

Abstract
The overactivation of signaling pathways, such as the PI3K and MAPK, which are crucial to cell growth and survival, is a common feature in many cancer types. Though a number of advances have been made in the development of molecular agents targeting these pathways, their application as monotherapies has not significantly improved clinical outcome. A novel liposomal preparation was developed, co-loaded with NCL-240, a small-molecule inhibitor of the PI3K/mTOR pathway, along with cobimetinib, a MEK/ERK pathway inhibitor. This combination drug-loaded nanocarrier, (N+C)-LP, was able to significantly enhance the cytotoxicity of these drugs against colon carcinoma cells in vitro demonstrating a clear synergistic effect (combination index of 0.79). The (N+C)-LP was also able to induce cell cycle arrest of the cells, specifically in the G1 phase thereby preventing their progression to the S-phase, typical of the action of MEK inhibitors. Analyzing the apoptotic events, it was found that this effect on cell cycle regulation is followed by the induction of apoptosis. The quantified distribution of apoptotic events showed that the (N+C)-LP induced apoptosis significantly by over 3–4 fold (P<0.001) compared to other treatment groups. The co-loaded liposomal preparation was also targeted to the transferrin receptor of cancer cells by modifying the surface of the liposome with transferrin. FACS analysis showed that transferrin-mediated targeting enhanced the association of liposomes to HCT 116 cells by almost 5-fold. This could potentially allow for cancer cell-specific effects in vivo thereby minimizing any non-specific interactions of the liposomes with non-cancerous cells. Taken together, this study clearly shows that the combined inhibition of the PI3K and MEK pathways correlates with a significant anti-proliferative effect, due to cell-cycle regulation leading to the induction of apoptosis.
Keywords: PI3K/AKT, MEK, NCL-240, cobimetinib, liposomes, colon carcinoma
Graphical Abstract
INTRODUCTION
Some of the hallmark capabilities of cancer cells involve their sustained proliferative signaling, evasion of growth suppressors, activation of metastasis, acquisition of replicative immortality, induction of angiogenesis and resistance to cell death mechanisms thus making them complex targets.1 More importantly, their ability to sustain proliferative signaling pathways by activating mutations in specific components of these pathways has been the subject of more recent approaches to effective cancer therapy.2 The PI3K/AKT and the MEK/ERK pathways are thought to play central roles in the intracellular transduction of proliferative signals in a variety of cancer types. Phosphoinositide 3-kinase (PI3K) belongs to a family of lipid kinases, which work by activating phosphatidylinositol in the plasma membrane of the cell leading to the recruitment and activation of AKT, a serine-threonine kinase.3 Activation of the PI3K/AKT pathway is critical to the survival, proliferation and growth of cancer. Similarly, mitogen-activated protein kinases (MAPKs) are also a ubiquitous family of enzymes, which link extracellular signals to gene expression pathways.4 The key players in the MAPK cascade involve the mitogen-activated protein kinase kinase kinase (RAF or MAP3K), mitogen-activated protein kinase kinase (MEK or MAP2K), and extracellular signal regulated protein kinase (ERK). This MAPK cascade is known to critically regulate a number of cellular activities such as migration, proliferation and cell cycle regulation. Therefore, targeting of these pathways with small molecule inhibitors represents a promising strategy to the development of effective cancer therapies.5, 6
Cobimetinib (formerly XL518/GDC-0973 from Exelixis/Genentech), is a potent small molecule MEK inhibitor.7, 8 Preclinical studies involving cobimetinib have shown the sustained MEK-mediated inhibition of melanoma, pancreatic as well as colon carcinoma tumors in vivo.9, 10 Recently, it was evaluated in combination with vemurafenib in a phase 3 clinical trial for patients with advanced or metastatic melanoma harboring a BRAF (V600) mutation.11 The study successfully showed a significant increase in progression-free survival.
Previously, our group had reported on the development and enhanced anti-tumor activity of NCL-240, a novel PI3K/AKT pathway inhibitor (DM-PIT1 analog).12 NCL-240 demonstrated robust inhibition of the TORC1/p70S6K/S6 pathway downstream from AKT in human glioblastoma (U87MG) and ovarian carcinoma (A2780) cells. Furthermore, NCL-240 also showed significant inhibition of cell migration in an A2780 wound healing assay. However, the high lipophilicity of this molecule (calculated log P = 5.3) makes its in vivo delivery challenging. In order to overcome this, our group had recently developed a formulation of NCL-240 in polyethyleneglycol (PEG)-based micelles. The NCL-240 micelles showed significant inhibition of A2780 tumors in a subcutaneous murine xenograft model.13
Though the PI3K/AKT and MEK/ERK are two distinct pathways, studies have clearly shown that there are a number of feedback loops between these two pathways.14 Blocking MAPK is known to increase activity of the PI3K and vice versa.15, 16 Interestingly, the activation of the PI3K pathway has been shown to drive the resistance to MAPK cascade inhibitors.17 Therefore in order to overcome these feedback loops, the use of combinational treatment regimens simultaneously targeting these two pathways may prove advantageous.10, 18, 19 With this in mind, the effects of combinational treatment with both NCL-240 and cobimetinib was investigated. Since oncogenic mutations in these pathways are present in a majority of colorectal carcinomas, the cytotoxic potential of these drugs was evaluated in HCT 116 colon carcinoma cells in vitro.20
