

Abstract
With the advent of safer and more efficient gene transfer methods, gene therapy has become a viable solution for many inherited and acquired disorders. Hematopoietic stem cells (HSCs) are a prime cell compartment for gene therapy aimed at correcting blood-based disorders, as well as those amenable to metabolic outcomes that can effect cross-correction. While some resounding clinical successes have recently been demonstrated, ample room remains to increase the therapeutic output from HSC-directed gene therapy. In vivo amplification of therapeutic cells is one avenue to achieve enhanced gene product delivery. To date, attempts have been made to provide HSCs with resistance to cytotoxic drugs, to include drug-inducible growth modules specific to HSCs, and to increase the engraftment potential of transduced HSCs. This review aims to summarize amplification strategies that have been developed and tested and to discuss their advantages along with barriers faced towards their clinical adaptation. In addition, next-generation strategies to circumvent current limitations of specific amplification schemas are discussed.
Keywords: Gene therapy, Hematopoietic stem cells, In vivo selection, Chemical Inducer of Dimerization, Chemo-selection, Lentivirus
Core tip: Though hematopoietic stem cell (HSC)-directed gene therapy is becoming a viable therapy for many disorders, optimization of clinical output needs improvement. One approach to circumvent lower efficiencies of gene transfer and/or engraftment is to apply in vivo amplification strategies. Here we review various modules that have been developed and tested to mediate amplification of HSCs after gene transfer.
INTRODUCTION
Hematopoietic stem cells (HSCs) are long-term, multipotent, self-renewing cells that reside in specialized bone marrow (BM) niches and are capable of generating and repopulating the entire spectrum of blood and lymphoid cells[1,2]. Due to these unique properties, HSCs are targets for therapy for a number of hematological malignancies and many inherited blood disorders including β-thalassemia, sickle cell anemia, chronic granulomatous disease, and severe combined immunodeficiencies (SCID-X1 and ADA-SCID) among others[3-8]. Additionally, HSC transplants have been used in attempt to correct other monogenic deficiencies, such as the mucopolysaccharidoses and Gaucher disease[9-11].
There are still numerous drawbacks of allogeneic transplantation despite its clinical utility. Often, HSCs are collected from the patient’s sibling, parents, or a matched donor. HLA-identical donors can be difficult to find and there are risks involved with the use of HLA-haploidentical or non-identical donors including rejection or poor engraftment of HSCs along with the occurrence of graft-versus-host disease (GVHD). Conditioning is also necessary for engraftment of HSCs, which can increase the risk of infections[12-14]. As a consequence, HSC allo-transplantation is still considered a fairly risky intervention and is applied with caution in the clinic.
Gene therapy targeting patient-derived HSCs is a viable solution for some monogenic diseases[15] (Figure 1A). Autologous transplantation has been well studied and detailed clinical protocols are available for this procedure[3]. Additionally, autologous transplantation does not have a risk of GVHD associated with it and immune reconstitution after ablation occurs in a shorter period of time[16,17]. Gene transfer into HSCs has been traditionally achieved by stable transduction of target cells using replication-incompetent retroviruses[15]. There the expression of transgenes can be driven by constitutive or tissue-specific promoters, giving a range of control over the intended therapeutic intervention. Next-generation strategies are also being developed to correct original nucleotide mutations with the use of gene-editing technologies, such as TALENs and CRISPR-Cas9, though these remain to be optimized for clinical application[18-20].
Figure 1.

General outline of ex vivo hematopoietic stem cell gene therapy and pre-selection methods. A: CD34+ cells are enriched by CliniMACS after apheresis of peripheral blood of patients following mobilization. These cells are then briefly activated ex vivo and can be modified, commonly by viral transduction, to express a desired therapeutic protein. Cells are then assessed for quality control metrics and engrafted into patients following ablation; B: Pre-selection of transduced cells. Cells can be engineered to express an inert surface marker that can be used to immuno-enrich for the transduced population prior to engraftment. This strategy can increase the chances of hematopoietic reconstitution from the transduced population. Alternatively, cells can be given resistance to cytotoxic drugs. Pre-treatment of the cells ex vivo with drugs can kill off the non-transduced population. Ex vivo treatment allows the use of drugs that would normally not be efficacious in the bone marrow environment at a tolerable dose.
Over 2000 clinical gene therapy trials have been conducted to date[4,15,21,22]. Most earlier trials employed onco-retroviral vectors, which have shown to be clinically disadvantageous because of their tendency to integrate close to genes that are important for cell growth and proliferation, enhancing their expression and increasing the likelihood of developing leukemias[4,15,23-25]. So far it appears that this genotoxicity and tendency towards insertional mutagenesis has been diminished with the introduction of HIV-1-derived, replication-incompetent, and self-inactivating lentiviral vectors (LVs), which do not show preferential integration near genes involved in cell growth and/or proliferation[4,26-30].
There are other caveats to using HSCs as target cells for gene therapy that are a result of their unique biology. HSCs can be more difficult to transduce than some other cell types, partially owing to the difficulty of culturing them ex vivo. Longer-term culturing ensures that the cells will differentiate. Transduction also requires transient activation of the cell cycle, especially with onco-retroviral-based vectors since their downstream integration requires a breakdown of the nuclear membrane. As a consequence of ex vivo manipulation and cell-cycle activation, transduced HSCs often have lower engraftment potential and reduced longevity once engrafted. These additional limitations have also been partially addressed with the use of LVs, which need shorter transduction times and do not require target cells to be fully cycling[31-35].
In spite of the progress made in HSC gene therapy with the implementation of recombinant LVs, there is ample room for additional improvements to increase therapeutic efficacy. Many active fields of research are geared towards optimizing gene therapy for HSCs. Efforts are under way to hone GMP-grade LV production to subsequently allow modulation of multiplicities of infection at a clinical level, whilst reducing the cost of gene therapy[36,37]. Improvements are also being made to protocols for ex vivo handling and culture of HSCs with studies demonstrating enhanced transduction with shorter culture times and less activation, which have resulted from better understanding of the biology of HSCs and their BM microenvironments[4,38,39]. In addition, in-depth studies of HSC biology have identified molecular targets for drugs that allow more efficient and safer mobilization of patient stem cells[40-42]. The gene therapy field has also sought out methods to provide extrinsic selective pressure for transduced cells, though clear clinical utility of any system has yet to be demonstrated, especially in the context of reconstitutive HSC-directed gene therapy.
Reconstitution of deficient gene products in some inherited blood diseases leads to innate positive selective pressure in vivo for mature cells derived from transduced progenitor cells, especially when the gene product is necessary for the development or function of those cells. For example, reconstitution of the common gamma chain (γc/CD132) in SCID-X1 allows immune cells to develop normally, thus progeny cells are derived from successfully transduced HSCs almost exclusively[17,43-45]. Selectivity for donor-derived late-stage erythrocytes has also been observed in β-thalassemia patients that have received allo-transplantations; and a similar trend has been observed with gene therapy in mouse models of the disorder[7,46,47]. HSC gene therapy may therefore become routine clinical practice some day for patients suffering from such hematological disorders that do not have matched donors, considering the innate advantage of such reconstituted cells.
There is also great potential of HSC-directed gene therapy for the treatment of non-hematological, monogenic enzyme deficiencies, such as those that lead to lysosomal storage disorders (LSDs) and other metabolic indications. In most cases, functional enzymes expressed after gene transfer into LSD patient cells have the potential to be secreted and subsequently taken up by other cells that do not have the transgene, a process termed metabolic cooperativity or “cross-correction”. This occurrence has been demonstrated for a number of LSDs including Gaucher, Farber, and Fabry diseases[48-51]. The current standard of care for many LSDs, enzyme replacement therapy (ERT), is actually a corollary of this phenomenon. HSC-directed gene therapy presents numerous putative advantages over conventional ERT, including sustained and continuous secretion of therapeutic enzyme by ubiquitously circulating cells, improvements in patient lifestyle by reducing the need for biweekly enzyme infusions, and overall cost savings. It is necessary to tailor HSC gene therapy for individual patients, however, which is incongruous with many current industrial business models, highlighting the necessity of shifting industrial focus from general to personalized therapeutics.
Our laboratory is currently pursuing first-in-man HSC-based gene therapy for Fabry disease and is concomitantly demonstrating the utility of gene therapy for amelioration of Farber disease. However, in these cases, as with many such target disorders, expression of the functional gene product imparts no innate growth advantages to transduced cells. Vector-encoded transgenes alone or in tandem that allow extrinsic selective pressure to be applied in vivo, leading to an increased percentage of vector-transduced cells over background could therefore be highly beneficial in the context of HSC gene therapy for LSDs and many other monogenic deficiencies. Additionally, application of positive selective pressure could result in cell populations that have higher transgene expression, resulting in an increased therapeutic benefit.
In this review, we aim to summarize the various strategies that have been employed to date in attempts to increase vector-transduced HSC numbers, thereby increasing the efficacy of HSC-targeted gene therapy. In addition, we will discuss putative next-generation strategies aimed at addressing current shortcomings of applying selective positive pressure on transduced HSCs.
