

Abstract
Store-operated Ca2+ channels (SOCs) are voltage-independent Ca2+ channels activated upon depletion of the endoplasmic reticulum Ca2+ stores. Early studies suggest the contribution of such channels to Ca2+ homeostasis in insulin-secreting pancreatic β-cells. However, their composition and contribution to glucose-stimulated insulin secretion (GSIS) remains unclear. In this study, endoplasmic reticulum Ca2+ depletion triggered by acetylcholine (ACh) or thapsigargin stimulated the formation of a ternary complex composed of Orai1, TRPC1, and STIM1, the key proteins involved in the formation of SOCs. Ca2+ imaging further revealed that Orai1 and TRPC1 are required to form functional SOCs and that these channels are activated by STIM1 in response to thapsigargin or ACh. Pharmacological SOCs inhibition or dominant negative blockade of Orai1 or TRPC1 using the specific pore mutants Orai1-E106D and TRPC1-F562A impaired GSIS in rat β-cells and fully blocked the potentiating effect of ACh on secretion. In contrast, pharmacological or dominant negative blockade of TRPC3 had no effect on extracellular Ca2+ entry and GSIS. Finally, we observed that prolonged exposure to supraphysiological glucose concentration impaired SOCs function without altering the expression levels of STIM1, Orai1, and TRPC1. We conclude that Orai1 and TRPC1, which form SOCs regulated by STIM1, play a key role in the effect of ACh on GSIS, a process that may be impaired in type 2 diabetes.
Keywords: beta cell (B-cell), calcium release-activated calcium channel protein 1 (ORAI1), insulin secretion, stromal interaction molecule 1 (STIM1), transient receptor potential channels (TRP channels)
Introduction
The prevalence of type 2 diabetes is increasing at an alarming rate, because of the combination of aging population, urbanization, physical inactivity, and the increasing prevalence of obesity (1). Type 2 diabetes originates from β-cell failure to compensate insulin resistance and secrete the necessary amount of insulin to maintain glucose and lipid homeostasis (2, 3). The fine-tuning of insulin secretion in response to glucose and other nutrients relies on a balance of signals controlling dynamic variations in the cytoplasmic Ca2+ concentration ([Ca2+]c) (4). The major pathway for [Ca2+]c increase involves metabolism of glucose and other nutrients, which leads to ATP production and closure of the ATP-sensitive potassium channels. This in turn results in membrane depolarization and opening of voltage-dependent Ca2+ channels (VDCCs),2 leading to a rapid elevation of [Ca2+]c, resulting in insulin exocytosis (4). Glucose-stimulated insulin secretion (GSIS) is modulated by several hormones and neurotransmitters, among which acetylcholine (ACh) plays a prominent role (5). ACh binding to M3 muscarinic receptors stimulates the phospholipase C, which generates two second messengers, diacylglycerol (DAG), and inositol 1,4,5-trisphosphate (IP3). DAG plays a key role in the activation of various protein kinase C isoforms, whereas IP3 binding to its receptor (IP3R) promotes the release of Ca2+ from the endoplasmic reticulum (ER) (5).
ER Ca2+ store depletion is known to trigger Ca2+ influx across the plasma membrane (PM) through a family of channels referred to as store-operated Ca2+ channels (SOCs) (6, 7). It has been widely accepted that the store-operated Ca2+ entry (SOCE) is a major and ubiquitous Ca2+ influx pathway in nonexcitable cells, necessary for the replenishment of intracellular Ca2+ stores (8). Orai1 was identified as the main protein forming SOCs that conducts the previously called Ca2+ release-activated current (ICRAC) (9). The activity of Orai1 channels is closely controlled by the ER membrane protein STIM1 (stromal interacting molecule 1), which functions as an ER Ca2+ sensor and translocates upon ER depletion to ER-PM regions of Orai1 clustering (10). Some transient receptor potential canonical (TRPC) channels are responsible for less selective store-operated currents carried by divalent and monovalent cations. Based on sequence similarity and function, the TRPC channel family is divided into two subgroups, TRPC1/4/5 as SOCs and TRPC3/6/7 as receptor-operated channels (11). Interestingly, TRPC1 has been shown to form STIM1-regulated SOCs together with Orai1 (12, 13).
Despite the prominent role of VDCCs in GSIS and [Ca2+]c dynamics, various aspects of the intracellular Ca2+ responses of β-cells to glucose and secretagogues are still unexplained, suggesting the involvement of additional plasmalemmal Ca2+ channels. Early studies indicate that the emptying of intracellular Ca2+ stores in β-cells induces SOCE (14–18). However the composition of the SOCE-mediated channels and their exact role in GSIS is unclear. A few studies performed in mouse models of insulin-secreting cells suggest that β-cells express STIM1 and Orai1 (19, 20), as well as several TRPC isoforms (18, 21). However, the role of STIM1, Orai1, and TRPCs in insulin secretion remains elusive. The aim of this study was to investigate the composition and potential role of SOCs during GSIS and in particular in response to ACh. We showed that Orai1 and TRPC1 form the channels responsible for SOC-mediated Ca2+ entry in β-cells and play a major role, together with the regulating protein STIM1, in the potentiating effect of ACh on GSIS. Finally, we demonstrated that prolonged exposure to supraphysiological glucose concentration impaired SOCs function, suggesting SOCs as therapeutic targets to improve β-cell function in type 2 diabetes.
Experimental Procedures
Materials and Plasmids
Acetylcholine, thapsigargin, SKF-96365, verapamil, and diazoxide were purchased from Sigma-Aldrich. BTP2 and xestospongin C were purchased from Calbiochem (EMD Millipore SAS, Molsheim, France). pmaxGFP vector was obtained from Amaxa Biosystems (Lonza). YFP-STIM1 (18861), YFP-STIM1-ΔK (18861), YFP-Orai1 (19756), and YFP-Orai1-E106D were purchased from Addgene (9, 22, 23). TRPC1 was a kind gift from Joo Young Kim (Yonsei University College of Medicine, Seoul, Korea) (12, 24), the mutant TRPC1 (F562A) (25) construct was a kind gift from Shmuel Muallem (University of Texas Southwestern Medical Center, Dallas, TX), and the N-terminal truncated fragment of human TRPC3 (amino acids 1–302 of hTRPC3) NTRPC3-YFP was a kind gift from Klaus Groschner (University of Graz, Graz, Austria) (26).
Cell Culture
The rat insulinoma cell line INS-1E (27) (kindly provided by Dr. Pierre Maechler (Centre Médical Universitaire, University of Geneva, Geneva, Switzerland) was maintained as previously described (28, 29). Islets of Langerhans from male Wistar rats (Janvier, France) were isolated by collagenase digestion and maintained as previously described (28, 29). Islets were dissociated in a 1:1 PBS-Trypsin-EDTA solution at 37 °C and mechanically dissociated by pipetting for 2 min. Rat care and euthanasia procedures were approved by the Cantonal Veterinary Office (Service de la Consommation et des Affaires Vétérinaires SCAV-EXPANIM, authorization number 2543).
Cell Transfection
INS1-E cells were transiently transfected with plasmids described above using Lipofectamine 2000 (Life Technologies) as previously described (28, 29). Cells were then cultured for a 36-h recovery period before being collected or treated as indicated. Empty pmaxGFP or pcDNA3-YFP vectors were used as control to verify whether transfection itself could affect the Ca2+ response.
Confocal Imaging of YFP-STIM1 Fluorescent Protein
For live cell imaging, INS-1E cells grown on glass-bottomed plates were placed in the on-stage incubator of a Zeiss LSM 710 Quasar Confocal inverted microscope (Zeiss, Germany). Photo excitation was achieved by illumination with the 488-nm line of the argon gas laser, and Z-stack time lapse image acquisition was performed using the accompanying Zen2009 software (Zeiss). Projection of Z-stack images was performed using ImageJ 1.48a software by maximum intensity projection of top to middle stack of the cell of interest.
Insulin Secretion and Human Growth Hormone (hGH) Secretion
Static insulin secretion assays were performed in INS-1E cells in KRBH buffer (130 mm NaCl, 4.8 mm KCl, 0.5 mm NaH2PO4, 5 mm NaHCO3, 2 mm CaCl2, 1.2 mm MgCl2, 10 mm HEPES, 2 mm CaCl2) as previously described (29). Perifusion experiments were performed using chambers developed in Prof. Raddatz's laboratory (30). Groups of 20–30 islets were perifused at 37 °C with KRBH solution (1.6 mm glucose) at a flow rate of 500 μl/min for 30 min to establish stable basal insulin secretion. Then the glucose concentration was raised to 16.7 mm glucose, and fractions were collected every minute. Insulin release and content were measured using the rat insulin enzyme immunoassay according to the manufacturer's instructions (Cayman Chemical, Adipogen AG, Liestal, Switzerland).
