

Abstract
We have screened our compound collection in an established cell based assay that measures the derepression of an epigenetically silenced transgene, the locus derepression assay. The screen led to the identification of 4-[4-(1-methylbenzimidazol-2-yl)piperazin-1-yl]sulfonylbenzenecarbohydroxamic acid (9b) as an active which was found to inhibit HDAC1. In initial structure activity relationships study, the 1-methylbenzimidazole ring was replaced by the isosteric heterocycles benzimidazole, benzoxazole, and benzothiazole and the position of the hydroxamic acid substituent on the phenyl ring was varied. Whereas compounds bearing a para substituted hydroxamic acid (9a-d) were active HDAC inhibitors, the meta substituted analogues (8a-d) were appreciably inactive. Compounds 9a-d selectively inhibited HDAC6 (IC50 = 0.1–1.0μM) over HDAC1 (IC50 = 0.9–6μM) and moreover, also selectively inhibited the growth of lung cancer cells vs. patient matched normal cells. The compounds induce a cell cycle arrest in the S-phase while induction of apoptosis is neglible as compared to controls. Molecular modeling studies uncovered that the MM-GBSA energy for interaction of 9a-d with HDAC6 was higher than for HDAC1 providing structural rationale for the HDAC6 selectivity.
Introduction
Aberrant regulation of gene expression is at the basis of several human diseases including many forms of cancer. This is partly the result of deregulation of DNA methylation or the post translational modification of histone tails. The cellular enzymes that mediate these modifications include DNA-methyltranferases (DNMTs), the recently described TET proteins that hydroxylate methylated DNA, histone acetyl transferases (HATs), histone deacetylases (HDACs), histone methyltransferases (HMTs), histone demethylases (HDMs) and histone kinases. Together, these enzymes influence transcription by maintaining tissue–specific epigenetic and transcriptional patterns as well as by acting as co-regulators for transcription factors. In disease, this regulation of the transcriptional process can be altered as a consequence of changes in the expression or function of epigenetic enzymes [1,2]. The most pharmacologically targeted in this class have been the HDACs.
HDACs influence the level of histone acetylation by removing the terminal ε-acetyl function of lysines. This activity condenses the chromatin into a closed structure resulting in the repression of transcription whereas the acetylation of histone lysine by HATs leads to an open chromatin structure thus allowing transcriptional initiation and gene expression. Currently there are 18 known human HDAC isoforms that are commonly divided into 4 classes, namely Class I (HDACs 1, 2, 3, and 8), Class II (HDACs, 4, 5, 6, 7, 9, and 10), Class III (SIRT 1–7) and class IV (HDAC 11). Whereas Class I, II and IV are zinc dependent, the class III enzymes require NAD+ for their activity. Most inhibitors of Class I, II and IV HDACs thus act by chelating active site zinc [3].
Since it was discovered that the antiproliferative activity of trichostatin A (TSA) was mediated through the inhibition of HDACs, [4] much effort has been invested in finding new inhibitors of this enzyme family. A diverse group of compounds discovered to date fall into four chemotypes: namely, hydroxamic acids, short fatty acids, cyclic tertrapeptides and benzamides of which several are currently in clinical development [5,6]. Of this first generation compounds, SAHA (Vorinostat), PXD101 (Belinostat), Romidepsin (Istodax), and RAS2410 (Resminostat) have received approval for the treatment of various cancers and were thought to target several or most members of the HDAC family. Although during the pre-clinical investigations these pan-HDAC inhibitors were deemed safe, in the clinical setting these compounds have exhibited various toxicities. For this reason there is an ongoing discussion of whether isoform-selective HDAC inhibitors would be more advantageous than pan-inhibitors [7]. Recent studies of the various isoform enzymes have revealed subtle differences in the amino acid make up of the active site which can be exploited in the development of isoform or class selective compounds [8]. In fact, this approach has been successful in achieving several class or isoform selective inhibitors, for example MS-275 (HDAC1), MGCD0103 (HDAC1,2), Tubacin and Tubastatin A (HDAC6), and PCI-34051 and SB-379278A (HDAC8) [9]. Furthermore, using molecular modeling and biochemical studies Dominguez and co-workers recently described a subset of small molecule hydroxamates as class IIa (HDAC4,5,7,9) specific inhibitors [10].
With the goal of increasing the diversity and selectivity of epigenetic modulators, we have screened our compound collection (Department of Pharmaceutical Chemistry, University of Innsbruck) against an established cell based assay that measures the derepression of an epigenetically silenced GFP transgene, the LDR or locus derepression assay [11,12]. This assay is sensitive to known HDAC inhibitors including TSA, depsipeptide and butyrate as well as to other epigenetic modulators such as 5-azadeoxycytidine, which activate the expression of the GFP transgene. The screen led to the identification of several active compounds with diverse molecular structures. One of the compounds, JIB-04, a pyridine hydrazone derivative modulates Jumonji histone demethylase activity and selectively inhibits cancer growth in vivo [13]. Among the active compounds was one which bears a hydroxamic acid functionality, (4-[4-(1-methylbenzimidazol-2-yl)piperazin-1-yl]sulfonylbenzenecarbohydroxamic acid, (9b), that was originally synthesized for studies of potential 11ß-hydroxysteroid dehydrogenase 1 inhibitors [14]. Here, we describe the activity of 9b and the synthesis and characterization of 9b analogs. We find that para-substituted hydroxamic acid analogs of 9b inhibit HDAC activity showing preference for HDAC6 over HDAC1 and have cancer selective antiproliferative properties. In addition to inhibition of HDAC activity, the compounds also induce a cell cycle arrest in cancer cells decreasing the cell number.
Materials and Methods
Chemistry
The synthesis of the mono-substituted 2-benzazolpiperazines 5a,b,d has been described in the literature by reacting 2-chlorobenzazols 1a,b,d with excess piperazine hydrate (4) [15,16].
Although it was reported that compounds 5a,b,d were obtained in high yields (70–90%), in our hands the major isolated product was the 1,4-diarylated piperazine in 80% yield. Compounds 5a,b,d, were then prepared in a 2-step reaction utilizing a mono-N-protected piperazine derivative (2) as described by Shafic et al (Fig 1 ) [17].
Fig 1. Synthesis of target compounds.

Reagents; i) 120°C, 3 h.; ii) 40% aqu. HBr, 70°C, 12 h; iii) Dry CH2Cl2, N(CH2CH3)3, 0°C; iv) 3-chlorosulfonylbenzoic acid or 4-chlorosulfonylbenzoic acid, N(CH2CH3)3, THF, 0°C to RT; v) (a) N-methyl morpholine, ethyl chloroformate, THF, 0°C to RT, (b) hydroxylamine HCl, KOH, EtOH, 0°C to RT.
Ethylcarboxylate piperazine derivatives 3a,b,d were obtained from 2-chlorobenzazoles 1a,b,d by nucleophilic substitution of the chlorine atom with ethyl N-piperazinecarboxylate 2. Subsequent removal of the ethylcarboxylate moiety with aqu. 40% HBr at 70°C gave the benzazol-2-yl piperazines 5a,b,d, in good yields. 2-Piperazin-1-yl-benzoxazole 5c was synthesized by the treatment of 2-chlorobenzoxazole 1c with piperazine hydrate 4 at 0°C in CH2Cl2 [18].
The sulfonamide derivatives 6a-d and 7a-d became accessible by reacting compounds 5a-d with either 3-chlorosulfonylbenzoic acid or 4-chlorosulfonylbenzoic acid, respectively. We adapted the mild and efficient one step conversion of carboxylic acids to hydroxamic acids reported by Reddy et al [19] for the synthesis of the target compounds 8a-d and 9a-d. Carboxylic acids 6/7a-d were first transformed to the acid anhydride by treatment with ethyl chloroformate at 0°C utilizing N-methyl morpholine as a base. Then freshly prepared hydroxylamine solution obtained by reacting hydroxylamine hydrochloride with potassium hydroxide in methanol was added to the acid anhydride mixture to obtain compounds 8a-d and 9a-d in high yields (Fig 1).
General Information
Infrared spectra (IR) were recorded from KBr pellets on a Mattson Galaxy Series FTIR 3000 spectrophotometer. 1H NMR spectra were recorded from DMSO-d6 solutions on a Varian Gemini 2000 (1H: 199.98 MHz) spectrometer. The centre of the solvent signal was used as internal standard, which was related to TMS with δ 2.49 ppm (1H). Assignments are based on chemical shift considerations. Melting points were determined on a Reichert Thermovar hot stage microscope and are uncorrected. Elemental analyses were performed at the "Institut für Physikalische Chemie", University of Vienna, Austria and the data for C, H, N are within ±0.4% of the calculated values. Reactions were monitored by TLC using Polygram SIL G/UV254 (Macherey-Nagel) plastic backed plates (0.25 mm layer thickness) and visualized using a UV lamp. Column chromatography was performed using Kieselgel 60 (0.040–0.063 mm).
2-Chlorobenzimidazole, 2-chlorobenzoxazole, 2-chlorobenzothiazole, 3-chlorosulfonyl benzoic acid and 4-chlorosulfonyl benzoic acid were purchased from Sigma-Aldrich Co. Abbreviations: CH2Cl2 (dichloromethane), DIPE (diisopropyl ether), EA (ethylacetate), EtOH (ethanol), MeOH (methanol), 2-PrOH (2-propanol), and THF (tetrahydrofuran).