EX VIVO PRE-SELECTION STRATEGIES
Resistance to cytotoxic drugs
Selection of genetically modified cells is a compilation of laboratory techniques commonly applied to acquire polyclonal cell lines after gene transfer. To achieve this, target cells are engineered to express proteins that confer resistance to drugs or proteins that allow selection by immune-affinity methods such as fluorescence- and magnetic-activated cell sorting (FACS and MACS, respectively)[52,53] (Figure 1B). Ideally, proteins expressed for enrichment should have low or no endogenous expression in target cells, and should have no effect on the biology of the transduced cells or their progeny. Traditionally, xenogenic enzymes have been used to confer cells with resistance to pan-toxic drugs in this context. For example, neomycin and hygromycin phosphotransferases (NeoR and HygR) derived from bacteria are commonly used to provide protection against neomycin and hygromycin B, respectively[54,55]. As such, first attempts at conferring resistance to cells for engraftment were made with these enzymes. However, the use of xenogenic enzymes in clinical protocols has been limited by their tendency to be highly immunogenic once such modified cells are engrafted[56-58]. To address this, mutants of various endogenous enzymes have been used to confer resistance to other cytotoxic drugs. These enzymes are discussed in the section below in the context of in vivo selection. However, most drugs require prolonged ex vivo culture to effectively enrich for the gene-modified population. Prolonged ex vivo handling of HSCs reduces their usefulness post-selection due to a loss of “stemness” and engraftment potential[31]. Thus, drug-mediated ex vivo pre-selection may not be ideal in current iterations for clinical purposes.
Cell-surface marking for immuno-enrichment
Transduced HSCs can also be enriched ex vivo with the use of cell-surface markers. Selectable cell-surface markers that have been studied for HSC marking and pre-selection include truncated forms of the human low-affinity nerve growth factor receptor (∆LNGFR)[59-62], the heat stable antigen (HSA/CD24)[48,50,63,64], the human lymphocyte antigen T1 (CD5)[65,66], and the human interleukin-2 receptor alpha chain (IL-2Rα/huCD25)[67]. In mouse allograft experiments, long-term engraftment of transduced and FACS-enriched BM cells along with hematopoietic cell marking has been demonstrated using CD24[64] and CD5[66] as selectable markers. However, it must be noted that those experiments did not include a control in which no pre-selection was applied prior to engraftment. This makes it difficult to unequivocally assess the contribution of pre-selection to the engraftment and repopulating ability of transduced cells.
Despite positive results in pre-clinical settings with the use of FACS for enrichment, it has more detrimental effects on cell survival, viability, and function than MACS, even though FACS can lead to higher purity[68]. It is also difficult to physically and/or temporally achieve enrichment of large numbers of clinically-applicable cells by FACS. As such, MACS and analogous schemas are preferred for enrichment prior to engraftment in patients. Over 90% purity has been achieved with MACS enrichment of ∆LNGFR-marked HSCs in vitro[60] and similarly marked lymphocytes in clinical trials[69-71]. In an allograft experiment with a mouse model of Fabry disease, BM mononuclear cells were transduced with a therapeutic vector capable of co-expressing α-galactosidase A and huCD25[67]. Pre-selection of transduced cells by MACS led to a long-term increase of huCD25-marked peripheral blood mononuclear cells when compared to controls. Therapeutic benefit of pre-selection was demonstrated by a higher α-galactosidase A activity in plasma and most organs. Additionally, the utility of pre-selection in long-term HSC marking has been demonstrated in some cases by secondary transplant experiments[64,67].
Since pre-selection strategies reduce the size of the transduced cell population[60], however, their application to HSC gene therapy can be critically limited if there are difficulties in collecting large numbers of patient HSCs. Ex vivo expansion of HSCs is currently not a viable solution in order to compensate for reduced cell numbers since over-activation can have detrimental effects on their “stemness” and engraftment potential[31]. In addition, pre-selection increases time of ex vivo manipulation, which increases costs and risks of contamination. It is therefore difficult to obtain a post-selection yield high enough to exert a therapeutic effect. Nevertheless, these studies demonstrate the benefit of enriching transduced cells ex vivo and that clinical translation may be augmented by higher yields of HSCs during their acquisition.
IN VIVO CHEMO-SELECTION STRATEGIES
Various proteins have been shown to grant variable degrees of chemoprotection in the context of cancer therapy, such as ATP-binding cassette, sub-family B, member 1 (ABCB1), dihydrofolate reductase (DHFR), and O6-alkylguanine DNA alkyltransferase (MGMT). Overexpression of these proteins in HSCs has been pursued with the aim of protecting the hematopoietic compartment from the severe toxicity of many cytotoxic drugs used in cancer chemotherapy[72].
Pan-resistance to chemotherapeutic agents using ABCB1
ABCB1 [also known as multidrug resistance protein 1 (MDR1); or P-glycoprotein 1 (P-gp1)] is a cell membrane transporter with broad specificity that pumps foreign compounds out of the cell and is also involved in lipid translocation[73,74]. ABCB1 mediates chemoresistance in cancer cells in which its expression is upregulated[75]. Overexpression of ABCB1 in murine BM was shown to confer protection to many chemotherapeutic agents such as vinblastin, doxorubicin, daunomycin, taxol, vincristine, etoposide, actinomycin D, colchicine, and paclitaxel[76,77]. Early studies with mouse allografts showed in vivo selection of hematopoietic cells derived from ABCB1-overexpressing HSCs, but it is unclear whether selection occurred at the stem cell level[78,79]. Later studies demonstrated successful selection of human HSC-derived cells in the BM of murine xenograft models[80,81]. In contrast to these outcomes, early autograft experiments in large animals and clinical trials demonstrated rather disappointing results. In a canine model, high toxicity was documented despite long-term ABCB1+-peripheral blood cell enrichment in the only surviving animal[82]. In a study involving non-human primates, there was low initial ABCB1 cell marking, drug-induced neutropenia, and no significant increase of neutrophil counts after drug treatment[83]. In clinical trials, selection after drug treatment has been low and predominantly transient, albeit with little or no toxicity[84-87]. The inefficacy of ABCB1-mediated selection may have been due to insufficient expression of the transgene in hematopoietic cells[88]. Onco-retrovirally-mediated expression of ABCB1 was found to be unstable due to cryptic splice sites within the cDNA[89]. This issue was resolved by introducing a silent mutation that inactivates that splice site, which subsequently increased expression of onco-retrovirally-delivered ABCB1[90,91]. Nevertheless, the robustness of this system must be reliably demonstrated in large animal models before it can be considered a feasible strategy to enrich HSCs after transplant for clinical gene therapy in patients.
Antifolate resistance using mutant DHFR
DHFR catalyzes the reduction of dihydrofolate to tetrahydrofolate, a precursor required for the de novo synthesis of purines and some amino acids. Antifolate drugs such as methotrexate (MTX) and trimetrexate (TMTX) inhibit DHFR activity, thus blocking cell proliferation and promoting apoptosis in dividing cells. HSCs and myeloid progenitor cells, however, can employ nucleotide salvage mechanisms to escape antifolate toxicity[92]. In order to overcome this, the nucleoside transport inhibitor nitrobenzylthioinosine 5’-monophosphate (NBTI/NBMPR-P) has been used in combination with MTX or TMTX[92]. Transplanted HSCs have been engineered to overexpress mutant forms of DHFR, such as DHFRL22Y, that are resistant to antifolate agents[93]. In vivo enrichment of transduced HSCs in murine allogeneic transplants has been demonstrated[94,95]. However, translation of this method into large animal models has been rather discouraging. In a study in rhesus macaques that used a recombinant onco-retrovirus to deliver DHFRL22Y, enrichment of cells derived from the transduced graft was only transient, indicating poor selection at the HSC level[96]. To address this problem, enrichment of CD34+ progenitor cells in a xenograft transplant of human embryonic stem cells (hESCs) into mice has been demonstrated, though this enrichment was only modest and no clinically established methods to transplant hESCs exist[97]. High toxicity and lethality has been documented in antifolate-mediated selection studies in dogs and rhesus macaques[96,98]. Additionally, antifolate toxicities are well documented when such compounds are indicated for treatment of cancer patients[99-102]. These toxicities and the lack of positive evidence suggest that DHFR-mediated in vivo selection may not be useful for HSC gene therapy targeting monogenic diseases. Instead, it may be better suited to prevent graft rejection after HSC transplants, because antifolates would spare highly-proliferating T lymphocytes arising from transduced donor HSCs while eliminating alloreactive recipient T cells as shown recently in vitro[103] and in a canine model[104].
Selectivity using O6BG-resistant MGMT
MGMT repairs DNA damage by removing adducts from the O6 position of guanine, and thus confers resistance to the cytotoxic effects of alkylating agents such as dacarbazine, temozolomide (TMZ), procarbazine, and nitrosoureas such as 1,3-bis-(2-chloroethyl)-1-nitrosourea (BCNU)[105,106]. MGMT is expressed at very low levels in the BM[107,108]. MGMT overexpression was attempted in murine[105,109,110] and human[105] HSCs to confer BCNU resistance. A modest increase in resistance to BCNU was achieved in murine progenitors, both in vitro and in vivo[105,109,110]. Human HSCs, however, had poor resistance in vitro[111]. In order to sensitize the HSC compartment to alkylating drugs to achieve better in vivo selection, BCNU or TMZ have been co-administered with O6-benzylguanine (O6BG), a pseudosubstrate that irreversibly inactivates endogenous MGMT[112]. O6BG-resistant mutants MGMTP140K and MGMTG156A have also been studied[113]. The former is more commonly used in selection strategies, despite having a modest reduction in its DNA-repair activity compared to the wild-type enzyme[113]. Indeed, MGMTP140K has been shown to mediate selection of transduced HSCs in murine and canine allograft and autograft models[114-117], and in murine xenograft models with human HSCs[30,118,119]. However, high dose administration of BCNU and/or TMZ has been shown to cause toxicity[120-122]. For example, selection experiments have shown up to 75% mortality in mice treated with TMZ[120] or BCNU[121], and 88% mortality in rhesus macaques treated with TMZ[122].