To specifically assess the exocytosis activity of transfected cells only, we used a hGH secretion reporter assay, as previously described (28). INS-1E cells were transiently co-transfected with a construct encoding the hGH, together with an empty vector (pmaxGFP) as control, or a plasmid encoding mutant version of Orai1 or TRPC1. 48 h later, hGH secretion experiments were performed as in static insulin experiments. The hGH (secreted and content) levels were determined using the hGH ELISA kit (Roche Applied Sciences, Switzerland), and exocytosis activity expressed as the ratio of secreted hGH over hGH content.
Cytosolic Ca2+ Monitoring
In most experiments, loading of the cells was performed at 25 °C in the dark in KRBH supplemented with 2 mm CaCl2, 20 mm glucose, and 3 μm Fura-2/AM plus 10% (w/v) Pluronic® F-127 (Life Technologies) for 40 min. Following loading, cells were washed and observed under a Zeiss inverted microscope (×40 oil immersion fluorescence objective). Fura-2-AM was excited at 340/380 nm with a Visichrome holographic monochromator, and emission fluorescence was monitored at 510 nm using a Hamamatsu Orca ER coded CCD camera. Images were treated with MetaFluor to evaluate the ratio of fluorescence emitted at 340 and 380 nm (Cellular Imaging Facility, University of Lausanne, Lausanne, Switzerland). To study the thapsigargin or acetylcholine-activated Ca2+ entry, loaded cells were washed in Ca2+-free KRBH containing 0.1 mm EGTA and depleted with 5 μm thapsigargin (Tg) or 100 μm ACh in the presence of the ATP-sensitive potassium channel opener diazoxide (D; 250 μm) and of the VDCCs blocker verapamil (V; 10 μm). Subsequently, 2 mm Ca2+ (in the presence of diazoxide + verapamil (D+V)) was added to the medium, and the peak amplitude of the fluorometric signal was measured as the ΔF (340/380 nm ratio). ER Ca2+ depletion was calculated as the area under the curve based on the ΔF normalized to basal line over time in 0 mm Ca2+.
PCR Amplification
Total RNA from INS-1E cells or freshly isolated rat islets were extracted using Tripure isolation reagent (Roche Diagnostics) and reverse-transcribed using the ImProm-2 reverse transcription System (Promega AG) as previously described (28, 29). PCR was performed using Titanium® Taq PCR kit (Takara Bio Europe/Clontech). Primer sequences for classic PCR are provided in Table 1.
TABLE 1.
Primer sequences for PCR amplifications
| Target mRNA | Amplicon bp | Sequence | |
|---|---|---|---|
| Rat TRPC1 | 240 | Sense | 5′-TTGCTTGCAGAAGGCTGCTT-3′ |
| Antisense | 5′-ACCAAACGTGCTGCAGGAAT-3′ | ||
| Rat ORAI1 | 593 | Sense | 5′-CAACGTCCACAACCTCAACTC-3′ |
| Antisense | 5′-AAAGCCTCTTCCTTCCACAC-3′ | ||
| Rat STIM1 | 613 | Sense | 5′-GGGAACAGATCAGCAGAGTTTC-3′ |
| Antisense | 5′-TGGGGCAGAGGGAAAATATC-3′ |
Western Blotting
The cells were washed once with 1× ice-cold PBS and directly lysed with Laemmli buffer. Lysates were resolved by SDS-PAGE and transferred to PVDF membrane, and Western blotting was performed as previously described (28, 29) using primary antibodies directed against Orai1 (catalog number O8264; Sigma-Aldrich), STIM1 (catalog number S6197; Sigma-Aldrich), TRPC1 (sc-133076; Santa Cruz Biotechnology, LabForce AG, Muttenz, Switzerland), and TRPC3 (Alomone Labs, Jerusalem, Israel). The membranes were revealed by enhanced chemiluminescence (EMD Millipore SAS) using the ChemiDocTM XRS+ System and analyzed using the accompanying proprietary program Image Lab (BETA2) version 3.0.01 (Bio-Rad).
Co-immunoprecipitation and Duolink Proximity Ligation Assay
After lysis, 2 μg of antibody was added to a volume of lysates containing 300 μg of proteins diluted with 600 μl of NET solubilization buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 5 mm EDTA, 0.05% Nonidet P-40 (v/v)) and incubated at 4 °C overnight with constant mixing. Then the protein-antibody complex was incubated for 1 h at 4 °C with constant mixing with 40 μl of protein A/G magnetic beads (Millipore). The immune complexes were collected with a magnetic stand and washed three times in NET, and samples were subjected to SDS-PAGE. The negative control were performed using lysates with beads (ctrl(−)) without any antibody. For technical reasons, it was not possible to perform immunoblotting of Orai1 because its molecular weight is approximately the same as the IgG light chain.
The Duolink in situ proximity ligation assay (Olink Bioscience, Uppsala, Sweden) is a sensitive method to detect protein-protein interaction in intact tissue using oligonucleotide-conjugated secondary antibodies that, when in close proximity, allow an in situ polymerization reaction (31). INS-1E cells were fixed for 5 min in −20 °C acetone and processed according to manufacturer's instructions and as previously described (32). The following antibodies were used to test protein-protein interaction: mouse anti-TRPC1 (1:500; Santa Cruz Biotechnology), polyclonal goat anti-Orai1 (1:500; sc-74778; Santa Cruz Biotechnology), and rabbit anti-STIM1 (1:1000; Sigma-Aldrich). The proximity ligation assay fluorescent signal was quantified using the ImageJ software (National Institutes of Health, Bethesda, MD) as follows: 30 images from 4 distinct experiments were converted to a 32-bit format, and the signal to noise ratio was determined by applying the Yen thresholding method. A binary image was then created, and the number of pixels of the duolink signal was measured. The data were normalized to the number of cells (nuclei imaged by DAPI staining) in each image.
Statistical Analysis
The data are presented as means ± S.E. Comparisons were performed by two-tailed unpaired Student's t test and between at least three groups with one-way or two-way analysis of variance completed by Fisher's least significant difference post hoc test for multiple comparisons. A p value of ≤0.05 was considered statistically significant.
Results
Inhibition of SOCs Leads to Impaired GSIS in isolated rat islets and INS-1E cells
To determine whether SOCs channels contribute to insulin secretion, 20–30 rat islets were placed in an islet perifusion chamber infused with KRBH supplemented with low (1.6 mm) followed by high (16.7 mm) glucose concentration, together or not with common SOCs inhibitors, SKF-96365 (SKF) or BTP2 (33). SKF and BTP2 impeded the first peak (5–12 min) and nearly fully blocked the second phase (13–30 min) of GSIS (Fig. 1, A and B), thereby reducing total insulin release by about 70 and 60%, respectively (Fig. 1, A and B). Further static insulin secretion experiments performed in INS-1E cells confirmed that SKF and BTP2 partially inhibited insulin secretion induced by 16.7 mm glucose (Fig. 1C). ACh or the SERCA pump inhibitor Tg led to ER Ca2+ depletion and activation of SOCs, and potentiated GSIS in β-cells (4, 15, 34). SKF or BTP2 fully blocked the potentiating effect of 100 μm ACh or 1 μm Tg in INS-1E cells (Fig. 1C). These data suggest that SOCs participate to GSIS, especially in potentiating mechanisms linked to ER Ca2+ release. Of note, SKF and BTP2 had no impact on insulin content in INS-1E cells (data not shown).
FIGURE 1.