Chemical Synthesis of Compounds
Synthesis of 2-Chloro-1-methyl-benzimidazole (1b): 2-Chlorobenzimidazole (10 g, 65.56 mmol) was dissolved in 80 ml of 2.5N NaOH (175 mM) and dimethyl sulfate (11.0 mL, 116 mM) was added drop wise under stirring at room temperature. After the addition the mixture was stirred for further 2 h and the precipitate formed was filtered by suction and the product washed several times with ice-water mixture till the filtered solution was neutral. Petrol ether was sucked through the solid product several times and then dried in vacuo to afford a light brown solid (8.76 g, 81% yield). 1H NMR (200 MHz, d 6-DMSO, δ): 3.78 (s, 3H, CH3), 7.26 (dq, J = 1.2Hz, J = 7.4Hz, BZI-H5/H6), 7.54–7.60 (m, 2H, BZI-H4/H7).
General method for synthesis of compounds 3a,b,d: A mixture of 2-chlorobenzazoles 1a, 1b, or 1d (1 equivalent) and ethoxycarbonylpiperazine 2 (3 equivalents) was heated at 120°C under stirring and the reaction followed by TLC (CH2Cl2/EA 7:3). After completion the reaction mixture was allowed to cool to room temperature and the mixture dissolved in 200 mL CH2Cl2. The organic phase was extracted with saturated NaHCO3 solution (3 x 100 mL), then with dist. H2O (2 x 50 mL), sat. NaCl solution (100 mL) and dried over anhy. Na2SO4. After filtration, the organic phase was evaporated to dryness. The precipitate formed after treatment of the residue with DIPE and stirring in the cold (ice/water mixture) was filtered and dried.
Ethyl 4-(1H-benzimidazol-2-yl)-piperazin-1-carboxylate (3a): 2-Chlorobenzimidazole (1.16 g, 5.88 mmol) and ethoxycarbonylpiperazine (2.85 g, 18.04 mmol) with a reaction time of 1h to yield a white product (1.06 g, 97%). 1H NMR (200 MHz, d 6-DMSO, d): 1.20 (t, J = 6.7Hz, 3H, CH3), 3.49–3.58 (m, 8H, 8x Pip-H), 4.07 (q, J = 6.7Hz, 2H, CH2), 6.39 (m, 2H, BZI-H5/6), 7.17 (m, 2H, BZI-H4/7), 11.39 (s, 1H, NH).
Ethyl 4-(1-methyl-benzimidazol-2-yl)-piperazin-1-carboxylate (3b): 2-Chloro-1-methylbenzimidazole (2.00 g, 12.04 mmol) and ethoxycarbonylpiperazine (3.80 g, 24.05 mmol) heated at 125°C for 1h to yield a light yellow product (2.80 g, 81%). 1H NMR (200 MHz, d 6-DMSO, d): 1.21 (t, J = 7.0Hz, 3H, CH3), 3.19–3.31 (m, 4H, 4x Pip-H), 3.55–3.62 (m, 7H, 4x Pip-H, N-CH3), 4.08 (q, J = 7,2Hz, 2H, CH2), 7.05–7.15 (m, 2H, BZI-H5/6), 7.31–7.43 (m, 2H, Bzi-H4/7).
Ethyl 4-(benzothiazol-2-yl)-piperazin-1-carboxylate (3d): 2-Chlorobenzimidazole (1.16 g, 6.82 mmol) and ethoxycarbonylpiperazine (3.24 g, 20.50 mmol) heated together for 5 h to yield a light yellow product (1.85 g, 93%). 1H NMR (200 MHz, d 6-DMSO, d): 1.20 (t, J = 7.0Hz, 3H, CH3), 3.40–3.65 (m, 8H, 8x Pip-H), 4.07 (q, J = 7.0Hz, 2H, CH2), 7.03–7.12 (m, 1H, BZT-H6), 7.24–7.32 (m, 1H, BZT-H5), 7.45–7.48 (m, 1H, BZT-H4), 7.75–7.79 (m, 1H, BZT-H7).
General method for synthesis of compounds 5a,b,d: Compound 5a, 5b, or 5d was treated with 48% HBr in water and the mixture heated at 70°C under stirring for 24 h. The reaction mixture was allowed to cool to room temperature and evaporated to dryness. The residue was dissolved in water (100 mL) and made basic with conc. NH3 and extracted with CH2Cl2 (4 x 100 mL). The combined organic phase was washed with dist. water (2 x 50 mL), sat. NaCl solution, dried over anhyd. Na2SO4, filtered and the solvent evaporated to dryness. The product was recrystallised from DIPE.
2-(Piperazin-1-yl)benzimidazole (5a): Heating ethyl 4-(1H-benzimidazol-2-yl)-piperazin-1-carboxylate 6a (2.00 g, 7.29 mmol) and 48% HBr (40 mL) yielded a white product (1.37 g, 93%). 1H NMR (200 MHz, d 6-DMSO, d): 3.01–3.60 (m, 4H, 4x Pip-H), 3.89–3.92 (m, 4H, 4x Pip-H), 7.27–7.33 (m, 2H, BZI-H5/6), 7.42–7.49 (m, 2H, BZI-H4/7), 9.18 (s, 1H, NH).
1-Methyl-2-(piperazin-1-yl)benzimidazole (5b): Ethyl 4-(1-methyl-benzimidazol-2-yl)-piperazin-1-carboxylate 6b (2.80 g, 9.71 mM) and 48% HBr (40.5 mL) to yield a light yellow product (1.93 g, 92%). 1H NMR (200 MHz, d 6-DMSO, d): 2.5–2.90 (m, 4H, 4x Pip-H), 3.11–3.21 (m, 4H, 4x Pip-H), 3.58 (s, 3H, N-CH3), 7.05–7.12 (m, 2H, BZI-H5/H6), 7.29–7.41 (m, 2H, BZI-H4/H7).
2-(Piperazin-1-yl)benzothiazole (5d): Ethyl 4-(benzothiazol-2-yl)-piperazin-1-carboxylate (6c) (1.50 g, 5.15 mM) and 48% HBr (24.5 mL) to yield a colourless product (1.08 g, 96%). 1H NMR (200 MHz, d 6-DMSO, d): 2.76 (m, 4H, 4x Pip-H), 3.43–3.48 (m, 4H, 4x Pip-H), 6.99–7.11 (m, 1H, BZT-H6), 7.21–7.31 (m, 1H, BZT-H5), 7.41–7.49 (m, 1H, BZT-H4), 7.71–7.75 (m, 1H, BZT-H7).
Synthesis of 2-(Piperazin-1-yl)benzoxazole (5c): 5.6g (65 mM) Piperazine (water free) and 3.27g (0.325 mol) triethylamine was dissolved in 50 ml of dry CH2Cl2 and the solution cooled to 0°C in an ice/salt bath. 2-Chlorobenzoxazole (1c) dissolved in 20 mL of dry CH2Cl2 was added dropwise to the solution under stirring keeping the solution at 0°C. After the addition, the mixture was stirred for a further 30 min at 0°C and then quenched with 200 mL of ice-water. After separation of the organic phase, the water phase was extracted with ethyl acetate (2 x 100 mL), the combined organic phases was washed with sat. NaCl solution, dried over anhyd. Na2SO4, filtered and evaporated to dryness. Compound 8d was isolated as a white solid over column chromatography using silica gel with CH2Cl2/MeOH (5:1) as eluent. Yield 4.96 g (75%). 1H NMR (200 MHz, d 6-DMSO, d): 3.48–3.53 (m, 4H, 4x Pip-H); 3.17 (s, 1H, NH); 2.75–2.80 (m, 4H, 4x Pip-H); 6.99 (dt, J = 1.6Hz, J = 8Hz, 1H, BZO-H5); 7.18 (dt, J = 1.2Hz, J = 7.6Hz, 1H, BZO-H6); 7.27 (dd, J = 1.4Hz, J = 7.6Hz, 1H, BZO-H7); 7.37 (dd, J = 0.6Hz, J = 7.8Hz, 1H, BZO-H4).
General synthesis of the carboxylic acid derivatives 6a-d and 7a-d: 2-(Piperazin-1-yl)benzazolyl derivatives 5a-d (1 equivalent) and triethylamine (1.5 equivalents) were dissolved in 25 mL dry CH2Cl2 and cooled to 0°C in an ice salt bath. Then the required 3 or 4-Chloro-sulfonylbenzoic acid (1.1 equivalents) dissolved in 15 mL dry THF was added dropwise. After the addition, the reaction mixture was stirred at 0°C for a further 1 h and then allowed to warm to room temperature and the course of the reaction followed by TLC (CH2Cl2/MeOH/konz. NH3: 898/100/2). After indicating the end of the reaction, the mixture was evaporated to dryness, the residue was dissolved in dist. water and made slightly acidic (pH 6) with glacial acetic acid and stirred for about 30 mins. The solid formed was filtered by suction, washed with water and then with petrol ether. The product was recrystallized from the appropriate solvent; the precipitate formed was filtered under suction and dried at 60°C under high vacuum.
3-[4( 1H -Benzimidazol-2-yl)-piperazin-1-yl]sulfonyl-benzoic acid (6a): 2-(Piperazin-1-yl)benzimidazole hydrobromide 5a (0.200 g. 1.06 mM), triethylamine (0.285 g, 4.22 mM) and 3-chlorosulfonylbenzoic acid (0.186 g, 1.27 mM) was reacted for overnight at RT to give colourless crystals (0.295 g, 72%). Compound 6a was recrystalissed from EtOH (m. pt. 335°C, decomposed). IR (KBr, v cm-1): 2994 (-OH), 1648 (C = O), 1613, 1560 (C = C, C = N), 1344 (-SO2-). 1H NMR (200 MHz, d 6-DMSO, d): 2.98–3.12 (M, 4H, 4x PIP-H), 3.55–3.66 (m, 4H, 4x PIP-H), 6.87–6.92 (m, 2H, BZI-H5,6), 7.13–7.18 (m, 2H, BZI-H4,7), 7.73–781 (m, 1H, Ar-H5), 7.98–8.02 (m, 1H, Ar-H6), 8.21–8.24 (m, 1H, Ar-H2,4).
