

Abstract
1H-detection can greatly improve spectral sensitivity in biological solid-state NMR (ssNMR), thus allowing the study of larger and more complex proteins. However, the general requirement to perdeuterate proteins critically curtails the potential of 1H-detection by the loss of aliphatic side-chain protons, which are important probes for protein structure and function. Introduced herein is a labelling scheme for 1H-detected ssNMR, and it gives high quality spectra for both side-chain and backbone protons, and allows quantitative assignments and aids in probing interresidual contacts. Excellent 1H resolution in membrane proteins is obtained, the topology and dynamics of an ion channel were studied. This labelling scheme will open new avenues for the study of challenging proteins by ssNMR.
Keywords: ion channels, membrane proteins, proton detection, side-chain protons, solid-state NMR spectroscopy
The recent advent of 1H-detection in biological solid-state NMR (ssNMR) spectroscopy can greatly increase spectral sensitivity,1 and thereby bears the potential to critically broaden the scope of ssNMR spectroscopy. The prevailing method to detect protons in solid proteins is perdeuteration, that is, the complete deuteration and subsequent reintroduction of exchangeable protons in protonated buffers. This labelling scheme largely removes line-broadening 1H–1H dipolar couplings and can provide spectra of extremely high quality.2 Moreover, it allows automated backbone assignments3 and probing contacts between backbone amino protons (HN), which are important to define protein folds.4
However, the absence of aliphatic side-chain protons in perdeuterated proteins curtails the potential of 1H-detection, given that side chains are important factors for protein structure and function. In addition, the availability of side-chain protons could facilitate the assignment of complex proteins. In general, to assign side-chain protons and use them for structural studies has remained a major difficulty for 1H-detected ssNMR spectroscopy. In principle, fully protonated proteins in combination with magic-angle spinning (MAS) frequencies of higher than 100 kHz2d could provide a future avenue to access side-chain protons, as suggested in recent studies with soluble and membrane proteins.5 However, it can be envisaged that the residual 1H linewidths and spectral crowding will remain a challenge for larger proteins. Moreover, for proteins such as membrane proteins with a substantial inhomogeneous contribution to the 1H linewidth, MAS frequencies higher than 100 kHz may not compensate for the sensitivity loss resulting from comparably small sample volumes. Excellent resolution and assignments of aliphatic protons have been reported with residual adjoining protonation (RAP), which relies on the random incorporation of protons into a deuterated protein matrix.6 Moreover, approaches such as ILV, proton cloud, or SAIL labelling can be used to probe contacts between side-chain protons of specific types of amino acids.7 Yet, such approaches employ isolated labels which may be very difficult to assign de novo and only give access to a selection of side-chain protons. To assign side-chain protons and exploit them for structural studies, even in larger proteins, a labelling scheme which 1) provides a high global 1H density and 2) mitigates spectral crowding nonetheless, could be very advantageous. Thus we explored “fractional deuteration” in 1H-detected ssNMR spectroscopy. This labelling scheme, based on protonated 13C-glucose and D2O in the growth medium, was previously proposed in solution NMR spectroscopy as an alternative to ILV labelling and in 13C-detected ssNMR spectroscopy for spectral editing.8 These studies reported that certain carbon atoms, such as Cα, are highly deuterated in fractionally deuterated proteins, while many side-chain carbon atoms retain sizeable 1H levels.
Herein we demonstrate that fractional deuteration provides access to well-resolved HN and side-chain protons of virtually all residues in one sample, and allows assignment and use of these protons for structural studies. Importantly, even though our approach works at much higher 1H levels, we observe an excellent resolution (0.07 ppm) for the HN protons in the fractionally deuterated (FD) membrane-embedded K+ channel KcsA, and it rivals the resolution in the perdeuterated channel. We outline our approach on ubiquitin and then use it to study KcsA, including its membrane topology and dynamics, as well as important channel–water interactions.