However, the administration of two different drugs in vivo may lead to their accumulation in different parts of the tumor due to the heterogeneous nature of cancer, preventing a dramatic improvement in tumor inhibition. It is therefore imperative that both these drugs are delivered to the same cancer cells so as to effect a better treatment outcome. To overcome the challenge of delivering two hydrophobic anti-cancer compounds together, a novel liposome-based formulation co-loaded with both NCL-240 and cobimetinib was developed. Furthermore, to better facilitate cancer cell-specific effects, these liposomes were targeted with transferrin since the transferrin receptor is frequently over-expressed in many cancer cell types.21
EXPERIMENTAL SECTION
Materials, Cell Culture and Animals
FITC-labeled mouse monoclonal anti-transferrin receptor antibody (ab47095) was purchased from Abcam (San Francisco, CA). FITC-labeled normal mouse IgG (sc-2855) (as negative control) was purchased from Santa Cruz Biotechnology (Dallas, TX). Nitrophenylcarbonyl-PEG3400-nitrophenylcarbonyl (NPC-PEG-NPC) was from Laysan Bio (Arab, AL). Eggphosphatidylcholine (ePC), cholesterol, cholesteryl hemisuccinate (CHEMS), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (PEG2000-DSPE) and (lissamine rhodamine) DPPE (rhodamine) were purchased from Avanti (Alabaster, AL). Dragendorff’s reagent, molybdenum blue, triethylamine (TEA), Sepharose CL-4B (40–165 μm) and human holo-transferrin (Tf) were purchased from Sigma-Aldrich (St. Louis, MO). The microBCA™ Protein Assay Kit, Whatman nucleopore polycarbonate membranes 19 mm at 0.2μ, 0.1μ and 0.05μ pore sizes and bovine serum albumin (BSA) were purchased from Fisher/Thermo Scientific (Waltham, MA). Spectra/Por pre-wetted 300,000 MWCO dialysis membrane was purchased from Spectrum Labs Inc. (Rancho Dominguez, CA). CellTiter-Glo® cell viability assay was purchased from Promega Corporation (Madison, WI). NCL-240 was synthesized at the National Chemical Laboratory (Pune, India).12 Cobimetinib was purchased from MedChem Express (Monmouth Jn, NJ). Hoechst33342, Yo-Pro-1 and propidium iodide were purchased from Life Technologies (Carlsbad, CA).
Streptomycin (25μg/mL)/Penicillin (10,000 U/mL) solution, Trypsin/EDTA and Cellstripper™ solution were purchased from Corning/Mediatech (Manassas, VA). Fetal bovine serum (FBS) was purchased from Atlanta Biologicals (Flowery Branch, GA). Human colon adenocarcinoma cells (HCT 116), human umbilical vein endothelial cells (HUVEC), F-12K and McCoy’s media were purchased from ATCC (Manassas, VA). HCT 116 cells were grown in McCoy’s media. HUVEC cells were grown in F-12K media supplemented with 0.1 mg/mL heparin and 0.03 mg/mL endothelial cell growth supplement. All media were supplemented with 10% FBS and 1% penicillin-streptomycin solution. Cells were all grown at 37°C with 5% CO2.
Preparation of Liposomes
A lipid film consisting of ePC, cholesterol, CHEMS and DOPE (64:24:6:6 mole %) (Table 1) as well as the drugs (at 2% w/w to the total lipid) was obtained by removal of chloroform by rotary evaporation followed by freeze drying on a Freezone 4.5L Freeze Dry System (Labconco, Kansas City, MO) for a minimum of 4 hours. The drugs were included at a mole ratio of 6:1 of NCL-240: cobimetinib. The film was then hydrated with phosphate buffered saline (PBS) pH 7.4 to maintain a lipid concentration of 10 mg/mL. The solution was vortexed and then extruded through 200 nm polycarbonate membranes. The extruded liposomes were then filtered through 0.2μm filters (Nalgene, Rochester, NY) to remove any unencapsulated drug, since these form aggregates due to their hydrophobic nature. Following this, PEG2000-DSPE and the Tf-PEG3400-DOPE were added to the respective liposomal formulations at 2 and 0.025 mole % by the post-insertion method. Briefly, a thin film of the PEG2000-DSPE obtained by the removal of organic solvent by drying under a stream of nitrogen gas followed by freeze drying for a minimum of 4 hours, was hydrated with the Tf-PEG3400-DOPE solution and/or PBS to form mixed micelles. These were then added to the liposomal solution and incubated at 37°C overnight to allow for complete incorporation of the PEG chains into the liposome membrane.
Table 1.
Composition of Liposomes
| Lipid | Mole Percentage % |
|---|---|
|
| |
| ePC | 64% |
| Cholesterol | 24% |
| Dioleoyl-sn-glycero-phosphoethanolamine (DOPE) | 6% |
| Cholesteryl hemisuccinate (CHEMS) | 6% |
| PEG2000-DSPE | 2 % |
| Tf-PEG3400-DOPE | 0.025% |
Characterization of Liposomes
Particle size and zeta potential analysis was carried out using a N4 Coulter particle size analyzer (Weste Lafayette, IN) and Zetaplus (Brookhaven Instruments Corporation, Holtsville, NY) respectively. For particle size analysis, 5μL of the liposomal solution was mixed with 990μL of 1mM KCl while for zeta potential, 50μL was mixed with 1.5mL of 1mM KCl. For analysis using transmission electron microscopy (TEM), the sample was stained using 1.5% phosphotungstic acid (PTA) by the negative staining technique. HPLC (Hitachi Elite LaChrom Pleasanton, CA) determination of liposomal drug content was carried out using a C18 column (XBridge, 4.6 × 250 mm, 5μm) (Waters Corporation, Milford, MA) and a mobile phase consisting of 60% acetonitrile and 40% (v/v) 10mM ammonium acetate at pH 4 (detection wavelength of 240 nm for both drugs) at a flow rate of 1 mL/min.