3-[4-(1-Methylbenzimidazol-2-yl)piperazin-1-sulfonyl]-benzoic acid (6b): 1-Methyl-2-(piperazin-1-yl)benzimidazole 5b (0.300 g. 1.38 mM), triethylamine (0.281 g, 2.76 mM) and 3-chlorosulfonylbenzoic acid (0.367 g, 1.67 mM) was reacted for 23 h at RT to give colourless crystals (0.523 g, 93%). Compound 6b was recrystallised from EtOH (m. pt. 291–293°C). IR (KBr, v cm-1): 2855 (-OH), 1714 (C = O), 1616, 1637 (C = C, C = N), 1349 (-SO2-). 1H NMR (200 MHz, d 6-DMSO, d): 3.14 (br s, 4H, 4x Pip-H), 3.33 (br. s, 4H, 4x Pip-H), 3.51 (s, 3H, N-CH3), 7.05–7.09 (m, 2H, BZI-H6,5), 7.29–7.39 (m, 2H, BZI-H4,7), 7.79–7.86, (m, 1H, Ph-H5), 8.02–8.06 (m, 1H, Ph-H6), 8.24–8.29 (m, 2H, Ph-H2,4).
3-(4-Benzoxazol-2-yl-piperazin-1-sulfonyl)-benzoic acid (6c): 2-(Piperazin-1-yl)benzoxazole 5c (0.300 g, 1.48 mM), triethylamine (0.299 g, 2.96 mM) and 3-chlorosulfonylbenzoic acid (0.391 g, 0.18 mM) was reacted for 23 h at RT to give colourless crystals (0.545 g, 95%). Compound 6c was recrystallized from EtOH (m. pt. 290–293°C, decomposses IR (KBr, v cm-1): 2867 (-OH), 1685 (C = O), 1640, 1581 (C = C, C = N), 1348 (-SO2-). 1H NMR (200 MHz, d 6-DMSO, d): 3.00–3.11 (m, 4H, 4x Pip-H), 3.69 (br s, 4H, 4x Pip-H), 6.95–7.03 (m, 1H, BZO-H5 or 6), 7.08–7.15 (m, 1H, BZO-H5 or 6), 7.23–7.27 (m, 1H, BZO-H4), 7.34–7.37 (m, 1H, BZO-H7), 7.73–7.8 (m, 1H, Ar-H5), 7.95–7.99 (m, 1H, Ar-H6), 8.19–8.23 (m, 1H, Ar-H2,4).
3-(4-Benzothiazol-2-yl-piperazin-1-sulfonyl)-benzoic acid (6d): 2-(Piperazin-1-yl)benzothiazole 5d (0.300 g. 1.37 mM), triethylamine (0.277 g, 2.73 mM) and 3-chlorosulfonylbenzoic acid (0.362 g, 1.64 mM) was reacted overnight at RT to give colourless crystals (0.515 g, 93%). Compound 6d was recrystallized from EtOH (m. pt. 375.5°C, decomposes). IR (KBr, v cm-1): 3441 (OH), 1704 (C = O), 1592, 1536 (C = N, C = C), 1350 (-SO2-). 1H NMR (200 MHz, d 6-DMSO, d): 3.06 (br. s., 4H, 4x Pip-H), 3.66 (br. s., 4H, 4x Pip-H), 7.01–7.09 (m, 1H, BZT-H6), 7.21–7.29 (m, 1H, BZT-H5), 7.43 (d, J = 7.8Hz, BZT-H4), 7.65–7.75 (m, 2H, 2x Ar-H5,6), 7.87 (d, J = 7.6 Hz, 1H, BZT-H7), 8.18–8.22 (m, 2H, 2x Ar-H2,4).
4-[4( 1H -Benzimidazol-2-yl)-piperazin-1-sulfonyl]-benzoic acid (7a): 2-(Piperazin-1-yl)benzimidazole hydrobromide 5a (0.400 g, 1.06 mM), triethylamine (0.570 g, 4.22 mM) and 4-chlorosulfonylbenzoic acid (0.373 g, 1.27 mM) was reacted overnight at RT to give colourless crystals (0.498 g, 92%). Compound 7a was recrystallized from EtOH (m. pt. > 350°C). IR (KBr, v cm-1): 2894 (-OH), 1638 (C = O), 1610 (C = N, C = C). 1H NMR (200 MHz, d 6-DMSO, d): 2.98–3.12 (m, 4H, 4x Pip-4H), 3.55–3.66 (m, 4H, 4x Pip-4H), 6.87–6.92 (m, 2H, BZI-H5,6), 7.11–7.16 (m, 2H, BZI-H4,7), 7.87 (ABd, J = 8.0 Hz, 2H, ArH-2,6), 8.52 (ABd, J = 8.2 Hz, 2H, ArH-3,5).
4-[4-(1-Methylbenzimidazol-2-yl)piperazin-1-yl]sulfonylbenzoic acid (7b): 1-Methyl-2-(piperazin-1-yl)benzimidazole 5b (0.300 g. 1.38 mM), triethylamine (0.281 g, 2.76 mM) and 4-chlorosulfonylbenzoic acid (0.367 g, 1.67 mM) was reacted for 3 days at RT to give colourless crystals (0.550 g, 99%). Compound 7b was recrystallised from EtOH (m. pt. 340°C, decomposition). IR (KBr, v cm-1): 2867 (-OH), 1704 (C = O), 1614, 1586 (C = C, C = N), 1354 (-SO2-), 1H NMR (200 MHz, d 6-DMSO, d): 3.14 (br.s, 4H, 4x Pip-H), 3.32 (br.s, 4H, 4x Pip-H), 3.51 (s, 3H, N-CH3), 7.04–7.09 (m, 2H, BZI-H5,6), 7.29–7.39 (m, 2H, BZI-H4,7), 7.90 (ABd, J = 8.4Hz, 2H, Ph-2,6), 8.19 (ABd, J = 8.4Hz, 2H, Ph-3,5).
4-(4-Benzoxazol-2-yl-piperazin-1-sulfonyl)-benzoic acid (7c): 2-(Piperazin-1-yl)benzoxazole 5c (0.200 g. 1.48 mM), triethylamine (0.199 g, 2.96 mM) and 4-chlorosulfonylbenzoic acid (0.260 g, 1.78 mM) was reacted overnight at RT to give colourless crystals (0.329 g, 85%). Compound 7c was recrystallized from EtOH (m. pt. > 350°C). IR (KBr, v cm-1): 2917 (-OH), 1689 (C = O), 1582 (C = C, C = N), 1346 (-SO2-). 1H NMR (200 MHz, d 6-DMSO, d): 3.09 (br s, 4H, 4x Pip-4H), 3.69 (br s, 4H, 4x Pip-4H), 6.96–7.03 (m, 1H, BZO-H5 or 6), 7.08–7.16 (m, 1H, BZO-H5 or 6), 7.25 (d, J = 8.0Hz, 1H, BZO-H7), 7.36 (d, J = 7.6 Hz, 1H, BZO-H4), 7.86 (ABd, J = 7.8 Hz, 2H, ArH2,6), 8.14 (ABd, J = 7.8Hz, 2H, ArH3,5).
4-(4-Benzothiazol-2-yl-piperazin-1-sulfonyl)-benzoic acid (7d): 2-(Piperazin-1-yl)benzothiazole 5d (0.300 g. 1.38 mM), triethylamine (0.277 g, 2.76 mM) and 4-chlorosulfonylbenzoic acid (0.362 g, 1.56 mM) was reacted for 23 h at RT to give colourless crystals (0.552 g, 98%). Compound 7d was recrystallized from EtOH (m. pt. 325–327°C). IR (KBr, v cm-1): 2992 (-OH), 1689 (C = O), 1593 (C = C, C = N), 1352 (-SO2-). 1H NMR (200 MHz, d 6-DMSO, d): 3.09 (br s, 4H, Pip-4H), 3.66 (br s, 4H, Pip-4H), 7.02–7.10 (m, 1H, BZT-H6), 7.21–7.29 (m, 1H, BZT-H5), 7.43 (d, J = 8.2Hz, 1H, BZT-H4), 7.74 (d, J = 7.4 Hz, 1H, BZT-H7), 7.87 (ABd, J = 8.0 Hz, 2H, ArH2,6), 8.5 (ABd, J = 8.4Hz, 2H, ArH3,5).
General synthesis of the hydroxamic acid derivatives 8a-d and 9a-d [19]
For the synthesis of compounds 8a-d and 9a-d a solutions of acids 6a-d and 7a-d were prepared to which is added a solution of hydroxylamine.
Preparation of acid anhydride
To a solution carboxylic acid derivatives 6a-d and 7a-d (1 equivalent) in 30 mL dry THF at 0°C is added ethylchloroformate (1.2 equivalents), N-methylmorpholine (1.3 equivalents) with the mixture stirred for a further 1 h and filtered. The reaction was controlled with TLC (PE/EtAc 3:7).
Preparation of free hydroxylamine
To a solution of potassium hydroxide (3 equivalents) in 25 mL dry EtOH was cooled to 0°C and hydroxylamine HCl (3 equivalents) was added in small portions After the addition the mixture was stirred for a further 1 h at 0°C and the precipitated KCl was filtered.