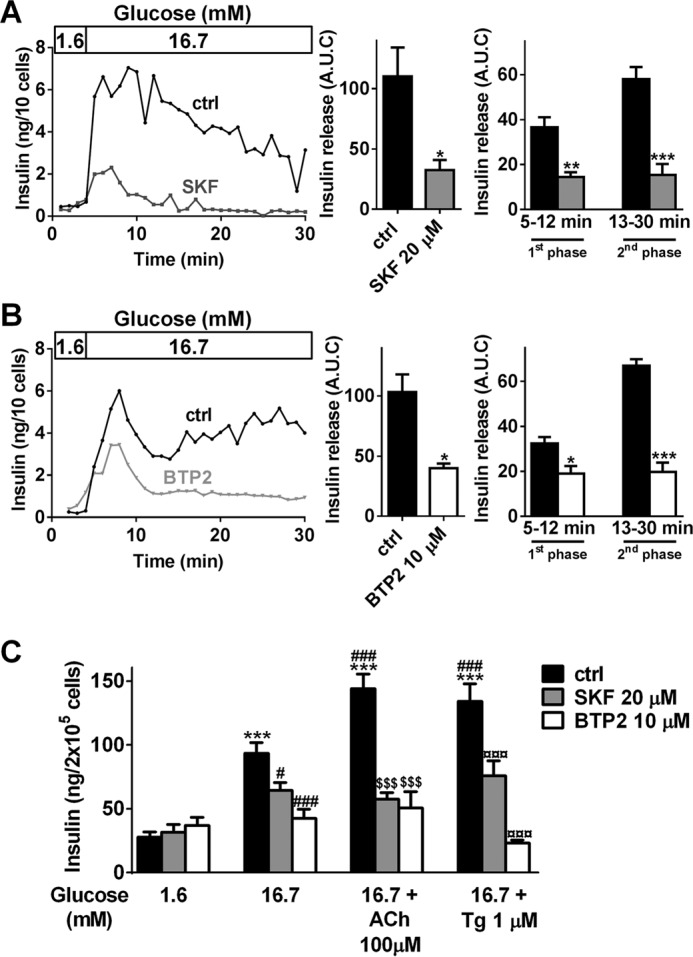
Inhibition of SOCs by SKF-96365 or BTP2 leads to impaired GSIS in β-cells. A and B, perifusion experiments of primary rat islets preincubated at 1.6 mm glucose and exposed to 16.7 mm glucose, in the absence (control, ctrl) or presence of 20 μm SKF-96365 (SKF, A) or 10 μm BTP2 (B). Left panels, representative traces of insulin release in the absence (ctrl, black traces) or presence of SKF (A, dark gray trace) or BTP2 (B, light gray trace). Middle panels, data are areas under the curve (A.U.C.) of insulin release from three to five independent experiments. *, p < 0.05 versus respective control insulin secretion. Right panels, quantification of insulin release during the first phase (5–12 min) or second phase (13–30 min). The data are the areas under the curve from three to five independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus respective ctrl insulin secretion. C, INS-1E cells were cultivated for 30 min in KRBH supplemented with 1.6, 16.7, or 16.7 mm glucose + 100 μm ACh or 16.7 mm glucose + 1 μm Tg in the absence (ctrl, black bars) or presence of SKF (20 μm, gray bars) or BTP2 (10 μm, white bars). Supernatants were collected after 30 min of stimulation. The data are from five independent experiments. ***, p < 0.001 versus basal insulin secretion at 1.6 mm glucose; #, p < 0.05; ###, p < 0.001 versus insulin secretion at 16.7 mm glucose; $$$, p < 0.001 versus insulin secretion at 16.7 mm glucose + ACh; ¤¤¤, p < 0.001 versus insulin secretion at 16.7 mm glucose + Tg.
Acetylcholine and Thapsigargin Trigger SOCE in INS-1E and Dispersed Rat Islet Cells
To further investigate the role of SOCs in GSIS, we first studied the effect of ACh and Tg on [Ca2+]c at stimulating 20 mm glucose concentration in Fura-2-loaded β-cells. SOCs were activated by ER Ca2+ store depletion using 2 μm Tg or 100 μm ACh in the absence of external Ca2+ and then exposed to 2 mm Ca2+ to elicit SOCE in INS-1E cells (Fig. 2, A and B) or dispersed rat islet cells (Fig. 2, C and D) in high glucose conditions. To selectively study SOCE without interference from VDCCs, the cells were hyperpolarized with the ATP-sensitive potassium channel opener diazoxide (D) combined with the VDCCs blocker verapamil (V) (15, 35). As shown in Fig. 2, store depletion in Ca2+-free solution with Tg or ACh induced large Ca2+ release from the ER in INS-1E cells and in dispersed rat islet cells. When extracellular Ca2+ was reintroduced, both Tg- and ACh-mediated depletion stimulated SOCE in INS-1E cells (Fig. 2, A and B) and dispersed rat islet cells (Fig. 2, C and D). In these conditions, i.e. when VDCCs were blocked, SOCs inhibition by SKF or BTP2 completely abolished Tg and ACh-mediated Ca2+ entry (Fig. 2, A–D). In contrast, the SOCs inhibitors BTP2 or SKF had no significant effect on Ca2+ entry stimulated by acute exposure to high glucose in nondepleted INS-1E cells (Fig. 2E). As expected, this Ca2+ entry was reduced by 60% in the presence of D+V, confirming that high glucose-stimulated Ca2+ entry is essentially due to VDCCs activity (Fig. 2E). This last result also confirmed that the SOCs inhibitors did not affect VDCCs activity.
FIGURE 2.

Inhibition of SOCs by SKF-96365 or BTP2 leads to reduced Tg- and ACh-induced SOCE in INS-1E cells and primary rat islet cells. Typical recording of [Ca2+]i variations measured with Fura-2/AM in INS-1E (A and B) and in dispersed rat islets cells (C and D) at 20 mm glucose. ER Ca2+ content was depleted in Ca2+-free medium by application of 2 μm Tg (A and C) or 100 μm ACh (B and D) in the presence of D+V. SOCE was measured by the addition of 2 mm Ca2+ in the extracellular medium also in the presence of D+V. A and B, left panels, representative mean traces of [Ca2+]i variation in INS-1E cells exposed to Tg (A) or ACh (B) in the absence (ctrl, black traces) or presence of 20 μm SKF (dark gray traces) or 10 μm BTP2 (light gray traces). Right panels, quantification of the amplitude of SOCE (ΔF) upon extracellular addition of 2 mm Ca2+. n = 5–7 experiments, n = 30 investigated cells/experiment minimum. *, p < 0.05; **, p < 0.01 versus ctrl condition. C and D, left panels, representative mean traces of [Ca2+]i variation in dispersed rat islets cells exposed to Tg (C) or ACh (D) in the absence (ctrl, black traces) or presence of 10 μm BTP2 (light gray traces). Right panels, quantification of the amplitude of SOCE (ΔF) upon extracellular addition of 2 mm Ca2+. n = 4 experiments, n = 10 investigated cells/experiment minimum. ***, p < 0.001 versus ctrl condition. E, quantification of high glucose-induced Ca2+ influx in nondepleted cells in absence (ctrl) or presence of the indicated inhibitors. **, p < 0.01 versus ctrl condition. n = 5 experiments, n = 30 investigated cells/experiment minimum. F, left panel, representative mean traces of [Ca2+]i variation in INS-1E cells exposed to ACh in the presence or not (ctrl) of xestospongin C (XeC). Middle panel, quantification of ACh-induced ER Ca2+ depletion content. Right panel, quantification of the amplitude of SOCE (ΔF) upon extracellular addition of 2 mm Ca2+. n = 4 experiments, n = 20 investigated cells/experiment minimum. *, p < 0.05; ***, p < 0.001 versus ctrl condition.
ACh binding to M3 muscarinic receptors stimulates the PLCβ, which generates DAG and IP3. DAG plays a key role in the activation of TRPC3/6/7 channels referred to as receptor-operated channels, whereas IP3 binding to its receptor (IP3R) promotes the release of Ca2+ from ER activating SOCs (5). Inhibition of IP3R using 20 μm xestospongin C (36) reduced ACh-induced ER Ca2+ depletion by 70% and decreased the ACh-induced SOCE by 60% (Fig. 2F), suggesting that ACh-mediated extracellular Ca2+ influx required IP3R-mediated Ca2+ release in INS-1E cells.
STIM1, Orai1, and TRPC1 Form a Molecular Complex upon ER Ca2+ Depletion in β-Cells
TRPC1, STIM1, and Orai1 were expressed in INS-1E cells and freshly isolated rat islets at the mRNA and protein levels (Fig. 3A). In response to ER Ca2+ store depletion, STIM1 is known to aggregate and translocate to ER-PM region, where it interacts with and activates Orai1-TRPC1 (10, 37). To study the intracellular localization of STIM1 in INS-1E cells, we overexpressed a fluorescent YFP-STIM1 protein. As expected, YFP-STIM1 displayed a reticular localization in control condition (11 mm glucose, t0) and rapidly aggregated to discrete regions close to the PM in cells exposed to 1 μm Tg or 100 μm ACh (Fig. 3B). We also found that TRPC1 co-immunoprecipitated with Orai1 and STIM1 and that STIM1 co-immunoprecipitated with Orai1 and TRPC1, demonstrating the existence of a ternary complex between TRPC1, STIM1, and Orai1 in untreated (data not shown) and in Tg-treated INS-1E cells (Fig. 3C). Proximity ligation assays (31) further demonstrated an increased interaction between Orai1, TRPC1, and STIM1 in cells treated with ACh or Tg (Fig. 3D) compared with untreated cells, indicating that ER Ca2+ depletion promotes the formation of this ternary complex. Of note, as compared with negative controls (ctrl(−)) where only one antibody against Orai1, TRPC1, or STIM1 was used, INS-1E cells displayed little or no STIM1, Orai1, and TRPC1 interactions in normal conditions (ctrl) (Fig. 3D).
FIGURE 3.