Optimization of drug dosing by co-administering high doses of O6BG with low doses of BCNU or TMZ has partially ameliorated the cytotoxic effects of the alkylating drugs and allowed better engraftment of HSCs transduced at low MOIs[121,123,124]. The improvements in survival are thought to be a result of lowering the threshold of MGMT expression required for resistance, which allows partial fulfillment of conditions expected in clinical trials. This dose-adjusted protocol has conferred successful chemoprotection of the hematopoietic compartment in a canine[125] and a nonhuman primate model[126], with no significant toxicity reported in the former. Despite survival of the macaques in the latter study, administration of chemoselective agents led to substantial peripheral blood cell depletion and enrichment of different blood lineages was highly variable[126]. In the same study, use of a multi-cistronic vector to co-express C46, which is an anti-HIV transgene, MGMTP140K, and enhanced green fluorescent protein (eGFP) resulted in lower selective potential[126]. From a translational point of view, the risk-to-benefit ratio is currently not in favor of implementation of such chemoselective strategies though further adjustments to drug regimens can be done.
In the context of gene therapy for murine models of β-thalassemia[123], hemophilia A[124], and hemophilia B[127], amelioration of the disease phenotype has been enhanced with the use of bicistronic LVs encoding the therapeutic gene and an MGMTP140K-based selection module. Additionally, increased expression of the therapeutic gene after drug selection in secondary[123,124] and tertiary[127] recipients of serial BMT demonstrated enrichment at the HSC level. MGMT-mediated enrichment of eGFP+ BM and peripheral blood cells has also been demonstrated in a murine model[128]. Despite these promising results, there are still no reports of successful MGMT-mediated selection following gene therapy in large animals that we are aware of, wherein HSC selection may be less efficient because of their lower replication rates[129]. However, autologous MGMTP140K-transduced HSC transplants have been attempted in MGMThi, TMZ-resistant glioblastoma patients[130]. Drug selection resulted in no significant extra-medullary toxicity and all three participants surpassed the median survival (12 mo) for glioblastoma patients with unmethylated MGMT-promoter status. Despite these promising results in the context of chemoprotection of the hematopoietic compartment during glioblastoma treatment[130], it must be noted that gene-modified circulating blood cells were depleted from the patients with termination of treatment. As such, repeat administration of chemotherapy may be required for the use of this system for amplification of transduced HSCs for gene therapy.
Hypoxanthine-guanine phosphoribosyltransferase inactivation for 6-thioguanine resistance
While the strategies described above rely on the overexpression of a protein that confers chemoprotection to transduced cells, down-regulating endogenous enzymes necessary to activate cytotoxic drugs can achieve analogous outcomes. Hypoxanthine-guanine phosphoribosyltransferase (HPRT) is an enzyme involved in the purine nucleotide salvage pathway. HPRT can catalyze the addition of ribose 5-phosphate to the purine analog 6-thioguanine (6TG) to generate thioguanosine monophosphate (thio-GMP)[131]. Thio-GMP is then converted into thiodeoxyguanosine triphosphate (thio-dGTP), which can be incorporated into DNA inducing futile mismatch repair and consequent apoptosis. It has been shown that BM cells of HPRT-deficient mice are resistant to 6TG treatment[132]. Transplantation of HPRT-deficient BM into wild-type HPRT mice under 6TG selection resulted in good engraftment and long-term hematopoietic reconstitution in primary and secondary recipients[133]. Furthermore, transduction of murine hematopoietic progenitor cells with a LV encoding a short-hairpin RNA (shRNA) that targets HPRT can confer resistance to 6TG in vitro[134]. The same knockdown strategy has shown effective enrichment in murine allograft models[135] and human HSC xenograft models[136]. This approach has some advantages over other drug selection methods described above. 6TG can be used for both pre-conditioning and chemoselection, and the shRNA sequence is very short, which makes it easier to include in a dual-gene vector. Also, ample information about 6TG dosage and toxicity is available because it has been used in the clinic for decades[137]. A recent study suggests, however, that this method may be limited to enrichment of committed progenitor cells, which would decrease long-term efficacy with single-dosing regimens[136]. Also, hereditary HPRT deficiency is the cause of Lesch-Nyhan syndrome, which has been associated with megaloblastic anemia[138]. Therefore, it is necessary to carefully assess long-term consequences of HPRT deficiency in the hematopoietic lineage, especially in large animal models. Nevertheless, selective induction of an enzyme deficiency, as demonstrated by virally-induced HPRT knockdowns, may be a powerful method of introducing selective pressure following gene therapy.
ENGINEERED INDUCIBLE GROWTH AND SELECTION MODULES
Cytotoxic chemical inducers of dimerization
Discoveries relating to functional consequences of forcing proteins such as receptor tyrosine kinases into proximity with each other have allowed the use of protein engineering to confer specific biological characteristics to a subset of modified cells with exposure to various stimuli. For example, chemical inducers of dimerization (CIDs) are synthetic compounds that can be used to induce dimerization of proteins that are expressed as fusions to CID-binding domains (CBDs). With the use of CBDs, cell-fates can be made dependent upon the addition of CIDs (Figure 2A). One of the first examples of such a system utilized FK1012, a synthetic dimer of the immunosuppressant FK506 (Tacrolimus)[139]. Proteins that can modulate cell biology, including proliferation[140] and apoptosis[141], have been engineered from growth factor receptors and the FK506 binding domain from FK506-binding protein (FKBP12). However, FK1012 retains the ability to bind endogenous FKBP12[139]. This is undesirable from a clinical point of view because endogenous FKBP12 could sequester the drug, preventing its intended effect. In addition, FK1012 administration could affect the normal physiological role of FKBP12[142-145]. As a result, the use of these systems has been limited, though thorough clinical evaluation of the drug has yet to be completed. However, amplification protocols can be envisioned wherein the underlying toxicities of FK1012, which are expected to be similar to FK506, are exploited. Future systems can be developed using other cytotoxic agents with their respective binding targets as CBDs fused to survival or growth signaling factors[146]. Careful dosing can allow for simultaneous depletion of non-transduced cells and expansion of the transduced population (Figure 2A). Such studies have yet to be performed in clinically-relevant settings. Conversely, the application of such current systems remains risky as the continuous use of cytotoxic agents in general can have detrimental effects on patient quality of life. A potential solution is to aim for selective pressure to be applied to more mature cells, where dose reduction can be envisioned whilst clinical benefit is still achieved.
Figure 2.
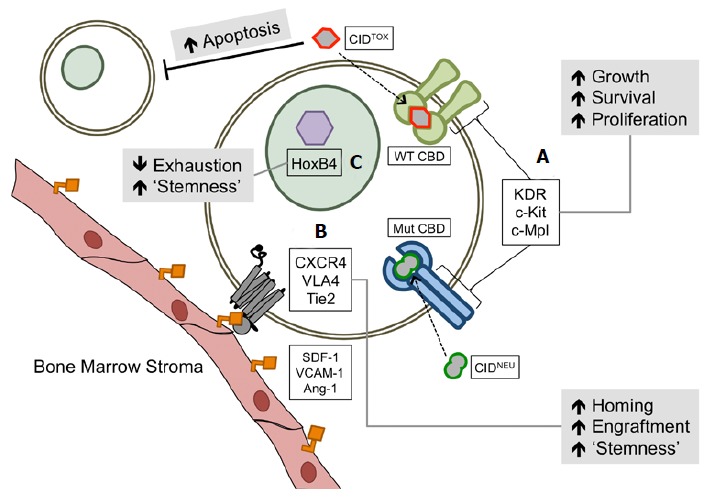
Summary of next-generation amplification modules. A: Fusion proteins comprised of chemical inducer of dimerizations (CBDs) such as FKBP12 (WT CBD) or F36V (Mut CBD) and receptors involved in hematopoietic stem cells (HSC) growth, proliferation, and survival. Activation of signaling by CIDs allows expansion of the transduced population. The use of cytotoxic CIDs (CIDTOX) can allow simultaneous depletion of the non-transduced population. FK1012 is a putative cytotoxic CID-binding domain. Examples of inert or neutral CIDs (CIDNEU) include AP20187 and AP1903; B: Controlled overexpression of HSC homing and adhesion molecules can increase the potential for therapeutic cells to survive and can promote long-term engraftment. Examples of such molecules include but are not limited to CXCR4, VLA4, and Tie2. Their corresponding ligands (SDF-1, VCAM-1, and Ang-1, respectively) are usually expressed on osteoblasts, osteoclasts, MSCs, and other cells that make up the bone marrow stroma; C: Downstream effectors of key signaling pathways involved in maintaining HSC phenotypes that are down-regulated during ex vivo handling of CD34+ cells can be reconstituted to prevent stem cell exhaustion and to increase long-term engraftment of transduced cells. HoxB4 is an example of a transcription factor that is activated in response to Wnt signaling and is key to maintenance of the stem phenotype of HSCs.