Preparation of target hydroxamic acid derivatives
The prepared acid anhydride in A was added dropwise to the freshly prepared hydroxylamine solution in EtOH at 0°C. The reaction was stirred further for 1 h and then at room temperature till the reaction declared complete after TLC control (PE/EtAc 3:7). After complete reaction (1 h.), the mixture was evaporated to dryness and the residue taken up in cooled distilled water and stirred for 30 min., the precipitate filtered under suction and washed with petrolether. The products were dried in high vacuum at 60°C.
3-[4( 1H -Benzimidazol-2-yl)-piperazin-1-sulfonyl]- N -hydroxy-benzamid (8a): Benzoic acid derivative 6a (0.270 g, 6.98 mM), N-methylmorpholine (0.092 g, 9.07 mM), ethylchloroformate (0.091 g, 8.38 mM), KOH (0.118 g, 2,09 mM) and hydroxylamine HCl (0.146 g, 2,094 mM) was reacted to give colourless crystals (0.205 g, 73%) of compound 8a (m. pt. 349–350°C). IR (KBr, v cm-1): 3423 (-OH), 2982–3068 (-NH), 1613 (C = O), 1344 (-SO2) 1H NMR (200 MHz, d 6-DMSO, d): 3.08 (br. s., 4H, 4x Pip-4H), 3,62 (br s, 4H, 4x Pip-4H), 6,95 (br s, 2H, BZI-H5,6), 7.19 (br s, 2H, BZI-H4,7), 7.74–7.82 (m, 1H, ArH), 8.00–8.04 (m, 1H, ArH), 8.21–8.24 (m, 2H, 2xArH), 10.40 (br s, 1H, OH), 11.2 (br s, 1H, NH). Anal. Calcd. for C18H19N5O4S (401.44): C, 53.85; H, 4.77; N, 17.45. Found: C, 54.20; H, 5.01; N, 17.32.
3-[4-(1-Methyl-Benzimidazol-2-yl)-piperazin-1-sulfonyl]- N -hydroxy-benzamid (8b): Benzoic acid derivative 6b (0.450 g, 1.12 mM), N-methylmorpholine (0.147 g, 1.46 mM), ethylchloroformate (0.146 g, 1.34 mM), KOH (0.188 g, 3.36 mM) and hydroxylamine HCl (0.233 g, 3.36 mM) was reacted to give colourless crystals (0.405 g, 87%) of compound 8b (m. pt. 167–169°C). IR (KBr, v cm-1): 3432–3349 (-OH), 2854–3068 (-NH), 1615 (C = O), 1597 (C = C, C = N), 1349 (-SO2-). 1H NMR (200 MHz, d 6-DMSO, d): 3.13 (br s, 4H, 4x Pip-4H), 3.34 (br s, 4H, 4x Pip-4H), 3.51 (s, 3H, N-CH3), 7.05–7.09 (m, 2H, BZM-H5,6), 7.29–7.40 (m, 2H, BZM-H4,7), 7.73–7.95 (m, 1H, ArH), 8.02–8.12 (m, 1H, ArH), 8.23–8.29 (m, 2H, 2x ArH), 9.25 (s, 1H, OH), 11.55 (s, 1H, NH). Anal. Calcd. for C19H21N5O4S (415.47): C, 54.93; H, 5.09; N, 16.86. Found: C, 55.20; H, 5.30; N, 16.90.
3-(4-Benzoxazol-2-yl-piperazin-1-sulfonyl)- N -hydroxy-benzamid (8c): Benzoic acid derivative 6c (0.450 g, 1,16 mM), N-methylmorpholine (0.153 g, 1,51 mM), ethylchloroformate (0.151 g, 1.39 mM), KOH (0.195 g, 3.48 mM)) and hydroxylamine HCl (0.242 g, 3.48 mM) was reacted to give colourless crystals (0.402 g, 86%) of compound 8c (m. pt. 257–262°C). IR (KBr, v cm-1): 1580 (C = C, C = N), 1348 (-SO2-). 1H NMR (200 MHz, d 6-DMSO, d): 3.09 (br s, 4H, 4x Pip-H), 3.70 (br s, 4H, 4x Pip-H), 6.96–7.03 (m, 1H, BZO-H5 or 6), 7.08–7.16 (m, 1H, BZO-H5 or 6), 7.24–7.27 (m, 1H, BZO-H7), 7.34–7.38 (m, 1H, BZO-H4), 7.69–7.81 (m, 1H, ArH), 7.87–8.08 (m, 2H, 2x ArH), 8.20–8.25 (m, 1H, ArH), 9.23 (br s, 1H, OH), 11.50 (br s, 1H, NH). Anal. Calcd. for C18H18N4O5S (402.42): C, 53.72; H, 4.51; N, 13.92. Found: C, 53.79; H, 4.57; N, 13.89.
3-(4-Benzothiazol-2-yl-piperazin-1-sulfonyl)- N -hydroxy-benzamid (8d): Benzoic acid derivative 6d (0.400 g, 0,98 mM), N-methylmorpholine (0.130 g, 1,27 mM), ethylchloroformate (0.128 g, 1.18 mM), KOH (0.166 g, 2.94 mM) and hydroxylamine HCl (0.206 g, 3.36 mM) was reacted to give colourless crystals (0.332 g, 80%) of compound 8d (m. pt. 207–210°C). IR (KBr, v cm-1): 2949–3298 (-NH, -OH), 1653 (C = O), 1591, 1532 (C = C, C = N), 1350 (-SO2-). 1H NMR (200 MHz, d 6-DMSO, d): 3.08 (br s, 4H, 4x Pip-H), 3:67 (br s, 4H, 4x Pip-H), 7.01–7.09 (m, 1H, BZT-H6), 7.21–7.29 (m, 1H, BZT-H5), 7.72–7.76 (m, 2H, BZT-H7, Ar-H), 7.41–7.45 (m, 1H, BZT-H4), 7.87–7.91 (m, 1H, Ar-H6), 8.04–8.08 (m, 2H, Ar-H2,4), 8.85–9.65 (br s, 1H, OH), 11.12–11.62 (br s, 1H, NH). Anal. Calcd. for C18H18N4O4S2 (418.49): C, 51.66; H, 4.34; N, 13.39. Found: C, 51.72; H, 4.32; N, 13.26.
4-[4(1H-Benzimidazol-2-yl)-pipreazin-1-sulfonyl]-N-hydroxy-benzamid (9a): Benzimidazol derivate 7a (0.400 g, 1.03 mM), N-methylmorpholine (0.136g, 1.34 mM), ethylchloroformate (0.134 g, 1.24 mM), KOH (0.174 g, 3.09 mM) and hydroxylamine HCl (0.215 g, 3.09 mM) was reacted to give colourless crystals (0.340 g, 82%) of compound 9a (m. pt. 382–383°C, decomposition). IR (KBr, v cm-1): 2859-3077(-NH, -OH), 1644 (C = O), 1611 (C = N), 1360 (-SO2-). ESI-MS (m/z): 402.14 [M+1]. 1H NMR (200 MHz, d 6-DMSO, d): 3.15 (br. s, 4H, 4x Pip-4H), 3.35 (br. s, 4H, 4x Pip-4H), 6.90–6.94(m, 2H, BZI-H5,6), 7.17–7.22 (m, 2H, BZI-H4,7), 7.90 (ABd, J = 8.4Hz, 2H, ArH-2/6), 8.15 (ABd, J = 8.4Hz, 2H, ArH-3/5), 9.23 (br.s, 1H, OH), 11.50 (br.s, 1H, NH). Anal. Calcd. for C18H19N5O4S (401.44): C, 53.85; H, 4.77; N, 17.45. Found: C, 54.07; H, 4.91; N, 17.48.
4-[4-(1-Methyl-Benzimidazol-2-yl)-piperazin-1-sulfonyl]- N -hydroxy-benzamid (9b): Benzoic acid derivative 7b (0.450 g, 1,12 mM), N-methylmorpholine (0.147 g, 1,46 mM), ethylchloroformate (0.146 g, 1,34 mM), KOH (0.189 g, 3,37 mM) and hydroxylamine HCl (0.234 g, 3,37 mM) was reacted to give colourless crystals (0.417 g, 89%) of compound 9b (m. pt. 269–271°C). IR (KBr, v cm-1): 3379 (-OH), 2855–3188 (-NH), 1615 (C = O), 1353 (-SO2). 1H NMR (200 MHz, d 6-DMSO, d): 3.14 (br s, 4H, 4x Pip-4H), 3.33 (br s, 4H, 4x Pip-4H), 3.52 (s, 3H, N-CH3), 6.98–7.14 (m, 2H, BZM-H5,6), 7.25–7.45 (m, 2H, BZM-H4,7), 7.84–7.93 (m, 2H, 2xArH), 7.98–8.02 (m, 1H, ArH), 8.17–8.21 (m, 1H, ArH), 9.29 (br s, 1H, OH), 11.50 (br s, 1H, NH). Anal. Calcd. for C19H21N5O4S (415.47): C, 54.93; H, 5.09; N, 16.86. Found: C, 55.18; H, 5.27; N, 16.95.