In Vitro Cytotoxicity Experiments
3,000 HCT 116 cells were seeded in each well of a 96-well plate 24 hours prior to the experiment. Formulations were sterile filtered and incubated with the cells for 48 hours following which the cell viability was measured using the Cell Titer Glo® cell viability assay according to the manufacturer’s protocol. The combination index (CI) was also calculated to check for synergism between the two drugs using the formula:
where: IC50ab1 e IC50ab2 are the IC50 values when the drugs are administered in combination, and IC50a e IC50b are the IC50 values when the drugs are administered as single agents.22
Analysis of Apoptosis and Cell Cycle Distribution
3,000 cells per well were seeded in Whatman black-walled polystyrene 96-well plates 24 hours prior to the experiment. Formulations were sterile filtered and incubated with the cells for 48 hours. Hoechst33342 (5μg/mL) followed by the markers, Yo-Pro (0.12μg/mL) for early apoptosis and propidium iodide (1μg/mL) for late apoptosis/necrosis were diluted in media and all added together directly onto the cells. After incubation at 37°C for 30 minutes, the stained cells were analyzed without washing using the iCyte imaging cytometer (Compucyte Corp., Westwood MA). A 40x objective lens was used with 0.25 μm spatial resolution in two-pass scanning. In the first pass, the 405 nm laser was used to excite Hoechst and fluorescence was collected through a 440/30 bandpass filter. In the second pass, the 488 nm argon laser was used, with a 515/30 bandwidth filter for green Yo-Pro fluorescence, and a 650 nm long pass filter for the red propidium iodide fluorescence. Cells were segmented using Hoechst fluorescence, and total cellular DNA fluorescence was quantified. For cell cycle distribution, live single cells were gated into the G1, S and G2 phases based on their combined DNA content and nuclear area. For analysis of apoptotic events, cells were gated based on their green Yo-Pro fluorescence and red propidium iodide signal and quantified by random segmentation.
Synthesis of pNP-PEG3400-DOPE
The pNP-PEG3400-DOPE was synthesized and purified by slight modifications to a previously established method.23 64.6 μmol NPC-PEG3400-NPC (or pNP-PEG3400-pNP) was first dissolved in 1mL chloroform and then reacted with 12.9 μmol DOPE in the presence of 38.8 μmol TEA. The reaction was incubated overnight at room temperature (RT) in an argon atmosphere under constant stirring. The reaction was monitored using thin-layer chromatography (TLC) in a mixture of chloroform-methanol as an eluent at an 80:20 volumetric ratio. Dragendorff’s reagent and molybdenum blue were used to visualize the PEG and DOPE respectively. After completion of the reaction, the solvent was removed from the mixture using rotary evaporation followed by freeze drying. The product was then dissolved in 0.001 M HCl, pH 3 at a lipid concentration of 200 mg/mL and separated on a column loaded with Sepharose CL-4B and eluted with 0.001M HCl. The various fractions collected were then analyzed for the presence of the product by TLC. Only those fractions containing the product were then pooled together and freeze dried. The resulting lyophilized product was dissolved in chloroform (at 5 mg/mL) and stored in −80°C until further use.
Synthesis of Transferrin-PEG3400-PE
A thin film containing 2 mg of pNP-PEG3400-DOPE was obtained by removing the chloroform by drying under a stream of nitrogen gas followed by freeze drying for a minimum of 4 hours. The film was hydrated with 1mL of a 15mg/mL solution of Tf in PBS pH 8.5. The reaction was incubated at RT overnight under constant stirring. Following this, the unreacted Tf was removed by dialyzing the reaction mixture against 2L of deionized water followed by dialysis against 2L of PBS pH 7.4 using an MWCO of 300,000 Da for 3 to 4 hours. The concentration of the product, Tf-PEG3400-DOPE was then determined using the microBCA™ assay kit. The product was stored at 4°C until further use.
Transferrin Receptor Characterization
For the characterization of the transferrin receptor, cells were detached using cell stripper solution (10 mins incubation at 37°C). Detached cells were then neutralized with complete media and centrifuged at 2000 RPM for 5 mins. Cell pellets were then resuspended in 3% BSA/PBS and counted. 200,000 cells were taken in each centrifuge tube. To the control, 100 μL 3% BSA/PBS was added; to the control antibody group, 100 μL of normal mouse IgG1-FITC was added (final dilution of 1:50 in cell suspension); to the transferrin antibody group 100 μL of anti-transferrin receptor IgG1-FITC was added (final dilution of 1:50 in cell suspension). These were incubated at 4°C for 30 mins following which the cell suspensions were centrifuged at 2000 RPM, 5 mins and washed with 3% BSA/PBS. Cell pellets were then resuspended in 3% BSA/PBS, kept on ice and immediately analyzed by flow cytometry (BD FACS Calibur®, Bedford, MA).
Cell Association Studies
The liposomes were labeled with 1 mole % rhodamine which was included along with the other lipids prior to film-hydration. 75,000 cells were seeded per well of a 12-well plate 24 hours prior to the experiment. Following this, the cells were treated with the formulations at a liposomal concentration of 0.1 mg/mL for 4 hours. The formulations were then washed off with PBS and the cells were trypsinized, washed and maintained as a cell suspension in PBS on ice. Cells were then immediately analyzed by flow cytometry for their fluorescent intensity. All samples were normalized with a control cell population based on their mean fluorescent intensity and subsequently analyzed.
Statistical Analysis
Data was generated in triplicates and expressed as mean +/− S.D. Statistical analyses were performed using one-way ANOVA followed by post-hoc analyses. Significance was determined by a P-value < 0.05 (denoted by *), P<0.01 (denoted by **) and P< 0.001 (denoted by ***).