Figure 1 shows 1H-detected two-dimensional (2D) CH and NH spectra of FD [13C,15N]-ubiquitin in aqueous (100 % H2O) buffers, acquired at 52 kHz MAS and 800 MHz 1H-frequency using MISSISSIPI water suppression9 and low-power PISSARRO decoupling.10 These spectra are of remarkable quality and feature a resolution as high as 0.05 and 0.07 ppm for aliphatic and exchangeable protons, respectively. The absence of CαHα correlations, which typically appear around δ${{_{{^{13}}{\rm C}}}}$=50–65 ppm and δ${{_{{^{1}}{\rm H}}}}$=3.0–5.0 ppm, is readily visible in the CH spectrum. A quantitative analysis using solution NMR spectroscopy (see section S1 in the Supporting Information) revealed, next to the absence of Hα protons (<2 % population), an interesting pattern of 1H depletion for the side chains in FD ubiquitin (see Figure 1 D and Table S1). The pyruvate-derived branched-chain amino acids (Ile, Leu, Val) exhibit very low (≤5 %) 1H levels at the Cβ-position, which is the same for the amino acids (Arg, Gln, Glu, Pro) derived from α-ketoglutarate (≤8 %). Amino acids that follow other pathways (such as Asn, Asp, His, Lys, Ser, Thr), however, retain much higher 1H levels (30–45 %; 90 % for Ser) at Cβ, with slightly reduced values for aromatic amino acids (Phe, Tyr). Most other carbon atoms, further away from the backbone, feature equally high 1H levels. Our data are in good agreement with the original solution NMR study, which also provides detailed biochemical explanations.8a Hence, many sites remain robustly protonated in FD proteins, yet feature a narrow 1H linewidth because the 1H network is, on average, starkly diluted. However, we like to emphasize that the local and global 1H density in FD proteins are much higher than those in RAP-labelled proteins.6 Broadening effects resulting from methylene isotopomers were not observed, probably because CH2 signals are broadened beyond detection. Methyl groups showed slight oval lineshapes because of isotopomers, but they did not significantly compromise the 1H resolution (0.05–0.08 ppm), presumably because CH3 signals are broader and less abundant than either CHD2 or CH2D signals (see Box I in Figure 1 A and Figure S2). Prominent features of the CH spectrum of FD ubiquitin are the unusually intense CαHN signals. As it can be readily shown with simulations (see Box II in Figure 1 A and section S2), this beneficial effect is caused by the absence of Hα protons in FD proteins.11
Figure 1.
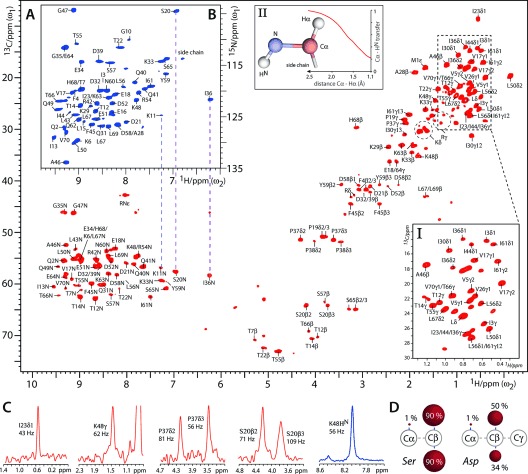
1H-detected ssNMR experiments in FD ubiquitin. A) 2D CH spectrum (in red). Box I shows an expansion of the methyl region. Box II shows simulations of the influence of CαHα dipolar couplings on the CP transfer from Cα to HN. B) 2D NH spectrum (blue). C) Cross-sections from the 2D CH (in red) and NH (blue) experiments. D) Examples of the protonation pattern in FD amino acids. See Table S1 for the complete list. Red spheres illustrate the 1H level at a given 13C.