4-(4-Benzoxazol-2-yl-piperazin-1-sulfonyl)- N -hydroxy-benzamid (9c): Benzoic acid derivative 7c (0.264 g, 0,681 mM), N-methylmorpholine (0.102 g, 0,89 mM), ethylchloroformate (0. 101 g, 0,82 mM), KOH (0.130 g, 2,04 mM) and hydroxylamine HCl (0.161 g, 2,04 mM) was reacted to give colourless crystals (0.225 g, 82%) of compound 9c (m. pt. 335–338°C). IR (KBr, v cm-1): 3400 (-OH), 2869–3094 (-NH), 1689 (C = O), 1582 (C = N), 1346 (-SO2-). 1H NMR (200 MHz, d 6-DMSO, d): 3,10 (br s, 4H, 4x Pip-4H), 3.70 (br s, 4H, 4x Pip-4H), 7.00 (t, J = 7.2Hz, 1H, BZO-H5 or 6), 7.13 (t, J = 7.2Hz, 1H, BZO-H5 or 6), 7.26 (d, J = 7.2Hz, 1H, BZO-H7), 7.36 (d, J = 7.4Hz, 1H, BZO-H4), 7.81–7.89 (m, 2H, 2x ArH), 7.97 (d, J = 8.2Hz, 1H, ArH), 8.15 (d, J = 7.6Hz, 1H, ArH), 9.25 (br s, 1H, OH), 11.45 (br s, 1H, NH). Anal. Calcd. for C18H18N4O5S (402.42): C, 53.72; H, 4.51; N, 13.92. Found: C, 53.83; H, 4.55; N, 13.91.
4-(4-Benzothiazol-2-yl-piperazin-1-sulfonyl)- N -hydroxy-benzamid (9d): Benzoic acid derivative 7d (400 mg, 0,99 mM), N-methylmorpholine (130,36mg, 1,29 mM), ethyl chloroformate (129,11 mg, 1,19 mM), KOH (166,87 mg, 2,97 mM) and hydroxylamine HCl (206.67 mg, 2.97 mM) was reacted to give colourless crystals (0.343 g, 82%) of compound 9d (m. pt. 324°C, decomposition). IR (KBr, v cm-1): 2858–3046 (-NH, -OH), 1689/1639 (C = O), 1593 (C = N),1543 (C = C),1350 (-SO2-) 1H NMR (200 MHz, d 6-DMSO, d): 3.09 (br s, 4H, 4x Pip-4H), 3.66 (br s, 4H, Pip-4H), 7.06 (t, J = 7.2Hz, 1H, BZT-H6), 7.26 (t, J = 7.4Hz, 1H, BZT-H5), 7.43 (d, J = 7.4Hz, 1H, BZT-H4), 7.74 (d, J = 7.4Hz, 1H, BZT-H7), 7.83–7.87 (m, 2H, 2x ArH), 7.97 (d, J = 8.6Hz, 1H, ArH), 8.15 (d, J = 7.8Hz, 1H, ArH), 10.40 (br s, 1H, OH), 11.20 (br s, 1H, NH). Anal. Calcd. for C18H18N4O4S2 (418.49): C, 51.66; H, 4.34; N, 13.39. Found: C, 51.79; H, 4.39; N, 13.40.
LDR studies and measures of GFP
LDR cells were maintained in DMEM with L-glutamine and high glucose, 1 mM sodium pyruvate, 0.1 mM MEM non-essential amino acids, 1% v/v penicillin-streptomycin and 10% heat inactivated FBS. Cells were seeded on glass bottom chambers at low-medium density and allowed to attach overnight. Compounds were then added for 24h at the indicated dose and cells were imaged under a fluorescent microscope equipped with a camera using a 20 or 40X objective. All images shown were exposed for identical time.
Purification of HDAC1 and Initial Histone Deacetylase Assay
For inhibition assays, partially purified HDAC1 (70–80 fold enriched relative to crude samples) from mouse A20 cells was used as enzyme source. HDAC1 activity was purified by a combination of anion exchange chromatography (Mono Q), affinity chromatography (Poly-lysin agarose, Heparin Sepharose) and TSK size exclusion chromatography (Jesacher and Loidl, unpublished). HDAC1 protein was identified by immunoblotting using anti mouse HDAC1 antibodies (collaboration with Zymed Laboratories, Inc., San Francisco, California, USA). HDAC activity was determined as described by Sendra et al., [20] using [3H]acetate-prelabeled chicken reticulocyte histones as substrate. 50 μL of mouse HDAC1 were incubated with different concentrations of compounds for 10 min on ice and 10 μL of total [3H] acetate pre-labeled chicken reticulocyte histones (4 mg/mL) were added, resulting in a concentration of 41 μM. This mixture was incubated at 37°C for 1 h. The reaction was stopped by addition of 50 μL of 1 M HCl/0.4 M acetylacetate and 1 mL ethylacetate. After centrifugation at 10000 x g for 5 min an aliquot of 600 μL of the upper phase was counted for radioactivity in 3 mL liquid scintillation cocktail.
MTS viability assays
Lung cancer cells (kindly provided by Dr. John D. Minna, [13, 21]) were maintained in RPMI 1640 media supplemented with 10% FBS. Immortalized normal human bronchial epithelial cells (also provided by Dr. John D. Minna [13, 21]) were cultured in KSFM supplemented with pituitary extract and EGF. For viability assays, cells were plated on 96 well plates and grown overnight. Cells were then exposed to increasing doses of the indicated compounds for 4 days and their viability assayed using the Promega Cell Titer 96 AQueous One kit per the manufacturer’s protocol. All doses were tested in 4–8 replicates. IC50 values were calculated using the in-house software DIVISA.
Cell cycle and apoptosis assays
For cell cycle analysis, cells were washed with PBS, fixed in 70% ethanol for 2 h, washed with PBS and resuspended in staining solution (0.1% Triton X-100, 0.2 mg/ml DNAse-free RNAse A and 20 mg/ml PI) and incubated at 37C for 15 min. Histograms were acquired on a BD FACSCalibur flow cytometer at UT Southwestern’s core facility. Apoptosis was measured using a FITC Annexin V Apoptosis Detection Kit I (BD Pharmingen) according to manufacturer’s protocol after treating cells with compounds as indicated in Figure legends.
Comparative HDAC inhibition assays
For the direct comparison of HDAC1 vs. HDAC6 inhibition, HDAC activity was analyzed using the HDAC Assay Kit from Millipore (17–356). Briefly, recombinant HDAC1 or recombinant HDAC6 (both purchased from Epigenetek, Purity of HDAC1 is more than 60% and purity HDAC6 is more than 85%). was incubated with the fluorometric HDAC substrate according to the manufacturer’s protocol in the presence of DMSO vehicle or increase compound doses. In a secondary activator reaction, the fluorophore is only cleaved from the deacetylated substrate, allowing for quantification. Fluorescence was quantified on a FLUOstar-Optima or a FLUOstar Omega plate reader (BMG Biosciences). To ensure the compounds do not interfere with the detection system control experiments with standard deacetylated substrate were performed in the presence of high doses of all compounds.
Computational methods
The crystal structure of human HDAC1 [22] (PDB ID: 4BKX) and a homology model of HDAC6 that is described elsewhere [23] was used for the docking study. The protein structure for docking was prepared using the Protein Preparation Wizard [24] within the Schrödinger software package Schrödinger Inc, New York, USA. The hydrogen bond network was optimized automatically and the protonation states were predicted with the PROPKA tool at pH 7.0. Ligands were prepared in MOE [25] (version 2012.10, Chemical Computing Group, Montreal, Canada). Conformational search for all structures has been carried out using the Low Mode MD sampling with a minimum RMSD between the conformations of 1 Å. This procedure was applied to obtain realistic conformations for all ligands containing flexible ring systems. Molecular docking studies have been performed on a Linux cluster using the Glide software (Schrödinger Inc, New York, USA) [24] in standard precision (SP) mode. Constraints to the Zn2+ ion were set and two conserved water molecules located inside the binding pocket were considered during the docking. Post-docking filtering has been carried out using the Python script distance_to_smarts (Revision 3.5, Schrödinger). Only docking poses with distances between both oxygen atoms of the hydroxamic acid of the ligand and the Zn2+ ion inside the binding pocket of the protein below 2.7 Å have been selected. Rescoring of the docking poses was done applying the MM-GBSA approach. The MMF94 force field and a GBSA solvation model within the MOE software package were used. Ligands were minimized inside the binding pocket until a gradient of 0.05 kcal/mol was reached. Protein heavy atoms were tethered using a force constant of 100 kcal/mol Å.
Results
Identification of 9b as an LDR active with HDAC inhibitory activity
We incubated LDR cells with compounds in our collection at 20μM and measured the activation of the silenced GFP gene. Fluorescence microscopy showed robust GFP expression in response to 4-[4-(1-methylbenzimidazol-2-yl)piperazin-1-yl]sulfonylbenzenecarbohydro xamic acid (9b) treatment (Fig 2 A). We confirmed that the signal was not coming from compound auto-fluorescence by co-inducing with estradiol which causes the nuclear translocation of the GFP chimeric protein [26]. Thus, 9b is a bona fide activator of the expression of GFP in the LDR system, suggesting that it may have activity as an epigenetic modulator (Fig 2 B).
Fig 2. Structure and locus de-repression assay activity of Trichostatin A (TSA) and 9b-9d.

A, Structure of 4-[4-(1-methylbenzimidazol-2-yl)piperazin-1-yl]sulfonylbenzenecarbohydroxa-mic acid, (compound 9b). B, LDR cells were assayed for GFP expression by fluorescence microscopy after overnight treatment with the indicated doses of the DMSO vehicle, the trichostatin A positive control or 9b-9d, followed by estradiol induction of the nuclear translocation of the GFP-estrogen receptor chimeric transgene. DAPI staining of the nucleus is shown for reference. The other compounds were inactive in the LDR assay.
Since the LDR assay is sensitive to hydroxamic acid HDAC inhibitors we hypothesized that 9b may be acting by this mechanism. Furthermore, compound 9b has the structural elements that have been described as a common template for HDAC inhibitors, comprising a cap group (1-methylbenzimidazole), a zinc binding enzyme inhibition group (CO(NHOH) functionality) and a spacer (piperazine-sulfonylbenzene moiety) that links the cap to the binding system [27]. Moreover, an HDAC inhibitor, R306465 (Fig 3) with juxtaposed flanking groups developed by Johnson & Johnson [28] shares significant sub-structural similarity to 9b and its analogs.