RESULTS
Characterization of Liposomes
The NCL-240-loaded (NCL-LP), cobimetinib-loaded (Cobi-LP) as well as the co-loaded ((N+C)-LP) liposomes all showed a uniform size distribution of ca 200 nm with a low polydispersity index reflecting a homogeneous particle size distribution (Table 2). The size range was further confirmed by analysis using transmission electron microscopy, which showed the roughly spherical/elliptical appearance of the nanoparticles when visualized with a PTA stain (Fig 1). Zeta potential analysis was then carried out on the nanoparticles. All the particles had an overall negative charge of around −30 mV due to the presence of neutral as well as anionic lipids. Furthermore, this overall negative surface charge along with the coating of PEG2000-DSPE, allows for the increased stability of the liposomes in solution preventing their flocculation and settling over time. In addition, fusogenic lipids such as CHEMS and DOPE were included to allow for release of the cargo from the late endosomes which are characterized by a drop in pH.24 It was found that at a lower pH of 5, the liposomes showed a significant drop in the zeta potential from −28 to −13 mV due to the pH sensitive nature of DOPE and CHEMS.
Table 2.
Physico-Chemical Characterization of Liposomes
| Formulation | Drug concentration per 10 mg/mL of liposome (μM)a | Encapsulation (%)a | Size (nm)b | PDIb | Zeta Potential (mV) | ||
|---|---|---|---|---|---|---|---|
| NCL-240 | Cobimetinib | NCL-240 | Cobimetinib | ||||
| NCL-LP | 361 | - | 98% | - | 218±42 | 0.062 | −28.4±1.7 |
| Cobi-LP | - | 53 | - | 86% | 204±51 | 0.086 | −29.4±2 |
| (N+C)-LP | 355 | 56 | 97% | 91% | 208±38 | 0.078 | −31.6±1.2 |
| Tf-NCL-LP | 361 | - | 98% | - | 205±59 | 0.145 | −28.2±4.2 |
| Tf-Cobi-LP | - | 53 | - | 86% | 201±59 | 0.219 | −30.6±3.9 |
| Tf-(N+C)-LP | 355 | 56 | 97% | 91% | 212±95 | 0.145 | −29.8±4.5 |
Measured by reverse-phase HPLC
Measured by dynamic light scattering. Data represent the mean particle size +/− the average particle size distribution.
Figure 1.
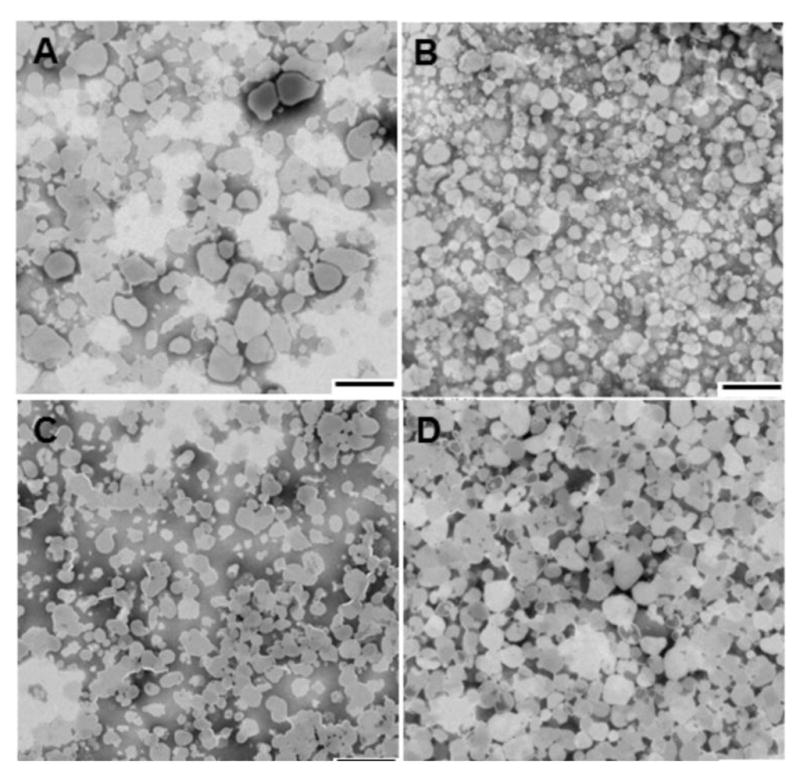
Transmission electron microscopy images of (A) Blank liposomes, (B) NCL liposomes, (C) Cobi liposomes and (D) NCL+Cobi liposomes by negative staining technique. (scale bar = 500 nm)
Next, the liposomes were characterized for their drug loading efficiency using HPLC. In order to effectively separate both drug peaks, a mobile phase consisting of 60% (v/v) acetonitrile and 40% (v/v) 10mM ammonium acetate (pH 4) was used. Cobimetinib had a retention time of about 3 minutes, while that of NCL-240 was 10 minutes. The concentration of encapsulated cobimetinib was about 53–56 μM and NCL-240 was about 355–361 μM for a 10 mg/mL liposomal lipid concentration. This translated to an encapsulation efficiency of about 90–98% for both drugs as due to their highly hydrophobic nature, they were able to assemble preferentially into the lipid bilayer of the liposomes. The concentrations of the liposome-encapsulated drugs as well as their loading efficiencies are summarized in Table 2. In addition, the long-term stability of these liposomes was also evaluated on storage at 4°C in PBS pH 7.4, by checking their size after 3 months. Any significant drug leakage from the liposomes should result in the formation of precipitates due to their hydrophobic nature. The stable size of the liposomes was confirmed within the 180–200 nm range with the lack of any aggregates present in solution using DLS (Supplementary Table 1).