In Figure 2 A we show assignments in FD ubiquitin, and they are based on dipolar transfer. Acquisition details can be found in section S2. Backbone connectivities were established with three-dimensional (3D) CαNH, Cα(CO)NH, and CCH experiments. In the 3D Cα(CO)NH, Cα polarization was prepared by a selective CP step (see Figure S4).2g, 12 These experiments were sufficient for backbone assignments, given that extensive chemical-shift data are available for ubiquitin.13 Side-chain assignments were performed with a 3D CCH experiment which included a 13C-13C DREAM14 double-quantum mixing block, optimized for one-bond transfer. Importantly, the efficient transfer from Cα to HN in the CCH experiments, and hence the presence of intense CαCβHβ and CβCαHN correlations, allow the use of HN as anchors to connect backbone and side chains, thereby greatly facilitating the assignment process. Moreover, the assignments of side-chain types are greatly simplified by the pattern of robustly protonated and deprotonated Cβ sites (see Table S1). CαCβHβ correlations were only detectable for residues with Hβ levels of greater than 20 %, which much reduced ambiguity. In total, we could assign the Hβ signals for 24 of the 28 residues with 1H levels of greater than 20 % (ignoring the mobile residues M1, T9, L27).13b Other inaccessible Hβ signals were from surface-exposed, and likely mobile, residues (K11, N25, N60). Additional side-chain protons such as the Hγ of Thr and Lys could also be readily assigned. To identify the methyl groups of Leu, Ile, Val, and Met, we resorted to published assignments.2d, 13a Such side-chain protons could also be assigned with longer 13C-13C mixing.
Figure 2.
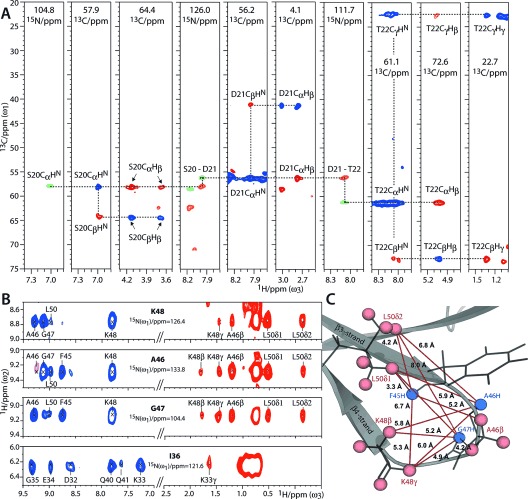
1H-detected assignments and structural studies in FD ubiquitin at 52 kHz MAS. A) Walk through S20–T22 (see section S3 for further walks). Signals from 3D CαNH (green), Cα(CO)NH (orange), and CCH (blue for positive; red for negative signals) experiments, are color-coded. B) Strips from a 3D NHH experiment. HN–HN contacts and inter-residual contacts between HN and side-chain protons are shown in blue and red, respectively. C) Illustration of the contacts shown in (B). HN–HN contacts are not shown for clarity.
Thanks to the high 1H density and resolution in FD proteins, the side-chain assignments can be readily exploited for structural studies, which are shown in Figures 2 B and C. We carried out a 3D NHH experiment with 1.5 ms 1H-1H DREAM mixing,2d, 7a in which we detected backbone–backbone HN–HN contacts, as well as backbone–side chain contacts between HN and aliphatic protons. Next to a large number of HN–HN contacts, many interresidual backbone–side chain contacts, of up to an 8 Å distance, could be assigned or identified (based on the X-ray structure PDB: 1UBQ). These contacts demonstrate that the high 1H density in FD proteins does not impede long-distance magnetization transfer. Unambiguous medium- and long-range 1H–1H contacts involved methyl groups and also methylene groups such as the CβHD and CγHD groups of Lys residues. Especially the latter contacts are noteworthy, since they are complementary to ILV labelling.