Fig 3. Structure of R306465, an HDAC inhibitor developed by Johnson & Johnson that shares structural motifs with 9a-9d series.
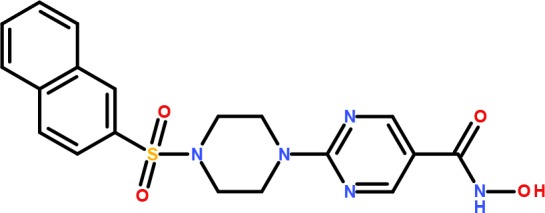
To evaluate this directly, we performed in vitro HDAC1 activity assays using a partially purified system with A20 cells as the source of enzyme. As can be seen in Fig 4, 9b is indeed an HDAC inhibitor with an IC50 of approximately 12μM in this assay, and its structure may be used as a scaffold to further diversify this activity. We therefore explored the structure/activity relationship of two elements of 9b by synthesizing and analyzing the effects of the replacement of the 1-methylbenzimidazole ring by the isosteric heterocycles benzimidazole, benzoxazole and benzothiazole and a variation of the position of the hydroxamic acid substituent on the phenyl ring (3- and 4- position (see Fig 1)).
Fig 4. Induction of GPF and inhibitory activity against partially purified HDAC1.

NS = not soluble,—negative, *The mean values of at least two independent experiments in which duplicate determinations were taken.
Locus derepression activity and ability of 9b analogues to inhibit HDAC1 activity
Using the LDR cells, we first assayed the 9b analogues 8a-d, 9a, 9c and 9d for their ability to induce the GFP transgene in our system. Indeed, a high level of GFP induction was observed upon treatment of the cells with compounds 9c, 9d (Fig 2B) which bears a hydroxamic acid functionality at the four position. In this assay system, compounds 8a-d bearing the hydroxamic acid functionality in the three position of the phenyl ring did not induce GFP (Fig 4). The position of the hydroxamic acid thus appears critical to activity and is likely related to the ability of this group to reach into the catalytic site in order to chelate the zinc ion. In addition, compound 9a where X = NH was also inactive in the LDR assay likely due to poor cell permeability.
We then evaluated if the 9b analogues which were LDR actives were able to inhibit HDAC1 activity. For enzyme inhibition assays, partially purified HDAC1 from mouse A20 cells was used as enzyme source. Compared to 9b (IC50 = 12.4 μM), both 9c (IC50 = 5.7 μM) and 9d (IC50 = 4.8 μM) were two to three fold more potent in inhibiting HDAC1 activity (Table 1). Although compound 9a was inactive in the LDR assay, it is equipotent (IC50 = 12.4 μM) compared to 9b in the in vitro HDAC inhibition assay, again suggesting poor cell penetration in the LDR assay. As a control, we also measured LDR inactives 8a-d for their HDAC1 inhibitory potential and found that as expected they were 10–60 fold less potent in vitro than 9b (Fig 4).
Table 1. Antiproliferative activity (IC50 μM) of compounds 9a-d, TSA, Apicidin and Depsipeptide.
| Compd | HCC4017 | HBEC-30KT | Selectivity | HCC4018 | HBEC-34KT | Selectivity |
|---|---|---|---|---|---|---|
| 9a | > 10* | > 10 | - | >10 | >10 | - |
| 9b | 1.49 | > 10 | > 6.71 | 6.1 | > 10 | > 1.64 |
| 9c | 1.24 | > 10 | > 8.06 | 5.05 | > 10 | > 1.98 |
| 9d | 1.63 | 5.37 | 3.29 | 4 | 8.22 | 2.06 |
| TSA | 0.073 | 0.285 | 3.90 | 0.023 | 0.346 | 15.0 |
| Apicidin | 0.12 | 0.09 | 0.75 | 0.03 | 0.79 | 26.3 |
| Depsi. | 0.0065 | 0.0009 | 0.14 | 0.0042 | 0.0013 | 0.31 |
| Patient A | Patient B | |||||
*The mean values of at least three independent experiments in which duplicate determinations were taken within each experiment. For clarity, standard deviation has been omitted.
Anti-proliferative activity and cancer-selectivity of compounds 9a- 9d
Given the known anti-cancer qualities of HDAC inhibitors, we evaluated the most potent compounds for their ability to block cancer cell proliferation and for their selectivity for cancer vs. normal cells. To accomplish this, we performed standard MTS viability assays on two pairs of patient matched non-small cell lung cancer cells and their corresponding normal human epithelial lines. As can be seen in Table 1, compounds 9b, 9c and 9d all had anti-proliferative activity with IC50 values between 1–6 μM. Furthermore, these three compounds demonstrated cancer selectivity, affecting the cancer lines HCC4017 and HCC4018 several fold more potently than the patient matched HBEC normal lines HBEC30KT and HBEC34KT, respectively (Table 1). Again, we saw no cellular activity with 9a.
Compared to the known HDAC inhibitors TSA, apicidin and depsipeptide, compounds 9b,c and d had less potent antiproliferative activity (Table 1). On the other hand, compounds 9b-d showed better selectivity for patient A matched cells compared to the three known HDAC inhibitors. However, for cells derived from patient B TSA and apicidin exhibited significantly higher selectivity by a factor of 7–13 fold compared to compounds 9b-d, yet in both paired cell lines depsipeptide was more toxic to normal cells than to the patient-matched cancer cells.
Effect of compounds 9b and 9c on cell cycle progression and induction of apoptosis
Recently, HDAC inhibitors have emerged as promising chemotherapeutic agents, and the findings of several studies suggest that they can induce a range of anti-tumor activities including the induction of cell-cycle arrest, the stimulation of differentiation, and the induction of apoptosis in a variety of transformed cells in culture. [29] In agreement with this, we found that compounds 9b and 9c affect cell cycle progression. FACs analysis indicated that both 9b and 9c affect cell cycle distribution after just 18h of treatment. Indeed the higher doses markedly influenced S-phase progression and resulted in accumulation of cells in S/G2/M especially at later times (Fig 5A and 5B). Compound 9c had particularly robust effects on cell cycle phase distribution (Fig 5B). Of note, no subG0 population was observed in either 9b or 9c treated cells even after 64h. Consistent with this, Annexin V staining did not show any reproducible significant changes in the percent of dead cells (S1–S3 Figs).
Fig 5. A, B: 9b and 9c strongly alter cell cycle distribution of cancer cells.

HCC4017 lung cancer cells show defects in S-phase progression and accumulation in G2/M after treatment with 9b (in A) or 9c (in B), as indicated. Cell cycle histograms were collected on PI stained samples of floating and adherent cells, on a FACs Calibur.
Tissue-specific antiproliferative activity in tumor cell lines
To evaluate if the antiproliferative activity of the novel hydroxamic acid derived compounds was general across cancer types, selected compounds were submitted for testing in the tissue-specific panel of human tumor cell lines at the National Cancer Institute, Bethesda, USA. This screen utilizes 60 human tumor cell lines, representing leukemia, melanoma and cancers of the lung, colon, brain, ovary, kidney, prostate and breast [30]. This cell line panel offers the advantage of having available microarray gene expression data across the 60 cell lines which can be used to determine the uniqueness of a new lead for which the molecular target(s) might not be known. The antitumor activity is presented for each cell line by three parameters, namely log10 GI50 value (GI50 = molar concentration of the compound that inhibits 50% net cell growth), log10 TG value (TGI = molar concentration of the compound leading to total inhibition of net cell growth), and log10 LC50 value (LC50 = molar concentration of the compound leading to 50% net cell death) accompanied by MID which refers to the average inhibitory value across the 60 cell lines.
Compounds 9b-d which were active in the in vitro HDAC assay and showed inhibition of cancer vs. normal lung cells, were submitted for testing across the 60 cell line panel for antiproliferative activity at a single dose of 10 μm in the initial screening assay. Only compounds 9c and 9d exhibited significant growth inhibition at this dose (average 50% and 62% respectively). These results are summarized in S4–S6 Figs. The activity of these two agents was then tested over a five dose concentration curve. In this assay, compounds 9c (NSC 747073) and 9d (NSC 747072) inhibited the proliferation of subsets of leukemia, colon, CNS, melanoma, and prostrate cancer cells to a higher extent than cells from other cancers (Fig 6A and 6B). As shown in Table 2, of the two compounds, 9d was more potent overall by a factor of 2 (MID: = 3.16 μm), compared to 9c (MID: = 6.61 μM) consistent with its higher activity in the one dose assay.
Fig 6. Differential response of cancers to 9c and 9d.

A, GI50 values across the NCI-60 cell line panel in response to 9c. B, GI50 values across the NCI-60 cell line panel in response to 9d.