Tg- or ACh-dependent ER Ca2+ depletion increases STIM1/Orai1, STIM1/TRPC1, and Orai1/TRPC1 interaction in INS-1E cells. A, TRPC1, STIM1, and Orai1 are expressed in rat β-cells. TRPC1, STIM1, and Orai1 mRNAs were identified by RT-PCR in INS-1E cells (top panel) and primary rat islets (middle panel) (n = 3 experiments). PCR products of the predicted size for TRPC1, STIM1, and Orai1 were 120, 450, and 398 bp, respectively. The negative control (H2O) contained water instead of DNA. Bottom panel, representative Western blot experiments of TRPC1, Orai1, and STIM1 proteins expression in rat islets and in INS-1E cells (n = 3 experiments). B, representative conventional (top panel) and confocal imaging (bottom panel) of YFP-STIM1 signal in transfected INS-1E cells at t0 (ctrl) and after 4 min (t4min) of exposure to 1 μm Tg or 100 μm ACh. Scale bar, 10 μm. Inset represent 3-fold magnification of main images. C, representative co-immunoprecipitation experiments from INS-1E cells treated with 1 μm Tg during 5 min showing that TRPC1, Orai1, and STIM1 proteins formed a macromolecular complex in Tg-INS-1E-treated cells. Lysates (input lane) were incubated with antibodies against TRPC1, STIM1, or Orai1 (IP). Western blots of the immunoprecipitated proteins were probed with antibodies against TRPC1 (top panel) or STIM1 (bottom panel). D, proximity ligation assay revealed interactions (white spots) between STIM1, Orai1 and TRPC1 in INS-1E cells (DAPI nuclei labeling in blue) exposed to 1 μm Tg or 100 μm ACh for 5 min. Scale bar, 10 μm. Bottom panels, quantification of labeled DNA probe (white spots) normalized to the number of cells (DAPI-labeled nuclei). n = 4–6 experiments. *, p < 0.05; **, p < 0.01 versus ctrl condition.
STIM1, Orai1, and TRPC1 Are Critical in SOCE in INS-1E Cells
To assess the specific and respective role of Orai1, TRPC1, and STIM1 in SOCE, native or dominant negative constructs of Orai1, TRPC1, and STIM1 were expressed in INS-1E cells 36 h before performing the same SOCE protocol as in Fig. 2 with (Fig. 4, B–D) or without D+V (Fig. 4, A and E). We favored a dominant negative strategy to suppress the function (via point mutation in the ionic pore) of a target channel without affecting its endogenous expression, which might induce compensatory regulation of other channels. SOCE was analyzed in Ca2+ imaging experiments in transfected cells only identified by YFP or GFP tag. Of note, GFP/YFP-transfected cells displayed similar SOCE compared with untransfected cells (data not shown). YFP-Orai1 overexpression had no significant effect on SOCE as compared with pmaxGFP-transfected cells (GFP) (Fig. 4A). In contrast, SOCE was increased by 40% in YFP-STIM1-positive cells, and the co-overexpression of YFP-STIM1 and YFP-Orai1 tripled the SOCE amplitude (Fig. 4A), demonstrating a crucial role of the STIM1-Orai1 duet in SOCE. TRPC1 overexpression alone increased SOCE by 40%, and the co-overexpression of YFP-STIM1 and TRPC1 further increased SOCE by 30% compared with TRPC1 alone (Fig. 4A). STIM1 has been proposed to gate TRPC1 through its K-rich domain (38). The overexpression of STIM1 deleted in its K-rich domain (YFP-STIM1-ΔK) alone or the co-overexpression of YFP-STIM1-ΔK with TRPC1 did not increase SOCE (Fig. 4A), suggesting a regulatory role of STIM1 in TRPC1-mediated SOCE.
FIGURE 4.
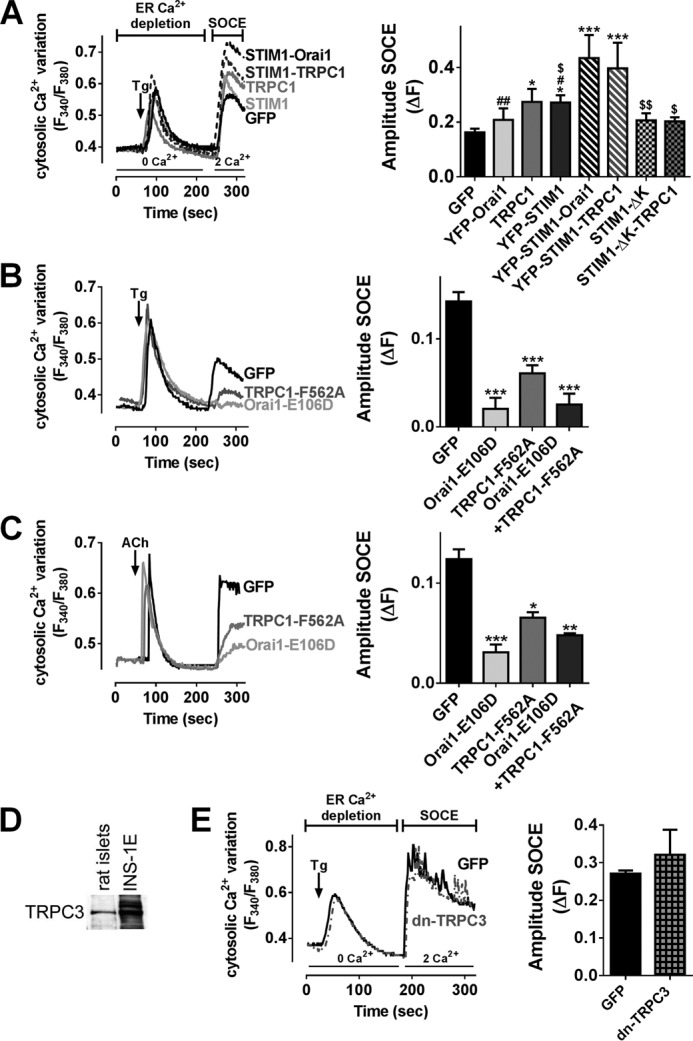
Orai1 and TRPC1 mediate Tg- and ACh-dependent Ca2+ entry in INS-1E cells. A, Fura-2/AM imaging in INS-1E cells transfected with the indicated plasmids in KRBH supplemented with 20 mm glucose. B and C, Fura-2/AM imaging in INS-1E cells transfected with the indicated plasmids in KRBH supplemented with 20 mm glucose in the presence of D+V. A, left panel, representative mean traces of [Ca2+]i variation in INS-1E cells exposed to Tg 2 μm in GFP-transfected cells (black trace), in transfected cells with YFP-STIM1 (light gray trace) or with TRPC1 (dark gray trace), in co-transfected cells with YFP-STIM1 and TRPC1 (dotted gray trace), or in co-transfected cells with YFP-STIM1 and YFP-Orai1 (dotted black trace). Right panel, quantification of the amplitude of SOCE (ΔF) upon extracellular addition of 2 mm Ca2+. n = 5–13 experiments, n = 10 investigated cells/experiment minimum. *, p < 0.05; ***, p < 0.001 versus GFP-transfected cells; #, p < 0.05; ##, p < 0.01 versus STIM1-Orai1-transfected cells; $, p < 0.05; $$, p < 0.01 versus STIM1-TRPC1-transfected cells. B and C, left panels, representative mean traces of [Ca2+]i variation in INS-1E cells exposed to Tg (B) or ACh (C) in GFP-transfected cells (black traces), in transfected cells with TRPC1-F562A (dark gray traces), or in transfected cells with Orai1-E106D (light gray traces). Right panels, quantification of the amplitude of SOCE (ΔF) upon extracellular addition of 2 mm Ca2+. n = 4 experiments, n = 10 investigated cells/experiment minimum. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus GFP-transfected cells. D, representative Western blot experiments of TRPC3 protein expression in rat islets and INS-1E cells (n = 3 experiments). E, Fura-2/AM imaging in INS-1E cells transfected with pmaxGFP (GFP) or dominant negative TRPC3 plasmid in KRBH supplemented with 20 mm glucose. Left panel, representative mean traces of [Ca2+]i variation in INS-1E cells exposed to Tg 2 μm in GFP-transfected cells (black trace) and in transfected cells with dominant negative TRPC3 (dotted gray trace). Right panel, quantification of the amplitude of SOCE (ΔF) upon extracellular addition of 2 mm Ca2+. n = 4 experiments, n = 25 investigated cells/experiment minimum.
Overexpression of Orai1-E106D, a dominant negative pore mutant for Orai1 (9), reduced Tg or ACh-induced SOCE to the same extend as BTP2 or SKF, suggesting that Orai1 is the predominant protein forming SOCs in β-cells (Fig. 4, B and C). Overexpression of TRPC1-F562A, a dominant negative pore mutant for TRPC1 (25), also reduced by 50% the SOCE triggered by Tg or ACh (Fig. 4, B and C). The combined inactivation of TRPC1 and Orai1 channels had no additive effect (Fig. 4, B and C), suggesting that TRPC1 and Orai1 are mutually dependent to mediate SOCE. We also observed that TRPC3 is expressed in rat β-cells (Fig. 4D). TRPC3 blockade using the dominant negative fragment of TRPC3 NTRPC3-YFP (26) had no effect on Tg-stimulated Ca2+ influx (Fig. 4E). These Ca2+ imaging data show that Orai1 and TRPC1, but not TRPC3, mediate SOCE, and that STIM1 constitutes the main trigger to activate Orai1/TRPC1 in INS-1E cells.