Free and Liposomal Combinations of Cobimetinib and NCL-240 Demonstrate Synergistic Cytotoxic Effects on HCT 116 Cells
The cytotoxic response of HCT 116 cells to treatments with NCL-240, cobimetinib as well as a combination of these drugs both in their free and liposomal forms was evaluated. Cells were treated at concentrations ranging from 60 nM to 60 μM with NCL-240 and from 10 nM to 10 μM with cobimetinib. The combinational treatment with NCL and cobimetinib clearly showed significant cytotoxic effects at all of the concentrations when administered in their liposomal form, as compared to the treatment with single drugs only (Fig 2). The free drug combinations showed significantly higher cytotoxicity only at the higher NCL/Cobi concentrations of 6/1 and 1.9/0.33 μM. Also, compared to the NCL-LP, the (N+C)-LP showed a significant 2.5-fold reduction in the IC50 values for NCL from 3.58 μM to 1.36 μM while free (N+C) treatment reduced the IC50 2-fold from 1.85 μM to 0.89 μM. As was expected, when comparing the cytotoxicity of the free drugs to their liposomal counterparts, the liposomal drugs showed lesser cytotoxicity due to their slower drug release as compared to the free drugs which are solubilized in methanol. More importantly, it is evident that both cobimetinib and NCL-240 retain their cytotoxic activity after encapsulation into liposomes. Next, the combination index (CI) of the drugs in combination both in their liposomal and free forms was determined. A CI of 1 indicates additive effects of the drugs while a CI of <1 shows a clear synergistic effect. It was found that when used in liposomes, the CI was 0.79 as opposed to a CI of 0.97 when used in their free drug forms clearly highlighting the increased synergistic effects of these drugs when combined together in liposomes.
Figure 2.

In vitro cytotoxicity analysis on HCT 116 cells showing enhanced cancer cell cytotoxicity of NCL and cobimetinib combinations
Cobimetinib Mediates Cell Cycle Arrest of HCT 116 Cells in G1
Next, using the iCyte imaging cytometer, the effect of the various drug-loaded liposomal formulations on the cell cycle distribution of the HCT 116 cells was analyzed. The DNA content of the cells was quantified based on their Hoechst signal, and after gating to eliminate multiple cells, DNA content histograms were obtained, and regions were defined corresponding to the G1, S and G2 DNA content levels. First a modified Kolmogorov-Smirnov (KS) test 25–27 was used to quantify the difference in the cell cycle distribution when compared to the control cell population (higher unsigned numbers indicate higher differences in the cell cycle distribution). The KS test numbers of the various formulations in triplicates are shown in Fig 3A. Interestingly, it was found that though treatment with NCL-240 did not seem to alter the cell cycle distribution of the cells, treatment with cobimetinib (both Cobi-LP and (N+C)-LP) showed a dramatic alteration confirmed by the higher KS test numbers. On further analysis, it was found that cobimetinib (both in Cobi-LP as well as (N+C)-LP) mediated cell cycle arrest specifically in the G1-phase, preventing their further progression into the S-phase. The semi-quantitative as well as quantitative cell cycle distributions are shown in Fig 3B.
Figure 3.

Analysis of cell cycle distribution showing cobimetinib-mediated cell cycle arrest in G1. (A) Histograms show the KS test which calculates difference in cell cycle distribution of the treatment groups compared to the control (the numerical value is an unsigned integer) followed by (B) the quantification of their distribution in the G1, S and G2 phases.
Treatment with (N+C)-LP Increases the Apoptotic Events in HCT 116 Cells
As was done earlier for the analysis of the cell cycle distribution, the apoptotic potential of the NCL-LP, Cobi-LP as well (N+C)-LP was evaluated using the iCyte imaging cytometer using the appropriate markers (Hoechst-DNA, YoPro-1- early apoptosis via membrane permeability and propidium iodide- late apoptosis/necrosis). Compared to the control, at a NCL/Cobi concentration of 6/1 μM, all the three liposomal treatment groups showed a significant inhibitory effect on the cell proliferation as seen by the low density of cells as well as an increase in apoptotic cells (Fig 4). Compared to the NCL-LP and Cobi-LP, the (N+C)-LP showed a dramatic increase in the number of early apoptotic cells shown in yellow as well as late apoptotic cells shown in magenta. In addition, the presence of apoptotic bodies or ‘apobodies’ was also clearly visualized in these cells (shown with arrows) further confirming their progression to apoptosis (Fig 4 inset). The quantified distribution of apoptotic events showed that the (N+C)-LP induced apoptosis significantly by over 3–4 fold (P<0.001) as compared to the other treatment groups. (Fig 4 lower right panel).
Figure 4.

Analysis of NCL-240- and cobimetinib-mediated apoptosis.
Images showing increased apoptosis of NCL+Cobi liposomes followed by the quantification of apoptotic events by random segmentation. (Apoptotic bodies shown with arrows) (Scale bar= 50 μm)
Characterization of Transferrin Receptor Expression and Cell Association of Transferrin-Targeted Liposomes
The next aim was to evaluate the application of a proof-of-concept targeting moiety. The expression pattern of the transferrin receptor in the HCT 116 cell line was characterized since it is a well-known fact that the transferrin receptor is over-expressed in a wide range of cancer types. Using a FITC-labeled transferrin receptor antibody it was found that compared to the HUVEC, a non-cancerous human cell line, the HCT 116 cells showed a 5–6 fold increase in the FITC fluorescence confirming their transferrin receptor over-expression (Fig 5A). As a negative control, a non-specific FITC-labeled antibody was used to negate the effect of non-specific antibody binding.
Figure 5.

(A) Transferrin receptor expression and (B) cell association of transferrin-targeted liposomes.
Next, the liposomes were fluorescently labeled with rhodamine and targeted with the transferrin conjugate (Tf-PEG3400-DOPE). On analysis by flow cytometry, it was found that compared to the untargeted PEGylated liposomes (PL), the Tf-targeted liposomes (Tf LP) showed a significant 5-fold increase in cell association further highlighting their ability to effectively target cancer cells (Fig 5B).