Table 2. Inhibitory concentration values across histological cancer types.
| NSC 747073 (9c) | NSC 747072 (9d) | |
|---|---|---|
| Cancer Type | GI50 (μM) | GI50 (μM) |
| Leukemia | 2.24 (4/4) b | 1.77 (5/5) |
| NSCL cancer a | 9.51 (3/9) | 3.77 (4/9) |
| Colon cancer | 6.05 (4/6) | 2.30 (6/7) |
| CNS cancer | 5.82 (4/6) | 2.77 (4/6) |
| Melanoma | 5.62 (6/8) | 2.66 (5/8) |
| Ovarian cancer | 7.66 (2/5) | 4.01 (3/3) |
| Renal Cancer | 8.24 (3/8) | 3.94 (3/8) |
| Prostate Cancer | 4.89 (2/2) | 2.48 (2/2) |
| Breast cancer | 8.71 (2/7) | 3.88 (4/7) |
| MID c | 6.61 | 3.16 |
aNon-Small Cell Lung cancer
bFigures in parenthesis refers to no. cell lines used for the assay/no inhibited by compound
cAverage value of inhibition across the 60 cell lines
Modeling of 8a-d and 9a-d in the catalytic site of HDACs
Molecular docking was carried out using the X-ray structure of human HDAC1 [22] and a homology model of human HDAC6 [23] to investigate the interaction of compounds 8a-d and 9a-d with these enzymes and explore potential specificity differences since these two HDACs have distinct biology and their catalytic sites show differences that can be targeted. The obtained docking poses were re-scored using the MM-GBSA approach. For both HDAC subtypes, the para-substituted inhibitors 9a-d showed favorable interactions (Fig 7 and Fig 8). The hydroxamic acid group is chelating the zinc ion and makes a hydrogen bond to His141 in HDAC1 and His611 in HDAC6. The aromatic ring of the inhibitors interacts with two conserved Phe residues (Phe150 and Phe 205 in HDAC1, Phe620 and Phe680 in HDAC6) whereas the sulfonamide group interacts with a conserved water molecule observed in HDAC crystal structures. Only in case of the para-substituted derivatives, the terminal bicyclic aromatic ring system makes favorable interaction with the rim of the catalytic pocket (Gly27, His28 and Pro29 in HDAC1, His500, Pro501 and Tyr570 in HDAC6). In case of HDAC6, a perfect fit between the benzimidazole, benzoxazole and benzothiazole group of 9a-d with the surface of the catalytic pocket is observed (Fig 8) which would predict stronger inhibition of HDAC6 over HDAC1. For the meta-substituted derivatives (8a-d), which are inactive or weakly active, the terminal group is extending in to the solvent (Fig 9).
Fig 7. Docking poses of compounds 9a-d in the HDAC1 catalytic domain.

Inhibitors are colored orange.The zinc ion is shown as a brown ball. Hydrogen bonds between HDAC1 and inhibitors are shown as blue lines and distances are given in Å. On the right side, the molecular surface is displayed and contoured according to the hydrophobic potential (magenta = hydrophilic, green = hydrophobic).
Fig 8. Docking poses of compounds 9a-d in the HDAC6 catalytic domain.

Inhibitors are colored in cyan. The zinc ion is shown as a brown ball. Hydrogen bonds between HDAC6 and inhibitors are shown as blue lines and distances are given in Å. On the right side the molecular surface is displayed and contoured according to the hydrophobic potential (magenta = hydrophilic, green = hydrophobic).
Fig 9. Docking results for compounds 8a-8d in HDAC1.

Due to the meta-substitution of the sulfonamide linker the terminal group of the inhibitors is not able to favorably interact with the rim of the pocket. The molecular surface is displayed and contoured according to the hydrophobic potential (magenta = hydrophilic, green = hydrophobic).
In case of HDAC1, the MM-GBSA energies were able to discriminate between actives (9a-d) and weakly active compounds (8a-d) (Table 3). For the most potent HDAC6 inhibitors favorable MM-GBSA energies were derived (Table 3).
Table 3. Inhibitory activities and MM-GBSA energies of the studied compounds.
| Compound | HDAC1 IC50 [μM] | HDAC1 pIC50 | MM-GBSA (kcal/mol) | HDAC6 IC50 [μM] | HDAC6 pIC50 | MM-GBSA(kcal/mol) |
|---|---|---|---|---|---|---|
| 8a | n.i. | - | -20.44 | - | - | n.c |
| 8b | 316 | 3.50 | -20.77 | - | - | n.c. |
| 8c | 216 | 3.67 | -20.89 | - | - | n.c. |
| 8d | 140 | 3.85 | -20.96 | - | - | n.c |
| 9a | 12.4 | 4.91 | -29.51 | 1.00 | 6.00 | -34.72 |
| 9b | 12.4 | 4.91 | -28.27 | 0.10 | 7.00 | -36.03 |
| 9c | 5.7 | 5.24 | -28.88 | 0.20 | 6.70 | -35.39 |
| 9d | 4.8 | 5.32 | -29.43 | 0.10 | 7.00 | -36.47 |
n.c. = not calculated, n.i. = no inhibition
The para-substituted compounds 9a-d show HDAC6 selectivity
To directly test if 9b and its analogs had greater potency to inhibit HDAC6 (a class IIb enzyme) compared to HDAC1 a (class I enzyme) as predicted in the virtual ligand binding assay, we performed parallel experiments for both enzymes using purified proteins and measured activity using a fluorophore system designed by Millipore (see Methods) which quantifies the amount of acetylated histone substrate. Although we found no differences in IC50 for HDAC6 vs. HDAC1 in the meta-substituted compounds (data not shown), a large difference in IC50 was apparent for the para-substituted compounds (Fig 10). In all cases, the potency against HDAC6 was 5–30 fold greater than that against HDAC1 (Table 4). However, compounds 9a-d are less potent as HDAC 6 inhibitor compared to Tubastatin A [31] and TSA [32].
Fig 10. Compounds 9a-9d inhibit HDAC6 over HDAC1.

Dose response curves of 9a-9d comparative inhibition of HDAC6 vs. HDAC1 activity using purified enzymes for in vitro assays measuring deacetylation of histone substrates. Assays were performed in parallel for both enzymes.
Table 4. Enzyme Inhibition Data of Compounds 9a-d.
Thus, we have characterized novel compounds 9a-d as HDAC6 selective inhibitors in vitro and shown that three of them (9b, 9c and 9d) have specific antiproliferative activity against cancer but not normal cells.
Discussion
Using an established cell based assay to screen for epigenetic modulators, we report the identification of a novel series of hydroxamic acid derivatives which possess activity as HDAC inhibitors selectively inhibiting HDAC6 over HDAC1 and having specific antiproliferative activity on human cancer cells vs. patient matched normal cells. Compounds with the hydroxamic acid functionality in the para-position of a benzene ring exhibited favorable HDAC inhibitory and antiproliferative activities. Although the potency of these compounds was lower compared to known HDACi TSA, apicidin and depsipeptide, they had comparable or greater selectivity of inhibition for cancer vs. normal cells and for HDAC6 vs HDAC1. In contrast, the meta substituted analogues were devoid of biological activity. The antiproliferative effect of 9b and 9c was at least in part mediated by robust changes in the cell cycle distribution leading to growth arrest of cancer cells exposed to these compounds, in line with known effects of HDAC inhibition.
Molecular modeling studies uncovered that the MM-GBSA energy data for interaction with HDAC6 was higher than for HDAC1 indicating that 4-[4-(1-methylbenzimidazol-2-yl)piperazin-1-yl]sulfonylbenzenecarbohydroxamic acid and its analogues might be selective HDAC6 inhibitors. Confirmatory studies showed that these compounds are indeed selective HDAC6 inhibitors compared to HDAC1 by a factor 6–40. In addition, 9b-9d demonstrated potent blocking of tumor cell viability while not affecting normal cells derived from the same patients.
Several HDACi are in preclinical development and clinical trials for the treatment of a wide range of diseases, including cancer. The in vivo toxicities associated with some of these compounds due to their molecular promiscuity may well prevent them from ever reaching clinical efficacy. The current hypothesis is that the development of selective inhibitors targeting only one member or a subclass of the HDAC family will lead to improved overall efficacy and fewer side effects. In view of this, the compounds described herein, particularly 9b, can serve as lead structures for the development of selective HDAC6 inhibitors with improved and more targeted activity. HDAC6 selective inhibitors have been described including Tubacin, Tubastatin A, ACY-1215 and several others [9, 33]. These inhibitors appear to primarily affect non-histone targets of HDAC6 including α-tubulin and HSP90 and have beneficial effects in disease models including cancer but also extending to neurodegeneration and immunity [31].
Among known HDAC inhibitors, R306465 developed by Johnson & Johnson [28] shares significant sub-structural similarity to 9b and its analogs. The in vitro specificity of R306465 suggested a preference for HDAC1 vs. HDAC8 and cellular studies determined that both histone and tubulin acetylation were increased by the compound though the effects on histone acetylation were stronger and detected at lower doses compared to tubulin acetylation [28]. Consistent with this, HDAC6 downstream target Hsp70 was not induced except by high doses of R306465 and c-raf was only degraded at similar high doses. Thus, R306465 appears to be selective for HDAC1 compared to HDAC6 at least in the tested cells. Remarkably, our study suggests that rearrangement of the chemical groups of R306465 into the 9b-d configuration switches the selectivity of the compounds towards HDAC6. Clinical studies with R306465 have been reported in abstract form and toxicities were moderate with some indication of target involvement and efficacy in regimens below the maximal tolerated doses [34]. This, taken together with our studies and the beneficial effects of HDAC6 inhibitors reported in other disease models [31, 33] indicates that 9b related compounds have clinical promise and should be further developed.
Supporting Information
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
Compounds 9c and 9d which exhibited significant growth inhibition were evaluated against the 60 cell panel at five concentration levels. This information can be at https://dtp.cancer.gov/.
(PDF)
Acknowledgments
We thank Jianjun Chang for technical assistance with proliferation assays. This project was partly funded by the NIH (R01CA12526901 to E.D.M., 1R21AI11622201 to I.D./E.D.M. P50-CA70907 to J.D.M., subaward to E.D.M.) by the Doctor’s Cancer Foundation (Nolan Miller Lung Cancer grant to E.D.M.), by the Friends of the Cancer Center, by the Welch Foundation (grants to E.D.M.) and by CPRIT (RP120717).