Functional Orai1 and TRPC1 Channels Are Required to Maintain INS-1E Cell Secretory Function
To further investigate the role of Orai1 and TRPC1 in β-cell function, we studied the effect of the dominant negative mutants Orai1-E106D or TRPC1-F562A on GSIS in INS-1E cells. To avoid confounding factors caused by transfection efficiency, the exocytosis activity was assessed in transfected cells only, using a reporter system with a plasmid encoding the hGH, which is secreted in the same granules as insulin (28). Orai1-E106D or TRPC1-F562A expression reduced glucose-induced hGH exocytosis by 50 and 30%, respectively, and fully blocked the potentiating effect of 100 μm ACh or 1 μm Tg (Fig. 5). Of note, selective inhibition of TRPC3 using Pyr3 at 10 μm (39) affected neither basal nor stimulated insulin secretion (data not shown). These results strengthen the idea of the specific nature of the store-dependent Ca2+ entry carried by TRPC1 and Orai1 in regulating GSIS and more, particularly in the potentiating effect of ACh.
FIGURE 5.
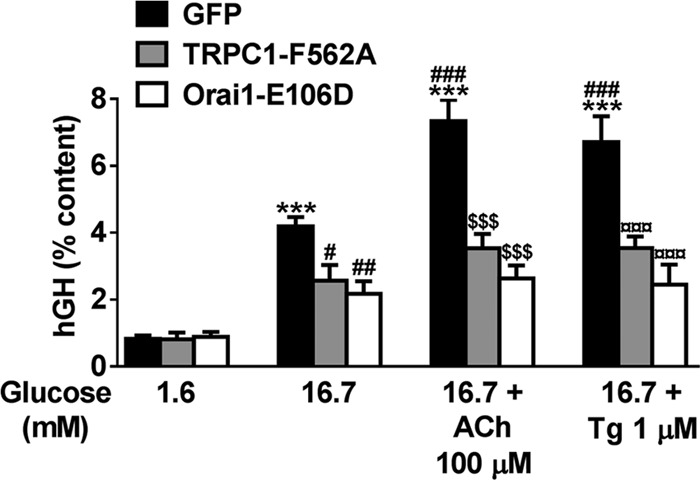
Orai1 and TRPC1 are involved in GSIS in INS-1E cells. INS-1E cells were co-transfected with plasmids coding for the hGH and one of the following plasmids: control pmaxGFP (GFP), TRPC1-F562A, or Orai1-E106D. Transfected cells were incubated for 30 min in KRBH supplemented with 1.6, 16.7, and 16.7 mm glucose + 100 μm ACh or 16.7 mm glucose + 1 μm Tg. hGH secretion (supernatant) was normalized by the hGH content (% content) and expressed as means ± S.E. of six independent experiments. ***, p < 0.001 versus basal insulin secretion at 1.6 mm glucose; #, p < 0.05; ##, p < 0.01; ###, p < 0.001 versus insulin secretion at 16.7 mm glucose; $$$, p < 0.001 versus insulin secretion at 16.7 mm glucose + ACh; ¤¤¤, p < 0.001 versus insulin secretion at 16.7 mm glucose + Tg.
Chronic High Glucose Leads to ER Ca2+ Depletion and Impaired Tg-induced SOCE in INS-1E Cells
Several lines of evidence underscore a role of chronic hyperglycemia referred to as “glucotoxicity” in dysfunctional insulin secretion and β-cell failure in type 2 diabetes (40). INS-1E cells were incubated in the presence of 30 mm glucose for 72 h (G30), a condition known to result in glucose desensitization and impaired GSIS (41, 42). To indirectly measure ER Ca2+ content, INS-1E cells were treated with 5 μm thapsigargin in the absence of extracellular Ca2+. The resulting [Ca2+]c rise directly depends on the Ca2+ concentration in this organelle (43). This first phase was followed by a second “descending” phase where [Ca2+]c returned to basal level (Fig. 6A), which directly reflects the Ca2+ buffering/export capacities of the cells (44, 45). As compared with cells maintained at control 11 mm glucose concentration (G11), G30 cells displayed decreased ER Ca2+ release (without changes in the kinetics of the Ca2+ release), suggesting reduced ER Ca2+ stores (Fig. 6A). In addition, the Tg-induced SOCE were decreased by 70% in G30 condition (Fig. 6B). Time course analysis revealed no effect of high glucose concentration on the protein levels of STIM1 (0.98 ± 0.02 at 72 h), Orai1 (0.92 ± 0.02 at 72 h), and TRPC1 (0.89 ± 0.06 at 72 h) (Fig. 6C), suggesting an effect of prolonged high glucose on SOC activity.
FIGURE 6.
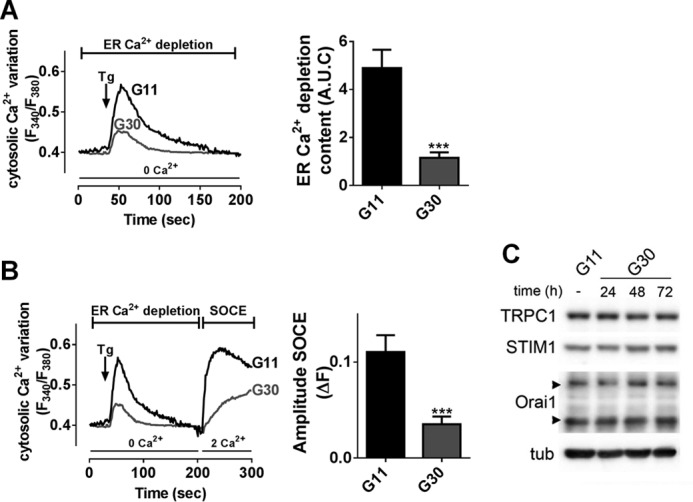
Prolonged high glucose concentration impairs Tg-induced Ca2+ influx. A and B, Fura-2/AM imaging in INS-1E cells maintained in 11 mm glucose (G11) or 30 mm glucose (G30) for 72 h in the presence of D+V. A, left panel, representative mean traces of ER Ca2+ depletion induced by 2 μm Tg in Ca2+-free medium. Right panel, quantification of Tg-induced ER Ca2+ release content (A.U.C.). B, left panel, representative mean traces of [Ca2+]i variation. Right panel, quantification of the amplitude of SOCE (ΔF) upon extracellular addition of 2 mm Ca2+. n = 4–7 experiments, n = 20 investigated cells/experiment minimum. ***, p < 0.001 versus G11 condition. C, representative Western blot of TRPC1, STIM1, Orai1, and α-tubulin (tub) in INS-1E cells cultivated in G11 or G30 condition for 24, 48, or 72 h. n = 4 experiments.
Discussion
GSIS relies mostly on Ca2+ entry through VDCCs. However, intracellular sequestration and release of Ca2+ from ER via pathways such as IP3R (4, 46) and ryanodine receptors (47–49) also have important roles in glucose-induced [Ca2+]i regulation and insulin secretion. Hence, in mouse β-cells, the production of IP3 induced by ACh triggers a rapid depletion of ER Ca2+ stores, which activates SOCs (14, 16, 17) and contributes to the amplifying pathway in GSIS (4). It was previously described that in excitable pancreatic β-cells, the modest SOCE modulates the membrane potential (15, 18) and is significant for the [Ca2+]c regulation, suggesting a potential role of such Ca2+ entry in insulin secretion in pancreatic β-cells. Currently, the nature of the channels mediating SOCE and their role in β-cell physiology is unclear. Here, we demonstrated the crucial role of SOCs in rat β-cell function and identified Orai1 and TRPC1 as the main pore-forming subunits of SOCs regulated by the Ca2+ sensor protein STIM1.
The concept of SOCE, proposed in 1986 (50), constitutes the predominant pathway of Ca2+ entry in nonexcitable cells. The original role of SOCE was to maintain ER Ca2+ levels. Although still under debate, there is a plethora of evidence that Orai1 forms the Ca2+ selective pore-forming unit of Ca2+ release-activated Ca2+ channels and that TRPCs, especially TRPC1 channels, form nonselective cation channels gated by store depletion. STIM1 serves as a Ca2+ sensor in the ER, which clusters proximally to the PM to activate Orai1 and/or TRPC channels when ER Ca2+ stores are depleted (11–13, 25, 51).