Investigation of the Cytotoxicity of Transferrin-Targeted Drug-Loaded Liposomes
The various drug-loaded liposomes were then targeted with transferrin by addition of the Tf-PEG3400-DOPE to preformed liposomes by the post-insertion method. As was done earlier, the cells were treated with varying concentrations of these liposomes from 60nM to 60μM for NCL and 10 nM to 10 μM for cobi. It was found that the transferrin-targeted NCL liposomes (Tf-NCL-LP), cobimetinib liposomes (Tf-Cobi-LP) as well as the co-loaded (Tf-(N+C)-LP) did not significantly increase the cytotoxicity of their untargeted counterparts (Fig 6). More importantly, it was seen that even after addition of the transferrin moiety, the synergistic relationship between the two co-loaded drugs was maintained in vitro. We hypothesize that the targeting will play a more crucial role in an in vivo setting allowing for cancer cell-specific effects.
Figure 6.

In vitro cytotoxicity analysis of transferrin-targeted liposomes on HCT 116 cells.
DISCUSSIONS AND CONCLUSIONS
As alluded to earlier, the dependence of cancer cells on their various cell signaling pathways has been critical to the development of a number of selective molecular-targeted therapies. However, there is increasing evidence to show that even strategically targeting crucial pathways with single agents may not be sufficient as the cancer cells are still able to get by, by acquiring somatic mutations thereby developing resistance to the primary treatment modality.28 Although, MEK and PI3K are very relevant molecular targets, targeting these using small molecule inhibitors has not led to any significant success in the clinic as yet. The ability to therefore effectively target multiple pathways crucial to the survival of cancer simultaneously, could represent a useful approach. Furthermore, it has been shown that resistance to a number of MEK pathway inhibitors could be mediated by activation of the PI3K/AKT/mTOR pathway.14, 17 The combined administration of NCL-240, a recently developed PI3K/AKT/mTOR pathway inhibitor, with cobimetinib, a MEK pathway inhibitor was therefore investigated. However, the high lipophilicity of both these drugs (calculated log P of 5.18 for NCL-240 and 5.96 for cobimetinib) could complicate dosing regimens in vivo. Furthermore, dosing of hydrophobic drugs separately may prove ineffective since the optimal concentrations of the drug may not end up reaching the tumor site causing heterogeneous drug distribution allowing the tumor cells to overcome treatment by the development of drug-resistance.29, 30 The liposomal delivery of such hydrophobic drugs represent an efficient way to deliver these potent molecules at their optimal dose ratios to tumor cells.31
Therefore a novel liposomal formulation co-loaded with both NCL-240 and cobimetinib was developed. The developed nanopreparations were in the 200 nm size range as the size of nanomedicines is also known to govern their effective extravasation into the tumor microspace by the enhanced permeability and retention effect as well as their efficient intracellular internalization.32–34 Using cytotoxicity assays, it was demonstrated that when NCL-240 and cobimetinib are administered as free drugs at a mole ratio of 6:1 respectively, they are able to significantly enhance their cytotoxicity on HCT 116 cells. These drugs were loaded into a single liposomal carrier at the same ratio with high loading efficiencies, as they were able to self-assemble into the lipid bilayers due to their hydrophobicity. Similar to the free drug combinations, the drug co-loaded (N+C)-LPs showed higher cytotoxicity than the single drug-loaded NCL-LP and Cobi-LP (Fig 2), Interestingly, calculation of the combination index (CI<1 indicates synergism) clearly showed synergistic effects of the liposomal form (CI of 0.79) over their combined administration as free drugs (CI of 0.97) highlighting the advantages of combining these drugs in a single liposomal carrier in vitro.
The effect of the various liposomal formulations on the regulation of cell cycle was then evaluated. It was found that though NCL-240 did not have a significant effect on the cell cycle distribution, both the Cobi-LP as well as the (N+C)-LP induced cell cycle arrest in G1, clearly preventing their progression to the S-phase (Fig 3). This result was consistent with a number of reports in literature, which demonstrated that this was typical of MEK inhibitors, since ERK activation is necessary for S-phase progression.35, 36
On analysis of the apoptotic events, progression of the cells to apoptosis following cell cycle arrest in G1 was confirmed. The (N+C)-LP was able to significantly enhance induction of apoptotic events by over 2–3 fold as compared to the other treatment groups (Fig 4). To be able to further the therapeutic applications of these liposomes, they were subsequently targeted with transferrin. The transferrin receptor is overexpressed in many cancer types due to its role in iron homeostasis and has been widely exploited to facilitate the cancer-specific delivery of a wide portfolio of small molecule as well as gene-based drugs into cancer cells.37–40 FACS analysis showed that the addition of transferrin-targeting enhanced the association of liposomes to HCT 116 cells almost 5-fold (Fig 5). This could potentially allow for cancer cell-specific effects in vivo thereby minimizing any non-specific interactions of the liposomes with non-cancerous cells.
In this work, the novelty of simultaneously delivering PI3K/AKT and MEK inhibitors using a liposomal formulation has been successfully demonstrated. To the best of our knowledge, the use of lipid nanoparticles to effectively target these two cell signaling pathways simultaneously has not been reported before. This study clearly shows that the combined inhibition of the PI3K and MEK pathways correlates with a significant anti-proliferative effect, due to cell-cycle regulation, leading to the induction of apoptosis. The highlighted in vitro efficiency of these co-loaded liposomes definitely warrants their further investigation on in vivo cancer models.
Supplementary Material
Acknowledgments
The authors would like to thank William Fowle and Dr. Rajiv Kumar at the Northeastern University Physio-Chemical Characterization Core for their help with the transmission electron microscopy. This work was supported by the FAPESP (São Paulo Research Foundation) grant 2014/03228-5 to Vananélia Geraldo and NIH grant U54CA151881 to Vladimir Torchilin.