In Figures 3 and 4, we show the potential of 1H-detection in more complex FD proteins using the K+ channel KcsA, a well-accepted model for ion-channel gating,15 as an example. FD [13C,15N]-KcsA in the closed-conductive state was reconstituted in E. coli lipids and aqueous (100 % H2O) buffers. Further details of the sample preparation are given in section S5. We acquired dipolar-based 2D NH and CH spectra of very high quality (Figure 3 A and Figure 4 A), featuring a resolution as high as 0.06 and 0.07 ppm for aliphatic and exchangeable protons, respectively. Remarkably, the HN resolution in FD KcsA is comparable to perdeuterated KcsA5c (see Figure S11) and the perdeuterated membrane protein OmpG3a (0.13–0.18 ppm). This resolution strongly suggests that the availability of side-chain protons in many FD membrane proteins comes at either very low or no cost at fast (>50 kHz) MAS, because the residual 1H linewidth is dominated by inhomogeneous contributions. Hence, fractional deuteration is highly advantageous for 1H-detection in non-microcrystalline proteins.
Figure 3.
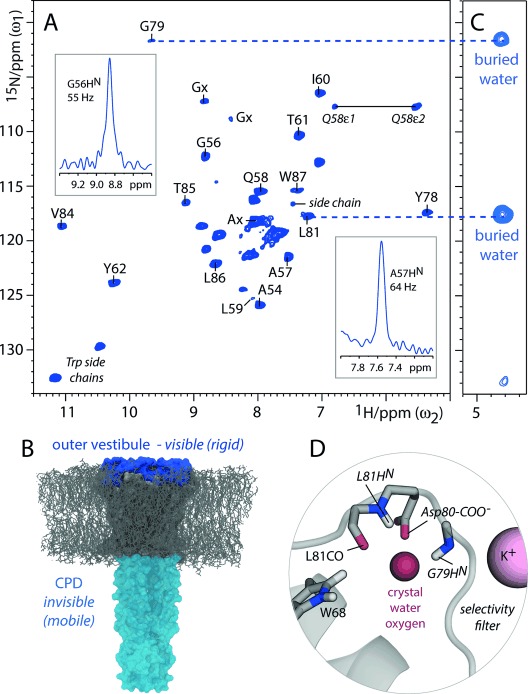
1H-detected studies with FD KcsA (closed-conductive). A) Dipolar-based 2D NH spectrum measured at 52 kHz MAS. B) The CPD is absent in this spectrum because of dynamics. C) Section of a 2D N(H)H spectrum using 0.75 ms 1H-1H DREAM mixing showing transfer of G79HN and L81HN to buried water behind the selectivity filter. The signals have negative intensity. D) Illustration of a KcsA structure (PDB: 1K4C).
Figure 4.
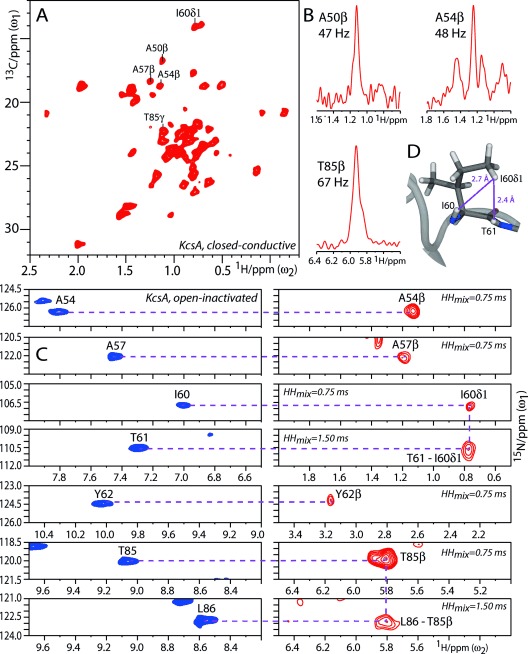
1H-detection of side chains in FD KcsA. A) Section of a 2D CH spectrum measured at 58 kHz MAS with closed-conductive KcsA. Cross-sections are shown in (B). C) Left: Sections from a 2D NH spectrum (blue); right: Sections from a 2D N(H)H spectrum (red) acquired with open-inactivated KcsA. D) The contact T61HN–I60Hδ1 is illustrated in the snapshot of an MD simulation. See section S3 for additional side-chain assignments.