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This project was partly funded by the NIH (R01CA12526901 to E.D.M., 1R21AI11622201 to I.D./E.D.M. P50-CA70907 to J.D.M., subaward to E.D.M.) by the Doctor’s Cancer Foundation (Nolan Miller Lung Cancer grant to E.D.M.), by the Friends of the Cancer Center, by the Welch Foundation (grants to E.D.M.), by the National Cancer Institut R01CA12526901 to E.D.M., and by CPRIT (RP120717). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Portel A, Esteller M. (2010) Epigenetic modifications and human Disease. Nat Biotech 28: 1057–1068. [DOI] [PubMed] [Google Scholar]
- 2. Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. (2012) Epigenetic protein families: a new frontier for drug discovery. Nat Drug Discov 11: 384–400. [DOI] [PubMed] [Google Scholar]
- 3. Lin H-Y, Chen C-S, Lin S-P, Weng J-R, Chen C-S. (2006) Targeting Histone Deacetylase in Cancer Therapy. Med Res Rev 26: 394–413. [DOI] [PubMed] [Google Scholar]
- 4. Yoshida M, Kijima M, Akita M, Beppu T. (1990) Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin. J Biol Chem 265: 17174–17179. [PubMed] [Google Scholar]
- 5. Ververis K, Hiong A, Karagiannis TC, Licciardi PV. (2013) Histone deacetylase (HDACIs): multitargeted anticancer agents. Biologics: Targets Therapy. 7: 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Grassadonia A, Cioffi P, Simiele F, Iezzi L, Zilli M, Natoli C. (2013) Role of hydroxamates-based histone deacetylase inhibitors (Hb-HDACIs) in the treatment solid malignancies. Cancers 5: 919–942. 10.3390/cancers5030919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ononye SN, van Heyst M, Falcone EM, Anderson AC, Wright D. (2012) Toward isozyme-selective inhibitors of histone deacetylase as therapeutic agents for the treatment of cancer. Pharm Pat. Anal. 1(2) 10.4144/ppa.12.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Silvestri L, Ballante F, Mai A, Marshall GR, Ragno R. (2012) Histone Deacetylase Inhibitors: Structure-Based Modeling and Isoform-Selectivity Prediction. J Chem Inf Model 52: 2215–2235. 10.1021/ci300160y [DOI] [PubMed] [Google Scholar]
- 9. Witt O, Deubzer HE, Milde T, Oehme I. (2009) HDAC family: What are the cancer relevant targets? Cancer Lett 277: 8–21. 10.1016/j.canlet.2008.08.016 [DOI] [PubMed] [Google Scholar]
- 10. Buürli RW, Luckhurst CA, Aziz O, Matthews KL, Yates D, Lyons KA, et al. (2013) Design, Synthesis, and Biological Evaluation of Potent and Selective Class IIa Histone Deacetylase (HDAC) Inhibitors as a Potential Therapy for Huntington’s Disease. J Med Chem 56: 9934–9954. 10.1021/jm4011884 [DOI] [PubMed] [Google Scholar]
- 11. Johnson RL, Huang W, Jadhav A, Austin CP, Inglese J, Martinez ED. (2008) A quantitative high-throughput screen identifies potential epigenetic modulators of gene expression. Anal Biochem 375: 237–248. 10.1016/j.ab.2007.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Best AM, Chang J, Dull AB, Beutler JA, Martinez ED. (2011) Identification of four potential epigenetic modulators from the NCI structural diversity library using a cell-based assay. J Biomed Biotech 868095: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang L, Chang J, Varghese D, Dellinger M, Kumar S, Best AM, et al. (2013) A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat Comm 4: 2035 10.1038/ncomms3035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hofer S, Kratschmar DV, Schernthanner B, Vuorinen A, Schuster D, Odermatt A, et al. (2013) Synthesis and biological analysis of benzazol-2-yl piperazine sulfonamides as 11β-hydroxysteroid dehydrogenase 1 inhibitors. Bioorg Med Chem Lett 23: 5397–5400. 10.1016/j.bmcl.2013.07.047 [DOI] [PubMed] [Google Scholar]
- 15. Orjales A, Mosquera R, Labeaga L, Rodes R. (1997) New 2-Piperazinylbenzimidazole Derivatives as 5-HT3 Antagonists. Synthesis and Pharmacological Evaluation. J Med Chem 40: 586–593. [DOI] [PubMed] [Google Scholar]
- 16. Walczyn´ski K, Guryn R, Zuiderveld OP, Timmerman H. (1999) Non-imidazole histamine H3 ligands. Part I. Synthesis of 2-(1-piperazinyl)- and 2-(hexahydro-1H-1,4-diazepin-1-yl)benzothiazole derivatives as H3-antagonists with H1 blocking activities, Il Farmaco 54: 684–694. [DOI] [PubMed] [Google Scholar]
- 17. Shafik RM, Eshba NH, El Semary MMA, Saudi MNS, El Bayaa RY. (2003) Piperazino and ethylenediamino analogs of benzimidazole: novel zwitterion-type candidates as possible nonsedating H1-blockers with potential antinociceptive activity. Alex J Pharm Sci 17: 137–144. [Google Scholar]
- 18. Sato Y, Yamada M, Yoshida S, Soneda T, Ishikawa M, Nizato T, et al. (1998) Benzoxazole Derivatives as Novel 5-HT3 Receptor Partial Agonists in the Gut. J Med Chem 41: 3015–3021. [DOI] [PubMed] [Google Scholar]
- 19. Reddy AS, Kumar MS, Reddy GR. (2000) A convenient method for the preparation of hydroxamic acids. Tetrahedron Lett 41: 6285–6288. [Google Scholar]
- 20. Sendra R, Rodrigo I, Salvador ML, Franco L. (1988) Characterization of pea histone deacetylases. Plant Mol Biol 11: 857–866. 10.1007/BF00019525 [DOI] [PubMed] [Google Scholar]
- 21. Chang J, Varghese DS, Gillam MC, Peyton M, Modi B, Schiltz RL, et al. (2012) Differential response of cancer cells to HDAC inhibitors trichostatin A and depsipeptide. Br J Cancer 106: 116–125. 10.1038/bjc.2011.532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Millard CJ, Watson PJ, Celardo I, Gordiyenko Y, Cowley SM, Robinson CV, et al. (2013) Class I HDACs share a common mechanism of regulation by inositol phosphates. Mol Cell 51: 57–67. 10.1016/j.molcel.2013.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schäfer S, Saunders L, Schlimme S, Valkov V, Wagner JM, Kratz F, et al. (2009) Pyridylalanine-containing hydroxamic acids as selective HDAC6 inhibitors. ChemMedChem 4: 283–290. 10.1002/cmdc.200800196 [DOI] [PubMed] [Google Scholar]
- 24. Suite 2012: Maestro version 9.3, Protein Preparation Wizard, Epik version 2.3, Glide version 5.8, Schrödinger, LLC, New York, NY, 2012. [Google Scholar]
- 25. Molecular Operating Environment (MOE), 2012.10; Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2012. [Google Scholar]
- 26. Martinez ED, Dull AB, Beutler JA, Hager GL. (2006) High-Content Fluorescence-Based Screening for Epigenetic Modulators. Methods in Enzymology 414: 21–26. [DOI] [PubMed] [Google Scholar]
- 27. Paris M, Porcelloni M, Binaschi M, Fattori D. (2008) Histone Deacetylase Inhibitors: From Bench to Clinic. J Med Chem 51: 1505–1529. 10.1021/jm7011408 [DOI] [PubMed] [Google Scholar]
- 28. Arts J, Angibaud P, Mariën A, Floren W, Janssens B, King P, et al. (2007) R306465 is a novel potent inhibitor of class I histone deacetylases with broad-spectrum antitumoral activity against solid and haematological malignancies. Br J Cancer 97: 1344–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Witt O, Deudzer HE, Milde T, Oehme I. (2009) HDAC family: What are the cancer relevant targets. Cancer Lett. 277, 8–21. 10.1016/j.canlet.2008.08.016 [DOI] [PubMed] [Google Scholar]
- 30. Monga M, Sausville EA. (2002) Developmental Therapeutics Program at the NCI: molecular target and drug discovery process. Leukemia 16:520–526. [DOI] [PubMed] [Google Scholar]
- 31. Butler KV, Kalin J, Brochier C, Vistoli G, Langley B, Kozikowski AP. (2010) Rational Design and Simple Chemistry Yield a Superior, Neuroprotective HDAC6 Inhibitor, Tubastatin. J Am Chem Soc 132: 10842–10846. 10.1021/ja102758v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Furumai R, Komatsu Y, Nishino N, Khochbin S, Yoshida M, Horinouchi S. (2001) Potent histone deacetylase inhibitors built from trichostatin A and cyclic tetrapeptide antibiotics including trapoxin. PNAS 98: 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dallavalle S, Pisano C, Zunino F. (2012) Development and therapeutic impact of HDAC6-selective inhibitors. Biochem Pharmacol. 84: 756–65. 10.1016/j.bcp.2012.06.014 [DOI] [PubMed] [Google Scholar]
- 34. Fong PC, Settatree S, Sinha R, Hardcastle A, Hellemans PW, Arts J, et al. (2007) A first-in-man phase I study of R306465, a histone deacetylases inhibitor exploring pharmacokinetics and pharmacodynamics utilizing an electrochemiluminescent immunoassay in patients with advanced tumors. Journal of Clinical Oncology, (2007) ASCO Annual Meeting Proceedings (Post-Meeting Edition). 25: No 18S (June 20 Supplement), 2007: 3578. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
Compounds 9c and 9d which exhibited significant growth inhibition were evaluated against the 60 cell panel at five concentration levels. This information can be at https://dtp.cancer.gov/.
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.