Footnotes
Size stability of liposomes.
Notes
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov. 2011;10(9):671–84. doi: 10.1038/nrd3504. [DOI] [PubMed] [Google Scholar]
- 3.Riehle RD, Cornea S, Degterev A. Lipid-mediated Protein Signaling. Springer; 2013. Role of phosphatidylinositol 3, 4, 5-trisphosphate in cell signaling; pp. 105–139. [DOI] [PubMed] [Google Scholar]
- 4.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, Stivala F, Libra M, Basecke J, Evangelisti C, Martelli AM, Franklin RA. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773(8):1263–84. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neuzillet C, Tijeras-Raballand A, de Mestier L, Cros J, Faivre S, Raymond E. MEK in cancer and cancer therapy. Pharmacol Ther. 2014;141(2):160–71. doi: 10.1016/j.pharmthera.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 6.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550–62. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 7.Miller CR, Oliver KE, Farley JH. MEK1/2 inhibitors in the treatment of gynecologic malignancies. Gynecol Oncol. 2014;133(1):128–37. doi: 10.1016/j.ygyno.2014.01.008. [DOI] [PubMed] [Google Scholar]
- 8.Wong H, Vernillet L, Peterson A, Ware JA, Lee L, Martini JF, Yu P, Li C, Del Rosario G, Choo EF, Hoeflich KP, Shi Y, Aftab BT, Aoyama R, Lam ST, Belvin M, Prescott J. Bridging the gap between preclinical and clinical studies using pharmacokinetic-pharmacodynamic modeling: an analysis of GDC-0973, a MEK inhibitor. Clin Cancer Res. 2012;18(11):3090–9. doi: 10.1158/1078-0432.CCR-12-0445. [DOI] [PubMed] [Google Scholar]
- 9.Hatzivassiliou G, Haling JR, Chen H, Song K, Price S, Heald R, Hewitt JF, Zak M, Peck A, Orr C, Merchant M, Hoeflich KP, Chan J, Luoh SM, Anderson DJ, Ludlam MJ, Wiesmann C, Ultsch M, Friedman LS, Malek S, Belvin M. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature. 2013;501(7466):232–6. doi: 10.1038/nature12441. [DOI] [PubMed] [Google Scholar]
- 10.Junttila MR, Devasthali V, Cheng JH, Castillo J, Metcalfe C, Clermont AC, Otter DD, Chan E, Bou-Reslan H, Cao T, Forrest W, Nannini MA, French D, Carano R, Merchant M, Hoeflich KP, Singh M. Modeling targeted inhibition of MEK and PI3 kinase in human pancreatic cancer. Mol Cancer Ther. 2015;14(1):40–7. doi: 10.1158/1535-7163.MCT-14-0030. [DOI] [PubMed] [Google Scholar]
- 11.Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, Mandala M, Demidov L, Stroyakovskiy D, Thomas L, de la Cruz-Merino L, Dutriaux C, Garbe C, Sovak MA, Chang I, Choong N, Hack SP, McArthur GA, Ribas A. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867–76. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- 12.Kommagalla Y, Cornea S, Riehle R, Torchilin V, Degterev A, Ramana CV. Optimization of the anti-cancer activity of phosphatidylinositol-3 kinase pathway inhibitor PITENIN-1: switching a thiourea with 1,2,3-triazole. Medchemcomm. 2014;5(9):1359–1363. doi: 10.1039/C4MD00109E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riehle RD, Cornea S, Degterev A, Torchilin V. Micellar formulations of pro-apoptotic DM-PIT-1 analogs and TRAIL in vitro and in vivo. Drug Deliv. 2013;20(2):78–85. doi: 10.3109/10717544.2013.766780. [DOI] [PubMed] [Google Scholar]
- 14.Saini KS, Piccart-Gebhart MJ. Dual targeting of the PI3K and MAPK pathways in breast cancer. APJOH. 2010;2:13–15. [Google Scholar]
- 15.Balmanno K, Chell SD, Gillings AS, Hayat S, Cook SJ. Intrinsic resistance to the MEK1/2 inhibitor AZD6244 (ARRY-142886) is associated with weak ERK1/2 signalling and/or strong PI3K signalling in colorectal cancer cell lines. Int J Cancer. 2009;125(10):2332–41. doi: 10.1002/ijc.24604. [DOI] [PubMed] [Google Scholar]
- 16.Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, Papa A, Nardella C, Cantley LC, Baselga J, Pandolfi PP. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118(9):3065–74. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Franklin RA, Montalto G, Cervello M, Libra M, Candido S, Malaponte G. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade inhibitors: how mutations can result in therapy resistance and how to overcome resistance. Oncotarget. 2012;3(10):1068. doi: 10.18632/oncotarget.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinelli E, Troiani T, D’Aiuto E, Morgillo F, Vitagliano D, Capasso A, Costantino S, Ciuffreda LP, Merolla F, Vecchione L, De Vriendt V, Tejpar S, Nappi A, Sforza V, Martini G, Berrino L, De Palma R, Ciardiello F. Antitumor activity of pimasertib, a selective MEK 1/2 inhibitor, in combination with PI3K/mTOR inhibitors or with multi-targeted kinase inhibitors in pimasertib-resistant human lung and colorectal cancer cells. Int J Cancer. 2013;133(9):2089–101. doi: 10.1002/ijc.28236. [DOI] [PubMed] [Google Scholar]
