1373. doi: 10.1892/07-057.1. [DOI] [PubMed] [Google Scholar]
- 18.Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346(25):1937–1947. doi: 10.1056/NEJMoa012914. [DOI] [PubMed] [Google Scholar]
- 19.Rozen S, Skaletsky HJ. Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S, editors. Bioinformatics Methods and Protocols: Methods in Molecular Biology. Totowa, NJ: Humana Press; 2000. pp. 365–386. [DOI] [PubMed] [Google Scholar]
- 20.Scott MC, Sarver AL, Gavin KJ, et al. Molecular subtypes of osteosarcoma identified by reducing tumor heterogeneity through an interspecies comparative approach. Bone. 2011;49(3):356–367. doi: 10.1016/j.bone.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shipp MA, Ross KN, Tamayo P, et al. Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat Med. 2002;8(1):68–74. doi: 10.1038/nm0102-68. [DOI] [PubMed] [Google Scholar]
- 22.Sorenmo K, Overley B, Krick E, et al. Outcome and toxicity associated with a dose-intensified, maintenance-free CHOP-based chemotherapy protocol in canine lymphoma: 130 cases. Vet Comp Oncol. 2010;8(3):196–208. doi: 10.1111/j.1476-5829.2010.00222.x. [DOI] [PubMed] [Google Scholar]
- 23.Starkey MP, Murphy S. Using lymph node fine needle aspirates for gene expression profiling of canine lymphoma. Vet Comp Oncol. 2010;8(1):56–71. doi: 10.1111/j.1476-5829.2009.00205.x. [DOI] [PubMed] [Google Scholar]
- 24.Staudt LM, Dave S. The biology of human lymphoid malignancies revealed by gene expression profiling. Adv Immunol. 2005;87:163–208. doi: 10.1016/S0065-2776(05)87005-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tamburini BA, Phang TL, Fosmire SP, et al. Gene expression profiling identifies inflammation and angiogenesis as distinguishing features of canine hemangiosarcoma. BMC Cancer. 2010;10(1):619. doi: 10.1186/1471-2407-10-619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tamburini BA, Trapp S, Phang TL, et al. Gene expression profiles of sporadic canine hemangiosarcoma are uniquely associated with breed. PLoS ONE. 2009;4(5):e5549. doi: 10.1371/journal.pone.0005549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomas R, Seiser EL, Motsinger-Reif A, et al. Refining tumor-associated aneuploidy through “genomic recoding” of recurrent DNA copy number aberrations in 150 canine non-Hodgkin lymphomas. Leuk Lymphoma. 2011;52(7):1321–1335. doi: 10.3109/10428194.2011.559802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Valli VE, Jacobs RM, Parodi AL, et al. Classification of Hematopoietic Tumors of Domestic Animals. Washington, DC: American Registry of Pathology; 2002. (2nd). [Google Scholar]
- 29.Valli VE, San Myint M, Barthel A, et al. Classification of canine malignant lymphomas according to the World Health Organization criteria. Vet Pathol. 2011;48(1):198–211. doi: 10.1177/0300985810379428. [DOI] [PubMed] [Google Scholar]
- 30.Valli VE, Vernau W, de Lorimier LP, et al. Canine indolent nodular lymphoma. Vet Pathol. 2006;43(3):241–256. doi: 10.1354/vp.43-3-241. [DOI] [PubMed] [Google Scholar]
- 31.Valli VEO. Veterinary pathologists achieve 80% agreement in application of WHO diagnoses to canine lymphoma. Cancer Ther. 2008;6:221–226. [Google Scholar]
- 32.Wilkerson MJ, Dolce K, Koopman T, et al. Lineage differentiation of canine lymphoma/leukemias and aberrant expression of CD molecules. Vet Immunol Immunopathol. 2005;106(3–4):179–196. doi: 10.1016/j.vetimm.2005.02.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.