Neutral CIDs
Reverse-engineering of proteins and drugs that bind to them have led to numerous other CIDs and their respective CBDs. Progress in understanding the modularity of protein signaling has yielded numerous opportunities to generate CBD-signaling domain fusions. From a gene therapy perspective, ideal CIDs are those that would have little to no effect on any cells other than the transduced population. Ideal CBDs should also have no effect on the biology of transduced cells without the presence of a CID. As well, engineered polypeptides with CBDs should be derived from endogenous proteins where possible to minimize the potential immunogenicity of the fusions. In addition, unexpected effects of CID-induced dimerization of CBD fusion proteins should not occur in cells derived from transduced HSCs. “Bump and hole” engineering of FK1012 and FKBP12 has yielded derivative CIDs such as AP20187 (B/B homodimerizer; Takara) and AP1903 (Rimiducid; Bellicum)[147]. Such CIDs are chemically modified to prevent them from binding the original CBD. The derivative CBDs, such as FKBP12F36V (F36V) for AP20187 and AP1903, have mutations that confer ability to bind the engineered CIDs[147] (Figure 2A). These modifications allow such CID-CBD systems to be acceptable for use in gene therapy, as outlined above.
Amplification of HSCs using neutral CIDs
Multiple studies have been performed in cell, mouse, and canine models that utilize AP20187 and protein receptors fused to F36V[148-154]. Our lab has previously shown the utility of a kinase insert domain receptor (KDR/CD309)-F36V fusion to control cell-fate in an AP20187 dependent manner, and we characterized the molecular mechanisms that are induced as a consequence of KDR dimerization in TF1 cells[150]. The utility of this system has yet to be demonstrated in an animal model of HSC engraftment. The characterization of HSC cells and their signaling components have yielded other targets for F36V fusion and CID-mediated control of cell fate, such as c-Kit (SCF receptor)[155], and c-Mpl (thrombopoietin receptor)[149,152,156] (Figure 2A). It must be noted that these signaling components are not fully unique to the HSC compartment and subsequent risk of unwanted proliferation can exist. That said, restricted expression of CBD-receptor fusions using HSC-specific promoters in gene transfer vehicles could reduce effects on non-target cells.
A long-term study in canines using an engineered thrombopoietin receptor, Mpl-F36V, has demonstrated the utility of intermittent use of AP20187 and AP1903 administration over the course of a number of years with no effect on the normal physiology of the dogs and no effect on the HSC compartment without drug administration[152]. However, this study only involved long-term monitoring of two dogs and did not test the utility of such a system in a reconstitutive gene therapy context. Studies in healthy human volunteers (see below) may need to be conducted to unravel the effects of long-term administration of AP20187 in order to inform the FDA prior to administration in patients.
Unlike AP20187, AP1903 has gained more momentum with respect to its translation into the clinic. It induces dimerization through the same CBD as AP20187, extending the utility of studies that use F36V fusions[150-156]. Most importantly, a phase I clinical trial in which a single infusion of AP1903 was administered to 28 healthy volunteers at a range of doses has already been conducted[157]. No relevant adverse effects were observed at any of the doses. Furthermore, donor T cells that are genetically modified with a CID-inducible caspase-9 (iCasp9) suicide system have been administered to leukemia patients to enhance immune reconstitution in recipients of allogeneic HSC transplants[158,159]. A single dose of AP1903 was sufficient to ameliorate the GVHD. T cell counts were reduced in as little as 30 min after drug administration followed by resolution of GVHD symptoms in 24 h. Further administration of AP1903 is being tested in patients receiving cell products that have been gene modified with iCasp9 (www.bellicum.com). The dosing for these patients has been informed by the pharmacokinetics observed in healthy volunteers[157]. Long-term follow-up of patients from one study has been published with up to 4-year occurrence-free survival of patients receiving iCasp9-modified T cells and AP1903[160]. However, it must be noted that repeated administration of the drug and associated safety profiles have not been published to the best of our knowledge. This is mainly because single-administration of the drug resolved symptoms in the studies conducted thus far and eliminated the need for further intervention[158,159]. Additionally, no information about biodistribution and pharmacogenomics of AP1903 in humans is available - that we are aware of. As such, the drug is still considered experimental and there is no prior knowledge about its efficacy in the bone marrow niche other than that which has been observed in animal models[152,161]. Further studies conducted in healthy volunteers to assess a full set of biological parameters can boost the clinical development of enrichment modules relevant to HSC-directed gene therapy.
Engineering alternative amplification modules
While FDA approval and consequential routine administration of existing CIDs in patients may be achievable in the future, there is value in stepping away from sole confinement to FKBP12-derived systems. Multiple other technologies exist that utilize either compounds known to be benign along with reverse engineered signaling domains or compounds and their respective binding domains derived from other species. For example, a recently demonstrated degron-system that uses an auxin-inducible domain derived from plants[162] could be engineered to enhance Wnt signaling in HSCs by targeting axin degradation in an auxin analog-dependent manner[163]. Mouse studies have shown no effect of this analog on normal physiology[164]. In addition, reverse engineering of G-protein-coupled receptors (GPCRs) have led to the development of a variety of Designer Receptors Activated by Designer Drugs (DREADDs)[165]. One DREADD has been designed to mediate chemotaxis of monocytes and neutrophils in a drug-dependent manner and highly promising results have been demonstrated in vivo[166]. This DREADD is activated in response to Clozapine-N-oxide (CNO), a benign metabolite of the clinically-approved anti-psychotic agent, Clozapine[167]. With our growing understanding of protein modularity, it is not difficult to envision the development of a similar DREADD that potently activates HSC-specific survival and proliferative signals. Development of systems that utilize compounds with known pharmacokinetics and biodistribution that can be fast-tracked for FDA approval can provide a significant boost to in vivo enrichment strategies for HSC-directed gene therapy.
MODULES TO ENHANCE ENGRAFTMENT OF THE TRANSDUCED HSC SUB-POPULATION
HSCs are housed in specialized compartments in the BM. They use cell surface receptors and ligands to anchor themselves in their niche and to modulate signals for long-term survival and self-renewal[4,168]. Roles of multiple signaling pathways have been elucidated; as well, cell surface proteins that can demarcate HSCs as single cells that can stably engraft and that are capable of reconstituting the entire hematopoietic system have been uncovered[169]. It is therefore not difficult to envision novel clinical roles for these cell surface proteins. For example, plerixafor (Mozobil, Sanofi) is a drug that was developed to disrupt the interaction of C-X-C chemokine receptor type 4 (CXCR4, fusin) and C-X-C motif chemokine 12 (CXCL12, or SDF-1)[170]. Greater numbers of CD34+ cells can be acquired with administration of plerixafor over the traditional mobilization strategy that uses granulocyte-colony stimulating factor[171,172]. Recently, experiments have shown the potential of another drug, Bortezomib (Velcade, PS-341), in HSC mobilization via disruption of very late antigen 4 and vascular cell adhesion molecules (VCAM-1) in mice[173].
Enhancement of engraftment can also be envisioned using information resulting from such aforementioned studies. Specifically, interactions that can be disrupted for HSC mobilization can also be used to increase engraftment potential. To this effect, studies have shown stage-specific roles for CXCR4 in BM homing and survival of engrafted HSCs[174,175] (Figure 2B). In addition, the benefit of virally-mediated CXCR4 transgene expression for HSC engraftment into a humanized mouse model has been shown with ex vivo-cultured human CD34+ cells[176,177]. Concerns of long-term side effects of CXCR4 expression, such as diminished repopulating potential of the transduced cells and off-target expression in non-HSCs, still remain. For example, high CXCR4 expression is associated with worse prognosis in acute myeloid leukemia, amongst other cancers[178,179]. Developments in transient cDNA introduction, conditional control of transcription, and/or robust, tissue-specific control of expression may minimize these concerns, though they remain to be tested. Should such technologies be developed in a clinically-relevant manner, multiple other cell surface targets for enhancing HSC homing, survival, and “stemness” could be utilized, such as Tie2, VLA4, and c-Kit[168,180-182] (Figure 2B). Other methods relying on expression of downstream components of survival and self-renewal signaling such as homeobox protein Hox-B4 (HoxB4) can also be envisioned to be employed in a controlled manner[183] (Figure 2C). Though introduction of transgenes may provide the ultimate solution to these challenges, other groups are focusing on development of small molecules and co-injection protocols as alternative methods; such as co-injection of BM-derived mesenchymal stem cells[184-186].
LINEAGE-SPECIFIC ENRICHMENT
As discussed previously, it can be conceptually difficult to target therapeutic HSCs for in vivo amplification due to the unique microenvironment in the BM niche in which they reside and because of their inherently quiescent nature. Whilst it is generally agreed that the most clinical benefit can be obtained by targeting long-term precursor cells, it is not conclusively proven to be so. Examples of gene therapy that result in enrichment of cells derived from transduced HSCs, such as in SCID-X1 and β-thalassemia, provide proof-of-concept for the clinical utility of targeting a subset of the mature hematopoietic compartment for selection[7,17,43-47]. Application of enrichment strategies in such a manner can overcome many of the hurdles of attempting to enrich the original HSC engraftment, such as bioavailability of the compounds used for enrichment and toxicities towards the core center of hematopoiesis. In addition, targeting mature cells, and more importantly, excluding the HSC compartment, reduces the chances of long-term complications in patients that have already gone through a risky intervention and reduces the likelihood of sporadic malignant disease arising from alterations in “stem-like” progenitor cells. One conceptual disadvantage of such a targeted system is the lack of persistence of selection, since mature hematopoietic cells will be replaced over time. Enrichment protocols can be developed, however, that utilize either drug dosing that has little or no toxicity in normal cells, or compounds that specifically act on cells that arise from transduced HSCs. As such, continuous administration of the respective agent can provide for prolonged enrichment with minimal or no side effects.