FD KcsA was grown in D2O and only water-exposed residues are visible in the NH spectrum, which we used to study the membrane topology.2c, 5c Intriguingly, the NH spectrum showed only around 25 signals, while KcsA features about 70 water-accessible residues, which, in particular, comprise the extracellular outer vestibule (residues 51–64 and 80–86) and the cytoplasmic domain (CPD; residues 118–160). To understand the composition of the NH spectrum, we performed 3D CαNH, 3D Cα(CO)NH, 3D NHH, 2D CH, and 2D C(C)H experiments (see section S3 for a detailed discussion of the assignments), supported by 13C- and 15N-chemical-shift data.15c,d We validated our sequential assignments by HN–HN contacts which we observed in a 3D NHH experiment (see Figure S7). Moreover, by using a slightly longer 13C to 1H CP contact time (700 μs), we obtained many CαHN+1 contacts in the 2D CH, which also allowed validation of sequential assignments (see Figure S8). Altogether, we could assign about 70 % of the HN signals, which all belonged to the outer vestibule, thus demonstrating that the CPD is too dynamic for dipolar transfer (Figure 3 B). Note that we did not observe marked sensitivity with scalar transfer, thus implying that CPD dynamics in lipid membranes are relatively slow (μs to ms). We also did not observe the CPD in open-inactivated KcsA (see Figure S10), where the CPD helices are loosely structured. The latter result, thus, excludes the possibility that the CPD is invisible in the 2D NH of the closed-conductive channel (Figure 3a) because of tight packing of CPD helices, which might possibly interfere with the reintroduction of protons. This finding is noteworthy given that the conformational flexibility of the CPD is important for KcsA activation gating.15b Furthermore, in Figures 3 C and D we used our 1H assignments to study buried water behind the conductive selectivity filter, as it is important for the gating mode.5c, 15a How this water is bound is not directly accessible in KcsA Xray structures, since protons are not resolved. By transferring magnetization from HN to buried water in 2D and 3D NHH experiments (see Figure S12) using DREAM mixing, we see that G79HN and L81HN contact buried water, thus strongly suggesting that both coordinate the water oxygen atom while water protons contact the nearby Asp80-COO− group and L81CO.
The assignment of the spectrum in Figure 3 A was greatly simplified by the availability of side-chain protons. As described for FD ubiquitin, we connected side-chain and backbone information through HN anchor protons (see Figure 4 C and Figure S8). We thus assigned de novo side chains by a 2D C(C)H experiment as well as 2D and 3D NHH experiments which included a short (750 μs) 1H-1H DREAM transfer. Note that longer (1.5 ms) 1H-1H mixing also allowed structural studies of side-chain protons (Figure 4 C,D). Only residues with Hβ levels of greater than 20 % showed (HN)CαHβ and NHNHβ correlations in these experiments, and we readily assigned residues such as A54, A57, T61, Y62, and T85.
In conclusion, we have introduced a labelling approach for 1H-detected ssNMR spectroscopy, which provides far-reaching access to very well resolved backbone and side-chain protons. Most importantly, for non-microcrystalline samples, our method greatly expands the power of the formidable perdeuteration approach without sacrificing much, if any, 1H resolution. We believe that our approach, which also avoids the use of expensive deuterated glucose, will significantly increase the impact of solid-state NMR spectroscopy, especially for membrane proteins or peptide assemblies such as fibrils, which usually cannot be obtained as microcrystalline preparations.
Acknowledgments
We acknowledge financial support from the NWO (700.26.121, 700.10.443, 722.012.002, and 723.014.003).
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1.Ishii Y, Tycko R. J. Magn. Reson. 2000;142:199–204. doi: 10.1006/jmre.1999.1976. [DOI] [PubMed] [Google Scholar]
- 2.pp. 3878–3881.