Here, we report that STIM1, TRPC1, and Orai1 mRNAs and proteins are present in rat β-cells. Overexpression of YFP-tagged STIM1 in rat β-cells confirms its translocation and aggregation to discrete regions close to the PM in response to ER Ca2+ store depletion by Tg or ACh. We further show that ER Ca2+ depletion stimulates the formation of a ternary complex composed of Orai1, TRPC1, and STIM1. This is in line with findings in mouse β-cells that STIM1 accumulates to subplasmalemmal region to co-cluster with Orai1 in response to ER depletion (19, 20). However, here we describe for the first time in β-cells that TRPC1 is an integral part of the STIM1/Orai1 complex. Overexpression experiments in Ca2+ imaging further confirm that both Orai1 and TRPC1 require STIM1 to form functional SOCs. Of note, the sole overexpression of Orai1 did not significantly increase SOCE, whereas the co-overexpression of YFP-STIM1 and YFP-Orai1 had a synergetic effect on SOCE. It is well described that the STIM1 to Orai1 ratio highly influences their activity. Some reports proposed that a ratio of 2/1 STIM1/Orai1 is optimal for maximal SOCE activity (52, 53). Thus, Orai1 overexpression alone may “dilute” the STIM1 binding to individual Orai1 channels, which could explain our data. In contrast, TRPC1 overexpression, alone or in combination with STIM1, increased SOCE, suggesting TRPC1 as an enhancer of SOCE, rather than the main SOC-forming partner. Specific blockade of Orai1 using the dominant negative Orai1 (Orai1-E106D) pore mutant abolished Tg-induced SOCE, whereas TRPC1 blockade using the TRPC1-F562A pore mutant only partially blocked Tg-induced SOCE, supporting the major role of Orai1. Moreover, concomitant inactivation of both channels had no further effect on SOCE, which is in accordance with our results and in line with previous reports (54, 55), supporting that TRPC1 and Orai1 are mutually dependent to form functional SOCs.
Although STIM1 interacts with and gates Orai1 via a cytosolic SOAR domain (amino acids 344–442) (12, 56), gating of TRPC1 by STIM1 following store depletion is achieved by a lysine-rich region in the STIM1 C terminus (684KK685) (38, 57). The overexpression of truncated STIM1 lacking the K-rich domain alone is not able to increase SOCE as the native STIM1. In addition, the co-overexpression of STIM1-ΔK with TRPC1 totally prevented the TRPC1-mediated SOCE. These data suggest a crucial role for STIM1 in store-dependent activation of TRPC1 channels in rat β-cells. Altogether these data suggest that Orai1/TRPC1/STIM1 may be key players to regulate the insulin secretion in β-cells.
In this study, SOCs were activated by ER Ca2+ store depletion using either the SERCA pump inhibitor thapsigargin or through ACh-mediated IP3 generation. We observed that ACh elicits robust IP3-induced Ca2+ release and triggers similar SOCE as thapsigargin in rat β-cells. However blockade of TRPC1 and Orai1 did not fully abolished ACh-induced SOCE, suggesting the involvement of other TRP-dependent Ca2+ channels. Besides SOCE, ACh may also induce receptor-operated Ca2+ entry through activation, by PKC- and/or DAG, of nonselective cation channels formed by other members of the TRPC family (58). This hypothesis is supported by studies showing that ACh triggers both SOC-dependent and SOC-independent inward currents in β-cells (14, 15, 17). We observed that TRPC3, which forms receptor-operated channels regulated by the DAG/PKC pathway (59, 60), is not involved in Tg-mediated Ca2+ entry and insulin secretion, supporting the hypothesis that TRPC1 is the main SOC-forming TRPC isoform (54, 55, 58). We also show that ACh-mediated Ca2+ entry requires IP3R-mediated Ca2+ release, suggesting that ACh-induced receptor-operated Ca2+ entry is minimal in β-cells. Altogether these data demonstrate that ACh triggers the opening of SOCs made of Orai1 and TRPC1 and regulated by the translocation of STIM1. We further demonstrated that blockade of Orai1 and TRPC1 via pharmacological inhibition or channel-dead mutant strategies impaired GSIS and fully blocked the potentiating effect of ACh and Tg on insulin secretion in INS-1E cells. SOCs inhibition altered mostly the second phase of insulin release, which is in line with the hypothesis that SOCs are mainly involved in the fine tuning of [Ca2+]c during the second phase where ER Ca2+ stores play a prominent role (61). Thus, we propose that SOCs contribute to the cholinergic amplifying pathway of insulin secretion (14, 16, 17), either through the SOC primordial role in refilling ER Ca2+ reservoirs or because this Ca2+ influx directly shapes [Ca2+]c oscillations (7) and pulsatile insulin secretion during the second phase. Our data also suggest a role for SOCs during the first phase of insulin release, when Ca2+ release from the ER is considered to play a minor role (62, 63). Of interest, recent reports indicate that STIM1 and Orai1 suppress L- and T-type Ca2+ channel activity in various cell types (64–66). We also previously showed that TRPC channels inhibit the L-type Ca2+ channel activity in the developing heart (30). In contrast, TRPC3 and TRPC6 channels have been shown, in vascular smooth muscle cells, to trigger sufficient depolarization to open VDCCs (67–69). Therefore, the Orai1/TRPC1/STIM1 complex may also contribute to insulin secretion via effects on VDCCs activity, which would have an effect on both phases of insulin release. The functional interaction between SOCs and VDCCs in β-cells deserves further investigation.
Other TRP channels have been shown to contribute to Ca2+ oscillations and GSIS, including Ca2+-permeable TRPM2 channel (70), monovalent cations TRPM4 (71) and TRPM5 channels (72), and nonselective cation TRPV4 channels (73). Our results and these previous reports ascertain the co-existence of both voltage-dependent and voltage-independent Ca2+ influx in β-cells.
SOCE is a ubiquitous and major mechanism for Ca2+ influx in mammalian cells. As a result, SOCs contribute to the function of all systems and organs and have physiological as well as pathophysiological relevance (7). In particular, accumulating evidence indicates altered SOCs function and/or expression associated with diabetic complications, including diabetes nephropathy, retinopathy, and peripheral neuropathies (74). It is well established that chronic exposure of isolated β-cells to hyperglycemic conditions results in glucose desensitization and impaired GSIS (41, 42). Although more studies are required to carefully assess the impact of glucose on ER Ca2+ levels, our data demonstrate for the first time that prolonged exposure to supraphysiological glucose concentration results in reduced ER Ca2+ release and decreased SOCE in INS-1E cells. Interestingly, this dysfunction seems independent of STIM1, Orai1, and TRPC1 expression, suggesting that hyperglycemia directly affects the activity of SOCs. SOCE being a key mechanism to replenish the ER Ca2+ reservoirs upon depletion, SOC dysfunction may also result in ER stress, which plays a key role in β-cell dysfunction and death (75). Further investigations are required to decipher the pathophysiological mechanisms by which SOCs are regulated in diabetes and to evaluate the therapeutic potential of the ternary TRPC1/Orai1/STIM1 complex.
Author Contributions
J. S. and F. A. designed the experiments. J. S., F. A., L. S., and L. L. G. performed the experiments. J. S., J.-A. H., E. R., and F. A. wrote the manuscript.
Acknowledgment
We thank Martine Lambelet (Laboratory of Vascular Surgery, Centre Hospitalier Universitaire Vaudois) for technical support.
This work was supported by Swiss National Science Foundation (SNSF) Grants 31003A-155897 (to J.-A. H.) and 310030-127633 (to E. R.). The authors declare that they have no conflicts of interest with the contents of this article.
- VDCC
- voltage-dependent Ca2+ channel
- ACh
- acetylcholine
- BTP2
- 3,5-bis(trifluoromethyl)pyrazole
- ER
- endoplasmic reticulum
- DAG
- diacylglycerol
- GSIS
- glucose-stimulated insulin secretion
- hGH
- human growth hormone
- IP3
- inositol 1,4,5-trisphosphate
- IP3R
- IP3 receptor
- PM
- plasma membrane
- SOCE
- store-operated Ca2+ entry
- SOC
- store-operated Ca2+ channel
- Tg
- thapsigargin
- TRPC
- transient receptor potential canonical
- D+V
- diazoxide + verapamil.