IMPDH2 mutants for MMF resistance in T/B cell progeny
Inosine monophosphate dehydrogenase 2 (IMPDH2) is the rate-limiting enzyme involved in the de novo biosynthesis of guanosine monophosphate (GMP)[187]. While most cells in the body have a salvage pathway, T cell activation and proliferation as well as B cell maturation are highly dependent upon this biosynthetic pathway[188,189]. Mycophenolic acid (MPA) is a potent, non-competitive, reversible inhibitor of IMPDH2[190-192]. Its prodrug, Mycophenolate Mofetil (MMF, Roche), is routinely used in the clinic as an immunosuppressant to control GVHD amongst other indications[193]. Mutants of IMPDH2 that have diminished binding affinity for MPA have been described[194,195]. The most potent amongst these is the combination of T333I and S351Y (IMPDH2IY). The utility of this double mutant has been demonstrated in the context of donor T cell selection, both in vitro and in mouse models using primary human T cells[196,197]. It should be noted that the total lymphocyte count in these experiments was dramatically lowered with MMF treatment, reducing the benefit of such a system with respect to T cell gene therapy[197]. The dosage of drug used in that study, however, was considerably higher than that used to treat patients for GVHD. The effect of low-dose MMF treatment on engrafted cells expressing IMPDH2IY has yet to be shown. In addition, it has been demonstrated that there is no biological effect of constitutive expression of this mutant enzyme on HSC differentiation[194]. Therefore, use of such an enrichment strategy could exclude HSCs and all hematopoietic progeny other than T and B cells from being affected. That said, previous work has only described the use of IMPDH2IY for application in T cell-related disorders, such as HIV treatment and prevention of GVHD[194,197]. To our knowledge, in vivo use of this enrichment module in HSC gene therapy has not yet been demonstrated.
CID-dependent enrichment of gene modified progeny
Numerous examples exist of receptor-CBD fusions that can provide a proliferative advantage to subsets of mature cells. Most recently, an erythropoietin receptor (EpoR)-F36V fusion has been developed for use in facilitating AP20187-dependent erythropoiesis[198]. The fusion is engineered with the minimal components of EpoR required for dimerization-induced signaling along with a myristoylation signal. The fusion is expressed under the control of an erythrocyte-specific promoter. The goal of that study was to design a system to replace the necessity of recombinant Epo administration in anemic patients. The authors have successfully demonstrated CID-dependent erythropoiesis in vivo in a mouse model[198]. Though this group has not shown the utility of their system in the context of HSC gene therapy, it can be postulated that it would be applicable for enhancement of treatment for disorders that affect erythropoiesis. It must be noted that such systems are hindered to date in their clinical translation due to their use of clinically-unavailable CIDs. Future studies utilizing clinically-available compounds and a variety of lineage specific growth signals are anticipated.
DISCUSSION
Safety of LV-mediated gene therapy
With the initial implementation of recombinant onco-retroviruses in gene therapy strategies, an emergent obstacle to be considered when genetically modifying long-term, stem-like cells is the potential for the development of malignancy[4,15,23-25]. As discussed above, LVs greatly diminish the likelihood of integration near known oncogenes or tumor suppressors[4,26-30]. However, multiple other mechanisms that can lead to gross cellular aberrations or changes in function of cells derived from transduced HSCs are still of concern. Though improbable, integrative modification of gene loci can lead to alternative splicing of putative oncogenes, or to the insertional inactivation of tumor suppressors[199,200]. Additionally, a comprehensive understanding of oncogenesis has yet to be achieved in all its different forms, especially within the complex network of cells in the hematopoietic system. As such, continuation of long-term studies investigating the effects of transplanting patients with transduced HSCs is a necessity, especially with new knowledge being acquired regarding the functional importance of intergenic “junk” DNA.
One strategy to circumvent putative side effects of HSC gene therapy is to include suicide modules (or “cell-fate control” systems) in the transfer vector. Suicide modules refer to elements of therapeutic vectors that are capable of inducing specific cell-death of transgenically modified cells. This is especially important when considering the inclusion of amplification modules, which have not been thoroughly tested in patients. These systems can be designed to induce cell death by providing a surface target for antibodies, by inclusion of an inducible component that activates the apoptotic pathway, or by being able to activate a normally non-toxic prodrug. For example, there are multiple CID-based systems that bring together components of the apoptotic-signaling pathway[158,159,201-203]. One of these, iCasp9, is currently being tested in patients receiving haploidentical donor T cell infusions amongst other indications, as discussed previously, though the CIDs being used have yet to acquire FDA approval[158,159]. Additionally, such systems have been tested for their ability to eliminate autologous HSC engraftments in rhesus macaques[204].
The ability to conditionally activate prodrugs has been a useful tool in molecular biology to induce killing of subsets of cells, though many of the enzymes used are derived from other species. For example, thymidine kinase (tk), derived from the herpes simplex virus (HSV-tk), can be used to render cells sensitive to the drug ganciclovir, a commonly employed laboratory technique[205]. HSV-tk has been used to ameliorate GVHD in patients receiving allogeneic transplants and in anti-tumor suicide gene therapy[71,206-209]. However, use of this system is limited by concerns of immunogenicity of non-human proteins that can cause elimination of otherwise useful cells[56,57]. Additionally, ganciclovir and acyclovir are commonly prescribed for viral infections following engraftment, and use of HSV-tk can lead to unintended elimination of transduced cells[210]. Our lab has developed a fusion protein comprised of the extracellular and transmembrane components of LNGFR (CD271) along with an engineered variant of human thymidylate kinase[211]. This module combines the advantages of being able to overexpress a cell surface marker for tracking transduced HSCs and their progeny, since CD271 expression is absent in circulating blood cells, and the ability to activate azidothymidine to a toxic form.
Stem cell exhaustion and clonal selection
One of the principal advantages of in vivo selection is the potential for an increase in therapeutic benefit from an initially lower number of transduced repopulating cells. Yet, proliferative stress on few selected HSCs can occur, resulting in a gross negative, long-term impact on HSC proliferation and lineage differentiation. This is termed “stem cell exhaustion” and can eventually lead to BM failure in recipients. Such concerns have been studied in mice[212] and dogs[213] that underwent serial MGMTP140K-expressing HSC transplantation under prolonged O6BG/BCNU treatment. Importantly, these studies revealed no apparent impairment in HSC repopulation, proliferation, or differentiation, suggesting that stem cell exhaustion may not be an issue, at least in the context of that mode of chemoselection. Further studies in long-lived, clinically-relevant models need to be conducted, however, especially in the context of HSC gene therapy, to demonstrate lack of long-term exhaustion within primary autologous recipients. Ex vivo selection may also be achievable without stem cell exhaustion with the co-expression of factors that maintain HSC “stemness”, such as HoxB4[183].
In vivo drug selection can also exacerbate clonal dominance, a phenomenon readily observed with the use of recombinant onco-retroviruses. Amplification strategies could augment the proliferative advantage of cells with proviral integration sites in or near proto-oncogenes, risking the development of hematopoietic malignancies. For example, analysis of tertiary MGMT-transduced BM recipients showed only 17 unique retroviral integration sites (RIS) following chemoselection[212]. Most RIS in that study were in or near genes involved in important cell regulatory processes, such as cell growth, cell development, and/or cell differentiation. However, this observation may be an artifact of the use of derivatives of murine stem cell retrovirus. In contrast, studies in canines and humanized mice that utilized recombinant LVs, and a mouse allograft study utilizing a foamy viral vector, showed no observable evidence of clonal dominance following chemoselection[30,213,214]. Taken together, current data suggests that stem cell exhaustion and clonal dominance are unlikely to occur with amplification strategies, especially with the use of recombinant LVs. Nevertheless, a persistent concern remains when considering HSC gene therapy in patients. As such, an excellent strategy to address issues surrounding in vivo amplification of transduced HSCs is to apply enrichment to mature hematopoietic cells, as discussed previously. However, more studies need to be conducted to demonstrate clinical efficacy and safety of amplification of mature cells subsequent to HSC gene therapy.
Future considerations
Many of the enrichment strategies that have been suggested or tested for use in gene therapy applications are designed to be expressed in a constitutively “on” manner. While this may increase the potency of the given strategy, unexpected secondary effects on normal physiology may occur. Therefore, conditional expression cassettes for clinical use should be developed in order to minimize unwanted expression of amplification modules. This is paramount when considering the use of highly engineered or trans-species proteins, which can, over time, elicit immune responses against target cells. In addition, to avoid similar issues at a genomic level, use of innate promoters, such as that derived from the elongation factor 1-alpha locus (EF1α), in contrast to virally-derived promoters, such as that derived from cytomegalovirus (CMV), should be considered for clinical application[215].
Greater focus in the gene therapy field is placed upon the improvement of gene delivery methods and, in the context of HSC gene therapy, efforts are being made to increase the efficacy of various aspects of HSC acquisition, engraftment, and patient care. However, there is limited research to develop next-generation strategies for other aspects of HSC gene therapy that can improve clinical efficacy of this treatment modality. Clinically-feasible strategies need to be developed that allow for selection or enrichment of transduced, therapeutic HSCs after engraftment. In addition, strategies that use other target cell types, such as MSCs, should also be considered for tandem gene therapeutics, to increase the efficiency of correction mediated by HSC gene therapy. More specifically, enrichment strategies that utilize clinically-approved compounds with known pharmacokinetics and pharmacodynamics, such as MMF, need to be developed. Such systems have the potential to be employed in the clinic more quickly and can allow for repeated administrations or continuous low dosing for long-term benefit. Dose adjustments can also be safely made to compensate for variability in patient pharmacogenomics. The development of modules that allow resistance to drugs used for the treatment of benign hematopoietic hyperplasias can encompass many of the aforementioned advantages.