- 2a.Chevelkov V, Rehbein K, Diehl A, Reif B. Angew. Chem. Int. Ed. 45 doi: 10.1002/anie.200600328. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2006;118 [Google Scholar]
- 2b.Linser R, Dasari M, Hiller M, Higman V, Fink U, Lopez del Amo JM, Markovic S, Handel L, Kessler B, Schmieder P, Oesterhelt D, Oschkinat H, Reif B. Angew. Chem. Int. Ed. 2006;50 doi: 10.1002/anie.201008244. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2011;123 [Google Scholar]
- 2c.Shi L, Kawamura I, Jung KH, Brown LS, Ladizhansky V. Angew. Chem. Int. Ed. 2011;50 doi: 10.1002/anie.201004422. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2011;123 [Google Scholar]
- 2d.Agarwal V, Penzel S, Szekely K, Cadalbert R, Testori E, Oss A, Past J, Samoson A, Ernst M, Bockmann A, Meier BH. Angew. Chem. Int. Ed. 2011;53 doi: 10.1002/anie.201405730. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2014;126 [Google Scholar]
- 2e.Lamley JM, Iuga D, Oster C, Sass HJ, Rogowski M, Oss A, Past J, Reinhold A, Grzesiek S, Samoson A, Lewandowski JR. J. Am. Chem. Soc. 2014;136 doi: 10.1021/ja5069992. [DOI] [PubMed] [Google Scholar]
- 2f.Schanda P, Triboulet S, Laguri C, Bougault CM, Ayala I, Callon M, Arthur M, Simorre JP. J. Am. Chem. Soc. 2014;136 doi: 10.1021/ja5105987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2g.Chevelkov V, Habenstein B, Loquet A, Giller K, Becker S, Lange A. J. Magn. Reson. 2014;242 doi: 10.1016/j.jmr.2014.02.020. [DOI] [PubMed] [Google Scholar]
- 3.pp. 12489–12497.
- 3a.Barbet-Massin E, Pell AJ, Retel JS, Andreas LB, Jaudzems K, Franks WT, Nieuwkoop AJ, Hiller M, Higman V, Guerry P, Bertarello A, Knight MJ, Felletti M, Le Marchand T, Kotelovica S, Akopjana I, Tars K, Stoppini M, Bellotti V, Bolognesi M, Ricagno S, Chou JJ, Griffin RG, Oschkinat H, Lesage A, Emsley L, Herrmann T, Pintacuda G. J. Am. Chem. Soc. 136 doi: 10.1021/ja507382j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b.Xiang SQ, Chevelkov V, Becker S, Lange A. J. Biomol. NMR. 2014;60 doi: 10.1007/s10858-014-9859-6. [DOI] [PubMed] [Google Scholar]
- 4.pp. 5905–5912.
- 4a.Linser R, Bardiaux B, Higman V, Fink U, Reif B. J. Am. Chem. Soc. 133 doi: 10.1021/ja110222h. [DOI] [PubMed] [Google Scholar]
- 4b.Knight MJ, Webber AL, Pell AJ, Guerry P, Barbet-Massin E, Bertini I, Felli IC, Gonnelli L, Pierattelli R, Emsley L, Lesage A, Herrmann T, Pintacuda G. Angew. Chem. Int. Ed. 2011;50 doi: 10.1002/anie.201106340. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2011;123 [Google Scholar]
- 5.pp. 11791–11801.
- 5a.Zhou DH, Shah G, Cormos M, Mullen C, Sandoz D, Rienstra CM. J. Am. Chem. Soc. 129 doi: 10.1021/ja073462m. [DOI] [PubMed] [Google Scholar]
- 5b.Marchetti A, Jehle S, Felletti M, Knight MJ, Wang Y, Xu ZQ, Park AY, Otting G, Lesage A, Emsley L, Dixon NE, Pintacuda G. Angew. Chem. Int. Ed. 2007;51 doi: 10.1002/anie.201203124. [DOI] [PubMed] [Google Scholar]
- 5c.Angew. Chem. 2012;124 [Google Scholar]
- Weingarth M, van der Cruijsen EA, Ostmeyer J, Lievestro S, Roux B, Baldus M. J. Am. Chem. Soc. 2012;136 doi: 10.1021/ja411450y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5d.Wang S, Parthasarathy S, Xiao Y, Nishiyama Y, Long F, Matsuda I, Endo Y, Nemoto T, Yamauchi K, Asakura T, Takeda M, Terauchi T, Kainosho M, Ishii Y. Chem. Commun. 2014;51 doi: 10.1039/c5cc04618a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asami S, Schmieder P, Reif B. J. Am. Chem. Soc. 2010;132:15133–15135. doi: 10.1021/ja106170h. [DOI] [PubMed] [Google Scholar]
- 7.pp. 915–918.