References
- 1.Wild S., Roglic G., Green A., Sicree R., and King H. (2004) Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care 27, 1047–1053 [DOI] [PubMed] [Google Scholar]
- 2.Poitout V., Amyot J., Semache M., Zarrouki B., Hagman D., and Fontés G. (2010) Glucolipotoxicity of the pancreatic beta cell. Biochim. Biophys. Acta 1801, 289–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robertson R. P., Harmon J., Tran P. O., and Poitout V. (2004) Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes 53, S119–S124 [DOI] [PubMed] [Google Scholar]
- 4.Gilon P., Chae H. Y., Rutter G. A., and Ravier M. A. (2014) Calcium signaling in pancreatic beta-cells in health and in type 2 diabetes. Cell Calcium 56, 340–361 [DOI] [PubMed] [Google Scholar]
- 5.Gilon P., and Henquin J. C. (2001) Mechanisms and physiological significance of the cholinergic control of pancreatic beta-cell function. Endocr. Rev. 22, 565–604 [DOI] [PubMed] [Google Scholar]
- 6.Putney J. W., Jr. (2001) Cell biology: channelling calcium. Nature 410, 648–649 [DOI] [PubMed] [Google Scholar]
- 7.Putney J. W. (2011) The physiological function of store-operated calcium entry. Neurochem. Res. 36, 1157–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parekh A. B., and Putney J. W. Jr. (2005) Store-operated calcium channels. Physiol. Rev. 85, 757–810 [DOI] [PubMed] [Google Scholar]
- 9.Prakriya M., Feske S., Gwack Y., Srikanth S., Rao A., and Hogan P. G. (2006) Orai1 is an essential pore subunit of the CRAC channel. Nature 443, 230–233 [DOI] [PubMed] [Google Scholar]
- 10.Zhang S. L., Yu Y., Roos J., Kozak J. A., Deerinck T. J., Ellisman M. H., Stauderman K. A., and Cahalan M. D. (2005) STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437, 902–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liao Y., Plummer N. W., George M. D., Abramowitz J., Zhu M. X., and Birnbaumer L. (2009) A role for Orai in TRPC-mediated Ca2+ entry suggests that a TRPC:Orai complex may mediate store and receptor operated Ca2+ entry. Proc. Natl. Acad. Sci. U.S.A. 106, 3202–3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuan J. P., Zeng W., Huang G. N., Worley P. F., and Muallem S. (2007) STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat. Cell Biol. 9, 636–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liao Y., Erxleben C., Abramowitz J., Flockerzi V., Zhu M. X., Armstrong D. L., and Birnbaumer L. (2008) Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM-regulated heteromeric Orai/TRPC model for SOCE/Icrac channels. Proc. Natl. Acad. Sci. U.S.A. 105, 2895–2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miura Y., Henquin J. C., and Gilon P. (1997) Emptying of intracellular Ca2+ stores stimulates Ca2+ entry in mouse pancreatic beta-cells by both direct and indirect mechanisms. J. Physiol. 503, 387–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Y. J., and Gylfe E. (1997) Store-operated Ca2+ entry in insulin-releasing pancreatic beta-cells. Cell Calcium 22, 277–286 [DOI] [PubMed] [Google Scholar]
- 16.Bertram R., Smolen P., Sherman A., Mears D., Atwater I., Martin F., and Soria B. (1995) A role for calcium release-activated current (CRAC) in cholinergic modulation of electrical activity in pancreatic beta-cells. Biophys. J. 68, 2323–2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mears D., and Zimliki C. L. (2004) Muscarinic agonists activate Ca2+ store-operated and -independent ionic currents in insulin-secreting HIT-T15 cells and mouse pancreatic beta-cells. J. Membr. Biol. 197, 59–70 [DOI] [PubMed] [Google Scholar]
- 18.Roe M. W., Worley J. F. 3rd, Qian F., Tamarina N., Mittal A. A., Dralyuk F., Blair N. T., Mertz R. J., Philipson L. H., and Dukes I. D. (1998) Characterization of a Ca2+ release-activated nonselective cation current regulating membrane potential and [Ca2+]i oscillations in transgenically derived beta-cells. J. Biol. Chem. 273, 10402–10410 [DOI] [PubMed] [Google Scholar]
- 19.Tamarina N. A., Kuznetsov A., and Philipson L. H. (2008) Reversible translocation of EYFP-tagged STIM1 is coupled to calcium influx in insulin secreting beta-cells. Cell Calcium 44, 533–544 [DOI] [PubMed] [Google Scholar]
- 20.Tian G., Tepikin A. V., Tengholm A., and Gylfe E. (2012) cAMP induces stromal interaction molecule 1 (STIM1) puncta but neither Orai1 protein clustering nor store-operated Ca2+ entry (SOCE) in islet cells. J. Biol. Chem. 287, 9862–9872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qian F., Huang P., Ma L., Kuznetsov A., Tamarina N., and Philipson L. H. (2002) TRP genes: candidates for nonselective cation channels and store-operated channels in insulin-secreting cells. Diabetes 51, S183–S189 [DOI] [PubMed] [Google Scholar]
- 22.Liou J., Kim M. L., Heo W. D., Jones J. T., Myers J. W., Ferrell J. E. Jr., and Meyer T. (2005) STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 15, 1235–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang S. L., Yeromin A. V., Hu J., Amcheslavsky A., Zheng H., and Cahalan M. D. (2011) Mutations in Orai1 transmembrane segment 1 cause STIM1-independent activation of Orai1 channels at glycine 98 and channel closure at arginine 91. Proc. Natl. Acad. Sci. U.S.A. 108, 17838–17843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heo D. K., Chung W. Y., Park H. W., Yuan J. P., Lee M. G., and Kim J. Y. (2012) Opposite regulatory effects of TRPC1 and TRPC5 on neurite outgrowth in PC12 cells. Cell. Signal. 24, 899–906 [DOI] [PubMed] [Google Scholar]
- 25.Kim M. S., Zeng W., Yuan J. P., Shin D. M., Worley P. F., and Muallem S. (2009) Native store-operated Ca2+ influx requires the channel function of Orai1 and TRPC1. J. Biol. Chem. 284, 9733–9741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poteser M., Graziani A., Eder P., Yates A., Mächler H., Romanin C., and Groschner K. (2008) Identification of a rare subset of adipose tissue-resident progenitor cells, which express CD133 and TRPC3 as a VEGF-regulated Ca2+ entry channel. FEBS Lett. 582, 2696–2702 [DOI] [PubMed] [Google Scholar]
- 27.Merglen A., Theander S., Rubi B., Chaffard G., Wollheim C. B., and Maechler P. (2004) Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology 145, 667–678 [DOI] [PubMed] [Google Scholar]
- 28.Allagnat F., Alonso F., Martin D., Abderrahmani A., Waeber G., and Haefliger J. A. (2008) ICER-1gamma overexpression drives palmitate-mediated connexin36 down-regulation in insulin-secreting cells. J. Biol. Chem. 283, 5226–5234 [DOI] [PubMed] [Google Scholar]
- 29.Allagnat F., Cunha D., Moore F., Vanderwinden J. M., Eizirik D. L., and Cardozo A. K. (2011) Mcl-1 downregulation by pro-inflammatory cytokines and palmitate is an early event contributing to beta-cell apoptosis. Cell Death Differ 18, 328–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sabourin J., Robin E., and Raddatz E. (2011) A key role of TRPC channels in the regulation of electromechanical activity of the developing heart. Cardiovasc. Res. 92, 226–236 [DOI] [PubMed] [Google Scholar]
- 31.Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K. J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L. G., and Landegren U. (2006) Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000 [DOI] [PubMed] [Google Scholar]
- 32.Alonso F., Boittin F. X., Bény J. L., and Haefliger J. A. (2010) Loss of connexin40 is associated with decreased endothelium-dependent relaxations and eNOS levels in the mouse aorta. Am. J. Physiol. Heart Circ. Physiol. 299, H1365–H1373 [DOI] [PubMed] [Google Scholar]
- 33.Bon R. S., and Beech D. J. (2013) In pursuit of small molecule chemistry for calcium-permeable non-selective TRPC channels: mirage or pot of gold? Br. J. Pharmacol. 170, 459–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thore S., Dyachok O., Gylfe E., and Tengholm A. (2005) Feedback activation of phospholipase C via intracellular mobilization and store-operated influx of Ca2+ in insulin-secreting beta-cells. J. Cell Sci. 118, 4463–4471 [DOI] [PubMed] [Google Scholar]
- 35.Dyachok O., and Gylfe E. (2004) Ca2+-induced Ca2+ release via inositol 1,4,5-trisphosphate receptors is amplified by protein kinase A and triggers exocytosis in pancreatic beta-cells. J. Biol. Chem. 279, 45455–45461 [DOI] [PubMed] [Google Scholar]
- 36.Kang G., Chepurny O. G., and Holz G. G. (2001) cAMP-regulated guanine nucleotide exchange factor II (Epac2) mediates Ca2+-induced Ca2+ release in INS-1 pancreatic beta-cells. J. Physiol. 536, 375–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liou J., Fivaz M., Inoue T., and Meyer T. (2007) Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc. Natl. Acad. Sci. U.S.A. 104, 9301–9306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang G. N., Zeng W., Kim J. Y., Yuan J. P., Han L., Muallem S., and Worley P. F. (2006) STIM1 carboxyl-terminus activates native SOC, Icrac and TRPC1 channels. Nat. Cell Biol. 8, 1003–1010 [DOI] [PubMed] [Google Scholar]