- 19.Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y, Chirieac LR, Kaur R, Lightbown A, Simendinger J, Li T, Padera RF, Garcia-Echeverria C, Weissleder R, Mahmood U, Cantley LC, Wong KK. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14(12):1351–6. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roper J, Sinnamon MJ, Coffee EM, Belmont P, Keung L, Georgeon-Richard L, Wang WV, Faber AC, Yun J, Yilmaz OH, Bronson RT, Martin ES, Tsichlis PN, Hung KE. Combination PI3K/MEK inhibition promotes tumor apoptosis and regression in PIK3CA wild-type, KRAS mutant colorectal cancer. Cancer Lett. 2014;347(2):204–11. doi: 10.1016/j.canlet.2014.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daniels TR, Bernabeu E, Rodriguez JA, Patel S, Kozman M, Chiappetta DA, Holler E, Ljubimova JY, Helguera G, Penichet ML. The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochim Biophys Acta. 2012;1820(3):291–317. doi: 10.1016/j.bbagen.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70(2):440–6. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 23.Torchilin VP, Levchenko TS, Lukyanov AN, Khaw BA, Klibanov AL, Rammohan R, Samokhin GP, Whiteman KR. p-Nitrophenylcarbonyl-PEG-PE-liposomes: fast and simple attachment of specific ligands, including monoclonal antibodies, to distal ends of PEG chains via p-nitrophenylcarbonyl groups. Biochim Biophys Acta. 2001;1511(2):397–411. doi: 10.1016/s0005-2728(01)00165-7. [DOI] [PubMed] [Google Scholar]
- 24.Simoes S, Moreira JN, Fonseca C, Duzgunes N, de Lima MC. On the formulation of pH-sensitive liposomes with long circulation times. Adv Drug Deliv Rev. 2004;56(7):947–65. doi: 10.1016/j.addr.2003.10.038. [DOI] [PubMed] [Google Scholar]
- 25.Young IT. Proof without prejudice: use of the Kolmogorov-Smirnov test for the analysis of histograms from flow systems and other sources. J Histochem Cytochem. 1977;25(7):935–941. doi: 10.1177/25.7.894009. [DOI] [PubMed] [Google Scholar]
- 26.Lampariello F. On the use of the Kolmogorov-Smirnov statistical test for immunofluorescence histogram comparison. Cytometry. 2000;39(3):179–188. doi: 10.1002/(SICI)1097-0320(20000301)39:3<179::AID-CYTO2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 27.Holme AL, Yadav SK, Pervaiz S. Automated laser scanning cytometry: A powerful tool for multi-parameter analysis of drug-induced apoptosis. Cytometry Part A. 2007;71(2):80–86. doi: 10.1002/cyto.a.20362. [DOI] [PubMed] [Google Scholar]
- 28.Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, Bencardino K, Cercek A, Chen CT, Veronese S, Zanon C, Sartore-Bianchi A, Gambacorta M, Gallicchio M, Vakiani E, Boscaro V, Medico E, Weiser M, Siena S, Di Nicolantonio F, Solit D, Bardelli A. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486(7404):532–6. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tredan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;99(19):1441–54. doi: 10.1093/jnci/djm135. [DOI] [PubMed] [Google Scholar]
- 30.Sriraman SK, Aryasomayajula B, Torchilin VP. Barriers to drug delivery in solid tumors. Tissue Barriers. 2014;2:e29528. doi: 10.4161/tisb.29528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4(2):145–60. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 32.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1):6387–92. [PubMed] [Google Scholar]
- 33.Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Deliv Rev. 2011;63(3):131–5. doi: 10.1016/j.addr.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 34.Sahay G, Alakhova DY, Kabanov AV. Endocytosis of nanomedicines. J Control Release. 2010;145(3):182–95. doi: 10.1016/j.jconrel.2010.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gysin S, Lee SH, Dean NM, McMahon M. Pharmacologic inhibition of RAF-->MEK-->ERK signaling elicits pancreatic cancer cell cycle arrest through induced expression of p27Kip1. Cancer Res. 2005;65(11):4870–80. doi: 10.1158/0008-5472.CAN-04-2848. [DOI] [PubMed] [Google Scholar]
- 36.Chambard JC, Lefloch R, Pouyssegur J, Lenormand P. ERK implication in cell cycle regulation. Biochim Biophys Acta. 2007;1773(8):1299–310. doi: 10.1016/j.bbamcr.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 37.Pun SH, Tack F, Bellocq NC, Cheng J, Grubbs BH, Jensen GS, Davis ME, Brewster M, Janicot M, Janssens B, Floren W, Bakker A. Targeted delivery of RNA-cleaving DNA enzyme (DNAzyme) to tumor tissue by transferrin-modified, cyclodextrin-based particles. Cancer Biol Ther. 2004;3(7):641–50. doi: 10.4161/cbt.3.7.918. [DOI] [PubMed] [Google Scholar]
- 38.Kobayashi T, Ishida T, Okada Y, Ise S, Harashima H, Kiwada H. Effect of transferrin receptor-targeted liposomal doxorubicin in P-glycoprotein-mediated drug resistant tumor cells. Int J Pharm. 2007;329(1–2):94–102. doi: 10.1016/j.ijpharm.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 39.Mendonca LS, Firmino F, Moreira JN, Pedroso de Lima MC, Simoes S. Transferrin receptor-targeted liposomes encapsulating anti-BCR-ABL siRNA or asODN for chronic myeloid leukemia treatment. Bioconjug Chem. 2010;21(1):157–68. doi: 10.1021/bc9004365. [DOI] [PubMed] [Google Scholar]
- 40.Lv Q, Li LM, Han M, Tang XJ, Yao JN, Ying XY, Li FZ, Gao JQ. Characteristics of sequential targeting of brain glioma for transferrin-modified cisplatin liposome. Int J Pharm. 2013;444(1–2):1–9. doi: 10.1016/j.ijpharm.2013.01.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.