- 7a.Huber M, Hiller S, Schanda P, Ernst M, Bockmann A, Verel R, Meier BH. ChemPhysChem. 12 doi: 10.1002/cphc.201100062. [DOI] [PubMed] [Google Scholar]
- 7b.Sinnige T, Daniels M, Baldus M, Weingarth M. J. Am. Chem. Soc. 2011;136 doi: 10.1021/ja412870m. [DOI] [PubMed] [Google Scholar]
- 7c.Wang S, Parthasarathy S, Nishiyama Y, Endo Y, Nemoto T, Yamauchi K, Asakura T, Takeda M, Terauchi T, Kainosho M, Ishii Y. PloS one. 2014;10 doi: 10.1371/journal.pone.0122714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.pp. 627–636.
- 8a.Rosen MK, Gardner KH, Willis RC, Parris WE, Pawson T, Kay LE. J. Mol. Biol. 263 doi: 10.1006/jmbi.1996.0603. [DOI] [PubMed] [Google Scholar]
- 8b.Otten R, Chu B, Krewulak KD, Vogel HJ, Mulder FA. J. Am. Chem. Soc. 1996;132 doi: 10.1021/ja907706a. [DOI] [PubMed] [Google Scholar]
- 8c.Nand D, Cukkemane A, Becker S, Baldus M. J. Biomol. NMR. 2010;52 doi: 10.1007/s10858-011-9585-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou DH, Rienstra CM. J. Magn. Reson. 2008;192:167–172. doi: 10.1016/j.jmr.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weingarth M, Bodenhausen G, Tekely P. J. Magn. Reson. 2009;199:238–241. doi: 10.1016/j.jmr.2009.04.015. [DOI] [PubMed] [Google Scholar]
- 11.Bayro MJ, Huber M, Ramachandran R, Davenport TC, Meier BH, Ernst M, Griffin RG. J. Chem. Phys. 2009;130:114506. doi: 10.1063/1.3089370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baldus M, Petkova AT, Herzfeld J, Griffin RG. Mol. Phys. 1998;95:1197–1207. [Google Scholar]
- 13.pp. 167–173.
- 13a.Schubert M, Manolikas T, Rogowski M, Meier BH. J. Biomol. NMR. 35 doi: 10.1007/s10858-006-9025-x. [DOI] [PubMed] [Google Scholar]
- 13b.Schanda P, Meier BH, Ernst M. J. Am. Chem. Soc. 2006;132 doi: 10.1021/ja100726a. [DOI] [PubMed] [Google Scholar]
- 14.Verel R, Baldus M, Ernst M, Meier BH. Chem. Phys. Lett. 1998;287:421–428. [Google Scholar]
- 15.pp. 121–124.
- 15a.Ostmeyer J, Chakrapani S, Pan AC, Perozo E, Roux B. Nature. 501 doi: 10.1038/nature12395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b.Uysal S, Cuello LG, Cortes DM, Koide S, Kossiakoff AA, Perozo E. Proc. Natl. Acad. Sci. USA. 2013;108 doi: 10.1073/pnas.1105112108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15c.van der Cruijsen EA, Nand D, Weingarth M, Prokofyev A, Hornig S, Cukkemane AA, Bonvin AM, Becker S, Hulse RE, Perozo E, Pongs O, Baldus M. Proc. Natl. Acad. Sci. USA. 2011;110 doi: 10.1073/pnas.1305563110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15d.Wylie BJ, Bhate MP, McDermott AE. Proc. Natl. Acad. Sci. USA. 2013;111 doi: 10.1073/pnas.1319577110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information