- 39.Kiyonaka S., Kato K., Nishida M., Mio K., Numaga T., Sawaguchi Y., Yoshida T., Wakamori M., Mori E., Numata T., Ishii M., Takemoto H., Ojida A., Watanabe K., Uemura A., Kurose H., Morii T., Kobayashi T., Sato Y., Sato C., Hamachi I., and Mori Y. (2009) Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc. Natl. Acad. Sci. U.S.A. 106, 5400–5405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poitout V., and Robertson R. P. (2002) Minireview: Secondary beta-cell failure in type 2 diabetes: a convergence of glucotoxicity and lipotoxicity. Endocrinology 143, 339–342 [DOI] [PubMed] [Google Scholar]
- 41.Eizirik D. L., Korbutt G. S., and Hellerström C. (1992) Prolonged exposure of human pancreatic islets to high glucose concentrations in vitro impairs the beta-cell function. J. Clin. Invest. 90, 1263–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Purrello F., Rabuazzo A. M., Anello M., and Patanè G. (1996) Effects of prolonged glucose stimulation on pancreatic beta cells: from increased sensitivity to desensitization. Acta Diabetol. 33, 253–256 [DOI] [PubMed] [Google Scholar]
- 43.Cardozo A. K., Ortis F., Storling J., Feng Y. M., Rasschaert J., Tonnesen M., Van Eylen F., Mandrup-Poulsen T., Herchuelz A., and Eizirik D. L. (2005) Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta-cells. Diabetes 54, 452–461 [DOI] [PubMed] [Google Scholar]
- 44.Lemmens R., Larsson O., Berggren P. O., and Islam M. S. (2001) Ca2+-induced Ca2+ release from the endoplasmic reticulum amplifies the Ca2+ signal mediated by activation of voltage-gated L-type Ca2+ channels in pancreatic beta-cells. J. Biol. Chem. 276, 9971–9977 [DOI] [PubMed] [Google Scholar]
- 45.Chen L., Koh D. S., and Hille B. (2003) Dynamics of calcium clearance in mouse pancreatic beta-cells. Diabetes 52, 1723–1731 [DOI] [PubMed] [Google Scholar]
- 46.Dyachok O., Tufveson G., and Gylfe E. (2004) Ca2+-induced Ca2+ release by activation of inositol 1,4,5-trisphosphate receptors in primary pancreatic beta-cells. Cell Calcium 36, 1–9 [DOI] [PubMed] [Google Scholar]
- 47.Santulli G., Pagano G., Sardu C., Xie W., Reiken S., D'Ascia S. L., Cannone M., Marziliano N., Trimarco B., Guise T. A., Lacampagne A., and Marks A. R. (2015) Calcium release channel RyR2 regulates insulin release and glucose homeostasis. J. Clin. Invest. 125, 1968–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Islam M. S. (2002) The ryanodine receptor calcium channel of beta-cells: molecular regulation and physiological significance. Diabetes 51, 1299–1309 [DOI] [PubMed] [Google Scholar]
- 49.Duman J. G., Chen L., Palmer A. E., and Hille B. (2006) Contributions of intracellular compartments to calcium dynamics: implicating an acidic store. Traffic 7, 859–872 [DOI] [PubMed] [Google Scholar]
- 50.Putney J. W. Jr., Aub D. L., Taylor C. W., and Merritt J. E. (1986) Formation and biological action of inositol 1,4,5-trisphosphate. Fed. Proc. 45, 2634–2638 [PubMed] [Google Scholar]
- 51.Cheng K. T., Liu X., Ong H. L., and Ambudkar I. S. (2008) Functional requirement for Orai1 in store-operated TRPC1-STIM1 channels. J. Biol. Chem. 283, 12935–12940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kilch T., Alansary D., Peglow M., Dörr K., Rychkov G., Rieger H., Peinelt C., and Niemeyer B. A. (2013) Mutations of the Ca2+-sensing stromal interaction molecule STIM1 regulate Ca2+ influx by altered oligomerization of STIM1 and by destabilization of the Ca2+ channel Orai1. J. Biol. Chem. 288, 1653–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoover P. J., and Lewis R. S. (2011) Stoichiometric requirements for trapping and gating of Ca2+ release-activated Ca2+ (CRAC) channels by stromal interaction molecule 1 (STIM1). Proc. Natl. Acad. Sci. U.S.A. 108, 13299–13304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheng K. T., Ong H. L., Liu X., and Ambudkar I. S. (2011) Contribution of TRPC1 and Orai1 to Ca2+ entry activated by store depletion. Adv. Exp. Med. Biol. 704, 435–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.DeHaven W. I., Jones B. F., Petranka J. G., Smyth J. T., Tomita T., Bird G. S., and Putney J. W. Jr. (2009) TRPC channels function independently of STIM1 and Orai1. J. Physiol. 587, 2275–2298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yuan J. P., Zeng W., Dorwart M. R., Choi Y. J., Worley P. F., and Muallem S. (2009) SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 11, 337–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zeng W., Yuan J. P., Kim M. S., Choi Y. J., Huang G. N., Worley P. F., and Muallem S. (2008) STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol. Cell 32, 439–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Putney J. W. (2005) Physiological mechanisms of TRPC activation. Pflugers Arch. 451, 29–34 [DOI] [PubMed] [Google Scholar]
- 59.Hofmann T., Obukhov A. G., Schaefer M., Harteneck C., Gudermann T., and Schultz G. (1999) Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 397, 259–263 [DOI] [PubMed] [Google Scholar]
- 60.Sabourin J., Antigny F., Robin E., Frieden M., and Raddatz E. (2012) Activation of transient receptor potential canonical 3 (TRPC3)-mediated Ca2+ entry by A1 adenosine receptor in cardiomyocytes disturbs atrioventricular conduction. J. Biol. Chem. 287, 26688–26701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Henquin J. C. (2009) Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia 52, 739–751 [DOI] [PubMed] [Google Scholar]
- 62.Rutter G. A., Tsuboi T., and Ravier M. A. (2006) Ca2+ microdomains and the control of insulin secretion. Cell Calcium 40, 539–551 [DOI] [PubMed] [Google Scholar]
- 63.Tengholm A., Hellman B., and Gylfe E. (2000) Mobilization of Ca2+ stores in individual pancreatic beta-cells permeabilized or not with digitonin or alpha-toxin. Cell Calcium 27, 43–51 [DOI] [PubMed] [Google Scholar]
- 64.Park C. Y., Shcheglovitov A., and Dolmetsch R. (2010) The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science 330, 101–105 [DOI] [PubMed] [Google Scholar]
- 65.Wang Y., Deng X., Mancarella S., Hendron E., Eguchi S., Soboloff J., Tang X. D., and Gill D. L. (2010) The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science 330, 105–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nguyen N., Biet M., Simard E., Béliveau E., Francoeur N., Guillemette G., Dumaine R., Grandbois M., and Boulay G. (2013) STIM1 participates in the contractile rhythmicity of HL-1 cells by moderating T-type Ca2+ channel activity. Biochim. Biophys. Acta 1833, 1294–1303 [DOI] [PubMed] [Google Scholar]
- 67.Wang Y., Deng X., Hewavitharana T., Soboloff J., and Gill D. L. (2008) Stim, ORAI and TRPC channels in the control of calcium entry signals in smooth muscle. Clin. Exp. Pharmacol. Physiol. 35, 1127–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Soboloff J., Spassova M., Xu W., He L. P., Cuesta N., and Gill D. L. (2005) Role of endogenous TRPC6 channels in Ca2+ signal generation in A7r5 smooth muscle cells. J. Biol. Chem. 280, 39786–39794 [DOI] [PubMed] [Google Scholar]
- 69.Reading S. A., Earley S., Waldron B. J., Welsh D. G., and Brayden J. E. (2005) TRPC3 mediates pyrimidine receptor-induced depolarization of cerebral arteries. Am. J. Physiol. Heart Circ. Physiol. 288, H2055–H2061 [DOI] [PubMed] [Google Scholar]
- 70.Uchida K., Dezaki K., Damdindorj B., Inada H., Shiuchi T., Mori Y., Yada T., Minokoshi Y., and Tominaga M. (2011) Lack of TRPM2 impaired insulin secretion and glucose metabolisms in mice. Diabetes 60, 119–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cheng H., Beck A., Launay P., Gross S. A., Stokes A. J., Kinet J. P., Fleig A., and Penner R. (2007) TRPM4 controls insulin secretion in pancreatic beta-cells. Cell Calcium 41, 51–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Colsoul B., Schraenen A., Lemaire K., Quintens R., Van Lommel L., Segal A., Owsianik G., Talavera K., Voets T., Margolskee R. F., Kokrashvili Z., Gilon P., Nilius B., Schuit F. C., and Vennekens R. (2010) Loss of high-frequency glucose-induced Ca2+ oscillations in pancreatic islets correlates with impaired glucose tolerance in Trpm5−/− mice. Proc. Natl. Acad. Sci. U.S.A. 107, 5208–5213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Skrzypski M., Kakkassery M., Mergler S., Grötzinger C., Khajavi N., Sassek M., Szczepankiewicz D., Wiedenmann B., Nowak K. W., and Strowski M. Z. (2013) Activation of TRPV4 channel in pancreatic INS-1E beta cells enhances glucose-stimulated insulin secretion via calcium-dependent mechanisms. FEBS Lett. 587, 3281–3287 [DOI] [PubMed] [Google Scholar]
- 74.Graham S., Yuan J. P., and Ma R. (2012) Canonical transient receptor potential channels in diabetes. Exp. Biol. Med. (Maywood) 237, 111–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eizirik D. L., and Cnop M. (2010) ER stress in pancreatic beta cells: the thin red line between adaptation and failure. Sci. Signal. 3, pe7. [DOI] [PubMed] [Google Scholar]