Footnotes
Supported by Canadian Institutes of Health Research Grant to Medin JA, No. MOP-123528.
Conflict-of-interest statement: Authors declare no conflict of interests for this article.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: August 8, 2015
First decision: September 22, 2015
Article in press: November 25, 2015
P- Reviewer: Fukuda S, Guo ZK, Hwang SM, Kwon S S- Editor: Qiu S L- Editor: A E- Editor: Wu HL
References
- 1.Spangrude GJ, Heimfeld S, Weissman IL. Purification and characterization of mouse hematopoietic stem cells. Science. 1988;241:58–62. doi: 10.1126/science.2898810. [DOI] [PubMed] [Google Scholar]
- 2.Bryder D, Rossi DJ, Weissman IL. Hematopoietic stem cells: the paradigmatic tissue-specific stem cell. Am J Pathol. 2006;169:338–346. doi: 10.2353/ajpath.2006.060312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Copelan EA. Hematopoietic stem-cell transplantation. N Engl J Med. 2006;354:1813–1826. doi: 10.1056/NEJMra052638. [DOI] [PubMed] [Google Scholar]
- 4.Tsuruta T. Recent Advances in Hematopoietic Stem Cell Gene Therapy. Available from: http://www.intechopen.com/books/innovations-in-stem-cell-transplantation/recent-advances-in-hematopoietic-stem-cell-gene-therapy.
- 5.Pai SY, Cowan MJ. Stem cell transplantation for primary immunodeficiency diseases: the North American experience. Curr Opin Allergy Clin Immunol. 2014;14:521–526. doi: 10.1097/ACI.0000000000000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Åhlin A, Fasth A. Chronic granulomatous disease - conventional treatment vs. hematopoietic stem cell transplantation: an update. Curr Opin Hematol. 2015;22:41–45. doi: 10.1097/MOH.0000000000000097. [DOI] [PubMed] [Google Scholar]
- 7.Angelucci E. Hematopoietic stem cell transplantation in thalassemia. Hematology Am Soc Hematol Educ Program. 2010;2010:456–462. doi: 10.1182/asheducation-2010.1.456. [DOI] [PubMed] [Google Scholar]
- 8.Talano JA, Cairo MS. Hematopoietic stem cell transplantation for sickle cell disease: state of the science. Eur J Haematol. 2015;94:391–399. doi: 10.1111/ejh.12447. [DOI] [PubMed] [Google Scholar]
- 9.Chinen Y, Higa T, Tomatsu S, Suzuki Y, Orii T, Hyakuna N. Long-term therapeutic efficacy of allogenic bone marrow transplantation in a patient with mucopolysaccharidosis IVA. Mol Genet Metab Rep. 2014;1:31–41. doi: 10.1016/j.ymgmr.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ito S, Barrett AJ. Gauchers disease--a reappraisal of hematopoietic stem cell transplantation. Pediatr Hematol Oncol. 2013;30:61–70. doi: 10.3109/08880018.2012.762076. [DOI] [PubMed] [Google Scholar]
- 11.Tsai P, Lipton JM, Sahdev I, Najfeld V, Rankin LR, Slyper AH, Ludman M, Grabowski GA. Allogenic bone marrow transplantation in severe Gaucher disease. Pediatr Res. 1992;31:503–507. doi: 10.1203/00006450-199205000-00019. [DOI] [PubMed] [Google Scholar]
- 12.Bergkvist K, Winterling J, Johansson E, Johansson UB, Svahn BM, Remberger M, Mattsson J, Larsen J. General health, symptom occurrence, and self-efficacy in adult survivors after allogeneic hematopoietic stem cell transplantation: a cross-sectional comparison between hospital care and home care. Support Care Cancer. 2015;23:1273–1283. doi: 10.1007/s00520-014-2476-9. [DOI] [PubMed] [Google Scholar]
- 13.Alshemmari S, Ameen R, Gaziev J. Haploidentical hematopoietic stem-cell transplantation in adults. Bone Marrow Res. 2011;2011:303487. doi: 10.1155/2011/303487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hilgendorf I, Greinix H, Halter JP, Lawitschka A, Bertz H, Wolff D. Long-term follow-up after allogeneic stem cell transplantation. Dtsch Arztebl Int. 2015;112:51–58. doi: 10.3238/arztebl.2015.0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Naldini L. Ex vivo gene transfer and correction for cell-based therapies. Nat Rev Genet. 2011;12:301–315. doi: 10.1038/nrg2985. [DOI] [PubMed] [Google Scholar]
- 16.Boulad F, Sands S, Sklar C. Late complications after bone marrow transplantation in children and adolescents. Curr Probl Pediatr. 1998;28:273–297. doi: 10.1016/s0045-9380(98)80030-3. [DOI] [PubMed] [Google Scholar]
- 17.Mukherjee S, Thrasher AJ. Gene therapy for PIDs: progress, pitfalls and prospects. Gene. 2013;525:174–181. doi: 10.1016/j.gene.2013.03.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meissner TB, Mandal PK, Ferreira LM, Rossi DJ, Cowan CA. Genome editing for human gene therapy. Methods Enzymol. 2014;546:273–295. doi: 10.1016/B978-0-12-801185-0.00013-1. [DOI] [PubMed] [Google Scholar]
- 19.Mandal PK, Ferreira LM, Collins R, Meissner TB, Boutwell CL, Friesen M, Vrbanac V, Garrison BS, Stortchevoi A, Bryder D, et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell. 2014;15:643–652. doi: 10.1016/j.stem.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cicalese MP, Aiuti A. Clinical applications of gene therapy for primary immunodeficiencies. Hum Gene Ther. 2015;26:210–219. doi: 10.1089/hum.2015.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ginn SL, Alexander IE, Edelstein ML, Abedi MR, Wixon J. Gene therapy clinical trials worldwide to 2012 - an update. J Gene Med. 2013;15:65–77. doi: 10.1002/jgm.2698. [DOI] [PubMed] [Google Scholar]
- 22.Gene Therapy Clinical Trials Worldwide Database. J Gene Med [accessed 2015 Jun 29] Available from: http//www.wiley.com/legacy/wileychi/genmed/clinical/
- 23.Noguchi P. Risks and benefits of gene therapy. N Engl J Med. 2003;348:193–194. doi: 10.1056/NEJMp020184. [DOI] [PubMed] [Google Scholar]
- 24.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, Lim A, Osborne CS, Pawliuk R, Morillon E, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 25.Trobridge GD. Genotoxicity of retroviral hematopoietic stem cell gene therapy. Expert Opin Biol Ther. 2011;11:581–593. doi: 10.1517/14712598.2011.562496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arumugam PI, Higashimoto T, Urbinati F, Modlich U, Nestheide S, Xia P, Fox C, Corsinotti A, Baum C, Malik P. Genotoxic potential of lineage-specific lentivirus vectors carrying the beta-globin locus control region. Mol Ther. 2009;17:1929–1937. doi: 10.1038/mt.2009.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schröder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521–529. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- 28.Montini E, Cesana D, Schmidt M, Sanvito F, Ponzoni M, Bartholomae C, Sergi Sergi L, Benedicenti F, Ambrosi A, Di Serio C, et al. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat Biotechnol. 2006;24:687–696. doi: 10.1038/nbt1216. [DOI] [PubMed] [Google Scholar]
- 29.Ronen K, Negre O, Roth S, Colomb C, Malani N, Denaro M, Brady T, Fusil F, Gillet-Legrand B, Hehir K, et al. Distribution of lentiviral vector integration sites in mice following therapeutic gene transfer to treat β-thalassemia. Mol Ther. 2011;19:1273–1286. doi: 10.1038/mt.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Phaltane R, Haemmerle R, Rothe M, Modlich U, Moritz T. Efficiency and safety of O⁶-methylguanine DNA methyltransferase (MGMT(P140K))-mediated in vivo selection in a humanized mouse model. Hum Gene Ther. 2014;25:144–155. doi: 10.1089/hum.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gothot A, van der Loo JC, Clapp DW, Srour EF. Cell cycle-related changes in repopulating capacity of human mobilized peripheral blood CD34(+) cells in non-obese diabetic/severe combined immune-deficient mice. Blood. 1998;92:2641–2649. [PubMed] [Google Scholar]
- 32.Miyoshi H, Smith KA, Mosier DE, Verma IM, Torbett BE. Transduction of human CD34+ cells that mediate long-term engraftment of NOD/SCID mice by HIV vectors. Science. 1999;283:682–686. doi: 10.1126/science.283.5402.682. [DOI] [PubMed] [Google Scholar]
- 33.Dorrell C, Gan OI, Pereira DS, Hawley RG, Dick JE. Expansion of human cord blood CD34(+)CD38(-) cells in ex vivo culture during retroviral transduction without a corresponding increase in SCID repopulating cell (SRC) frequency: dissociation of SRC phenotype and function. Blood. 2000;95:102–110. [PubMed] [Google Scholar]
- 34.Guenechea G, Gan OI, Inamitsu T, Dorrell C, Pereira DS, Kelly M, Naldini L, Dick JE. Transduction of human CD34+ CD38- bone marrow and cord blood-derived SCID-repopulating cells with third-generation lentiviral vectors. Mol Ther. 2000;1:566–573. doi: 10.1006/mthe.2000.0077. [DOI] [PubMed] [Google Scholar]
- 35.Barrette S, Douglas JL, Seidel NE, Bodine DM. Lentivirus-based vectors transduce mouse hematopoietic stem cells with similar efficiency to moloney murine leukemia virus-based vectors. Blood. 2000;96:3385–3391. [PubMed] [Google Scholar]
- 36.Merten OW, Charrier S, Laroudie N, Fauchille S, Dugué C, Jenny C, Audit M, Zanta-Boussif MA, Chautard H, Radrizzani M, et al. Large-scale manufacture and characterization of a lentiviral vector produced for clinical ex vivo gene therapy application. Hum Gene Ther. 2011;22:343–356. doi: 10.1089/hum.2010.060. [DOI] [PubMed] [Google Scholar]
- 37.Ausubel LJ, Hall C, Sharma A, Shakeley R, Lopez P, Quezada V, Couture S, Laderman K, McMahon R, Huang P, et al. Production of CGMP-Grade Lentiviral Vectors. Bioprocess Int. 2012;10:32–43. [PMC free article] [PubMed] [Google Scholar]
- 38.Dahlberg A, Delaney C, Bernstein ID. Ex vivo expansion of human hematopoietic stem and progenitor cells. Blood. 2011;117:6083–6090. doi: 10.1182/blood-2011-01-283606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duinhouwer LE, Tüysüz N, Rombouts EW, Ter Borg MN, Mastrobattista E, Spanholtz J, Cornelissen JJ, Ten Berge D, Braakman E. Wnt3a protein reduces growth factor-driven expansion of human hematopoietic stem and progenitor cells in serum-free cultures. PLoS One. 2015;10:e0119086. doi: 10.1371/journal.pone.0119086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gertz MA. Current status of stem cell mobilization. Br J Haematol. 2010;150:647–662. doi: 10.1111/j.1365-2141.2010.08313.x. [DOI] [PubMed] [Google Scholar]
- 41.Mohty M, Duarte RF, Croockewit S, Hübel K, Kvalheim G, Russell N. The role of plerixafor in optimizing peripheral blood stem cell mobilization for autologous stem cell transplantation. Leukemia. 2011;25:1–6. doi: 10.1038/leu.2010.224. [DOI] [PubMed] [Google Scholar]
- 42.Schroeder MA, DiPersio JF. Mobilization of hematopoietic stem and leukemia cells. J Leukoc Biol. 2012;91:47–57. doi: 10.1189/jlb.0210085. [DOI] [PubMed] [Google Scholar]
- 43.Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, Gross F, Yvon E, Nusbaum P, Selz F, Hue C, Certain S, Casanova JL, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288:669–672. doi: 10.1126/science.288.5466.669. [DOI] [PubMed] [Google Scholar]
- 44.Hacein-Bey-Abina S, Le Deist F, Carlier F, Bouneaud C, Hue C, De Villartay JP, Thrasher AJ, Wulffraat N, Sorensen R, Dupuis-Girod S, et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N Engl J Med. 2002;346:1185–1193. doi: 10.1056/NEJMoa012616. [DOI] [PubMed] [Google Scholar]
- 45.Cavazzana-Calvo M, Fischer A. Gene therapy for severe combined immunodeficiency: are we there yet? J Clin Invest. 2007;117:1456–1465. doi: 10.1172/JCI30953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu CJ, Hochberg EP, Rogers SA, Kutok JL, Biernacki M, Nascimento AF, Marks P, Bridges K, Ritz J. Molecular assessment of erythroid lineage chimerism following nonmyeloablative allogeneic stem cell transplantation. Exp Hematol. 2003;31:924–933. doi: 10.1016/s0301-472x(03)00227-3. [DOI] [PubMed] [Google Scholar]
- 47.Finotti A, Breda L, Lederer CW, Bianchi N, Zuccato C, Kleanthous M, Rivella S, Gambari R. Recent trends in the gene therapy of β-thalassemia. J Blood Med. 2015;6:69–85. doi: 10.2147/JBM.S46256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Migita M, Medin JA, Pawliuk R, Jacobson S, Nagle JW, Anderson S, Amiri M, Humphries RK, Karlsson S. Selection of transduced CD34+ progenitors and enzymatic correction of cells from Gaucher patients, with bicistronic vectors. Proc Natl Acad Sci USA. 1995;92:12075–12079. doi: 10.1073/pnas.92.26.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Medin JA, Takenaka T, Carpentier S, Garcia V, Basile JP, Segui B, Andrieu-Abadie N, Auge N, Salvayre R, Levade T. Retrovirus-mediated correction of the metabolic defect in cultured Farber disease cells. Hum Gene Ther. 1999;10:1321–1329. doi: 10.1089/10430349950018003. [DOI] [PubMed] [Google Scholar]
- 50.Medin JA, Tudor M, Simovitch R, Quirk JM, Jacobson S, Murray GJ, Brady RO. Correction in trans for Fabry disease: expression, secretion and uptake of alpha-galactosidase A in patient-derived cells driven by a high-titer recombinant retroviral vector. Proc Natl Acad Sci USA. 1996;93:7917–7922. doi: 10.1073/pnas.93.15.7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Platt FM, Lachmann RH. Treating lysosomal storage disorders: current practice and future prospects. Biochim Biophys Acta. 2009;1793:737–745. doi: 10.1016/j.bbamcr.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 52.Mortensen RM, Kingston RE. Selection of transfected mammalian cells. Curr Protoc Mol Biol. 2009;Chapter 9:Unit9.5. doi: 10.1002/0471142727.mb0905s86. [DOI] [PubMed] [Google Scholar]
- 53.Strair RK, Towle MJ, Smith BR. Recombinant retroviruses encoding cell surface antigens as selectable markers. J Virol. 1988;62:4756–4759. doi: 10.1128/jvi.62.12.4756-4759.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Southern PJ, Berg P. Transformation of mammalian cells to antibiotic resistance with a bacterial gene under control of the SV40 early region promoter. J Mol Appl Genet. 1982;1:327–341. [PubMed] [Google Scholar]
- 55.Blochlinger K, Diggelmann H. Hygromycin B phosphotransferase as a selectable marker for DNA transfer experiments with higher eucaryotic cells. Mol Cell Biol. 1984;4:2929–2931. doi: 10.1128/mcb.4.12.2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Berger C, Flowers ME, Warren EH, Riddell SR. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood. 2006;107:2294–2302. doi: 10.1182/blood-2005-08-3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Traversari C, Marktel S, Magnani Z, Mangia P, Russo V, Ciceri F, Bonini C, Bordignon C. The potential immunogenicity of the TK suicide gene does not prevent full clinical benefit associated with the use of TK-transduced donor lymphocytes in HSCT for hematologic malignancies. Blood. 2007;109:4708–4715. doi: 10.1182/blood-2006-04-015230. [DOI] [PubMed] [Google Scholar]
- 58.Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, Forman SJ. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16:1245–1256. doi: 10.1016/j.bbmt.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mavilio F, Ferrari G, Rossini S, Nobili N, Bonini C, Casorati G, Traversari C, Bordignon C. Peripheral blood lymphocytes as target cells of retroviral vector-mediated gene transfer. Blood. 1994;83:1988–1997. [PubMed] [Google Scholar]
- 60.Fehse B, Uhde A, Fehse N, Eckert HG, Clausen J, Rüger R, Koch S, Ostertag W, Zander AR, Stockschläder M. Selective immunoaffinity-based enrichment of CD34+ cells transduced with retroviral vectors containing an intracytoplasmatically truncated version of the human low-affinity nerve growth factor receptor (deltaLNGFR) gene. Hum Gene Ther. 1997;8:1815–1824. doi: 10.1089/hum.1997.8.15-1815. [DOI] [PubMed] [Google Scholar]
- 61.Stockschläder M, Haiss M, Exner S, Schmah O, Veelken H, Follo M, Rüger R, Finke J. Expansion and fibronectin-enhanced retroviral transduction of primary human T lymphocytes for adoptive immunotherapy. J Hematother Stem Cell Res. 1999;8:401–410. doi: 10.1089/152581699320162. [DOI] [PubMed] [Google Scholar]
- 62.Verzeletti S, Bonini C, Marktel S, Nobili N, Ciceri F, Traversari C, Bordignon C. Herpes simplex virus thymidine kinase gene transfer for controlled graft-versus-host disease and graft-versus-leukemia: clinical follow-up and improved new vectors. Hum Gene Ther. 1998;9:2243–2251. doi: 10.1089/hum.1998.9.15-2243. [DOI] [PubMed] [Google Scholar]
- 63.Pawliuk R, Kay R, Lansdorp P, Humphries RK. Selection of retrovirally transduced hematopoietic cells using CD24 as a marker of gene transfer. Blood. 1994;84:2868–2877. [PubMed] [Google Scholar]
- 64.Pawliuk R, Eaves CJ, Humphries RK. Sustained high-level reconstitution of the hematopoietic system by preselected hematopoietic cells expressing a transduced cell-surface antigen. Hum Gene Ther. 1997;8:1595–1604. doi: 10.1089/hum.1997.8.13-1595. [DOI] [PubMed] [Google Scholar]
- 65.García-Hernández B, Sánchez-García I. Retroviral vector design for gene therapy of cancer: specific inhibition and tagging of BCR-ABLp190 cells. Mol Med. 1996;2:125–133. [PMC free article] [PubMed] [Google Scholar]
- 66.García-Hernández B, Castellanos A, López A, Orfao A, Sánchez-García I. Murine hematopoietic reconstitution after tagging and selection of retrovirally transduced bone marrow cells. Proc Natl Acad Sci USA. 1997;94:13239–13244. doi: 10.1073/pnas.94.24.13239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Qin G, Takenaka T, Telsch K, Kelley L, Howard T, Levade T, Deans R, Howard BH, Malech HL, Brady RO, et al. Preselective gene therapy for Fabry disease. Proc Natl Acad Sci USA. 2001;98:3428–3433. doi: 10.1073/pnas.061020598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li Q, Zhang X, Peng Y, Chai H, Xu Y, Wei J, Ren X, Wang X, Liu W, Chen M, et al. Comparison of the sorting efficiency and influence on cell function between the sterile flow cytometry and immunomagnetic bead purification methods. Prep Biochem Biotechnol. 2013;43:197–206. doi: 10.1080/10826068.2012.719846. [DOI] [PubMed] [Google Scholar]
- 69.Borchers S, Provasi E, Silvani A, Radrizzani M, Benati C, Dammann E, Krons A, Kontsendorn J, Schmidtke J, Kuehnau W, et al. Genetically modified donor leukocyte transfusion and graft-versus-leukemia effect after allogeneic stem cell transplantation. Hum Gene Ther. 2011;22:829–841. doi: 10.1089/hum.2010.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Casucci M, Perna SK, Falcone L, Camisa B, Magnani Z, Bernardi M, Crotta A, Tresoldi C, Fleischhauer K, Ponzoni M, et al. Graft-versus-leukemia effect of HLA-haploidentical central-memory T-cells expanded with leukemic APCs and modified with a suicide gene. Mol Ther. 2013;21:466–475. doi: 10.1038/mt.2012.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weissinger EM, Borchers S, Silvani A, Provasi E, Radrizzani M, Beckmann IK, Benati C, Schmidtke J, Kuehnau W, Schweier P, et al. Long term follow up of patients after allogeneic stem cell transplantation and transfusion of HSV-TK transduced T-cells. Front Pharmacol. 2015;6:76. doi: 10.3389/fphar.2015.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sorrentino BP. Gene therapy to protect haematopoietic cells from cytotoxic cancer drugs. Nat Rev Cancer. 2002;2:431–441. doi: 10.1038/nrc823. [DOI] [PubMed] [Google Scholar]
- 73.Eckford PD, Sharom FJ. The reconstituted P-glycoprotein multidrug transporter is a flippase for glucosylceramide and other simple glycosphingolipids. Biochem J. 2005;389:517–526. doi: 10.1042/BJ20050047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.van Meer G, Halter D, Sprong H, Somerharju P, Egmond MR. ABC lipid transporters: extruders, flippases, or flopless activators? FEBS Lett. 2006;580:1171–1177. doi: 10.1016/j.febslet.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 75.Sharom FJ. ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics. 2008;9:105–127. doi: 10.2217/14622416.9.1.105. [DOI] [PubMed] [Google Scholar]
- 76.Mickisch GH, Aksentijevich I, Schoenlein PV, Goldstein LJ, Galski H, Stahle C, Sachs DH, Pastan I, Gottesman MM. Transplantation of bone marrow cells from transgenic mice expressing the human MDR1 gene results in long-term protection against the myelosuppressive effect of chemotherapy in mice. Blood. 1992;79:1087–1093. [PubMed] [Google Scholar]
- 77.Hanania EG, Fu S, Roninson I, Zu Z, Deisseroth AB. Resistance to taxol chemotherapy produced in mouse marrow cells by safety-modified retroviruses containing a human MDR-1 transcription unit. Gene Ther. 1995;2:279–284. [PubMed] [Google Scholar]
- 78.Podda S, Ward M, Himelstein A, Richardson C, de la Flor-Weiss E, Smith L, Gottesman M, Pastan I, Bank A. Transfer and expression of the human multiple drug resistance gene into live mice. Proc Natl Acad Sci USA. 1992;89:9676–9680. doi: 10.1073/pnas.89.20.9676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sorrentino BP, Brandt SJ, Bodine D, Gottesman M, Pastan I, Cline A, Nienhuis AW. Selection of drug-resistant bone marrow cells in vivo after retroviral transfer of human MDR1. Science. 1992;257:99–103. doi: 10.1126/science.1352414. [DOI] [PubMed] [Google Scholar]
- 80.Schwarzenberger P, Spence S, Lohrey N, Kmiecik T, Longo DL, Murphy WJ, Ruscetti FW, Keller JR. Gene transfer of multidrug resistance into a factor-dependent human hematopoietic progenitor cell line: in vivo model for genetically transferred chemoprotection. Blood. 1996;87:2723–2731. [PubMed] [Google Scholar]
- 81.Schiedlmeier B, Schilz AJ, Kühlcke K, Laufs S, Baum C, Zeller WJ, Eckert HG, Fruehauf S. Multidrug resistance 1 gene transfer can confer chemoprotection to human peripheral blood progenitor cells engrafted in immunodeficient mice. Hum Gene Ther. 2002;13:233–242. doi: 10.1089/10430340252769761. [DOI] [PubMed] [Google Scholar]
- 82.Licht T, Haskins M, Henthorn P, Kleiman SE, Bodine DM, Whitwam T, Puck JM, Gottesman MM, Melniczek JR. Drug selection with paclitaxel restores expression of linked IL-2 receptor gamma -chain and multidrug resistance (MDR1) transgenes in canine bone marrow. Proc Natl Acad Sci USA. 2002;99:3123–3128. doi: 10.1073/pnas.052712199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hibino H, Tani K, Ikebuchi K, Wu MS, Sugiyama H, Nakazaki Y, Tanabe T, Takahashi S, Tojo A, Suzuki S, et al. The common marmoset as a target preclinical primate model for cytokine and gene therapy studies. Blood. 1999;93:2839–2848. [PubMed] [Google Scholar]
- 84.Hesdorffer C, Ayello J, Ward M, Kaubisch A, Vahdat L, Balmaceda C, Garrett T, Fetell M, Reiss R, Bank A, et al. Phase I trial of retroviral-mediated transfer of the human MDR1 gene as marrow chemoprotection in patients undergoing high-dose chemotherapy and autologous stem-cell transplantation. J Clin Oncol. 1998;16:165–172. doi: 10.1200/JCO.1998.16.1.165. [DOI] [PubMed] [Google Scholar]
- 85.Cowan KH, Moscow JA, Huang H, Zujewski JA, O’Shaughnessy J, Sorrentino B, Hines K, Carter C, Schneider E, Cusack G, et al. Paclitaxel chemotherapy after autologous stem-cell transplantation and engraftment of hematopoietic cells transduced with a retrovirus containing the multidrug resistance complementary DNA (MDR1) in metastatic breast cancer patients. Clin Cancer Res. 1999;5:1619–1628. [PubMed] [Google Scholar]
- 86.Moscow JA, Huang H, Carter C, Hines K, Zujewski J, Cusack G, Chow C, Venzon D, Sorrentino B, Chiang Y, et al. Engraftment of MDR1 and NeoR gene-transduced hematopoietic cells after breast cancer chemotherapy. Blood. 1999;94:52–61. [PubMed] [Google Scholar]
- 87.Abonour R, Williams DA, Einhorn L, Hall KM, Chen J, Coffman J, Traycoff CM, Bank A, Kato I, Ward M, et al. Efficient retrovirus-mediated transfer of the multidrug resistance 1 gene into autologous human long-term repopulating hematopoietic stem cells. Nat Med. 2000;6:652–658. doi: 10.1038/76225. [DOI] [PubMed] [Google Scholar]
- 88.Devereux S, Corney C, Macdonald C, Watts M, Sullivan A, Goldstone AH, Ward M, Bank A, Linch DC. Feasibility of multidrug resistance (MDR-1) gene transfer in patients undergoing high-dose therapy and peripheral blood stem cell transplantation for lymphoma. Gene Ther. 1998;5:403–408. doi: 10.1038/sj.gt.3300588. [DOI] [PubMed] [Google Scholar]
- 89.Sorrentino BP, McDonagh KT, Woods D, Orlic D. Expression of retroviral vectors containing the human multidrug resistance 1 cDNA in hematopoietic cells of transplanted mice. Blood. 1995;86:491–501. [PubMed] [Google Scholar]
- 90.Knipper R, Kuehlcke K, Schiedlmeier B, Hildinger M, Lindemann C, Schilz AJ, Fauser AA, Fruehauf S, Zeller WJ, Ostertag W, et al. Improved post-transcriptional processing of an MDR1 retrovirus elevates expression of multidrug resistance in primary human hematopoietic cells. Gene Ther. 2001;8:239–246. doi: 10.1038/sj.gt.3301384. [DOI] [PubMed] [Google Scholar]
- 91.Cmejlova J, Hildinger M, Cmejla R, Fuchs O, Kalabova D, Baum C, Jelinek J. Impact of splice-site mutations of the human MDR1 cDNA on its stability and expression following retroviral gene transfer. Gene Ther. 2003;10:1061–1065. doi: 10.1038/sj.gt.3301967. [DOI] [PubMed] [Google Scholar]
- 92.Allay JA, Spencer HT, Wilkinson SL, Belt JA, Blakley RL, Sorrentino BP. Sensitization of hematopoietic stem and progenitor cells to trimetrexate using nucleoside transport inhibitors. Blood. 1997;90:3546–3554. [PubMed] [Google Scholar]
- 93.Lewis WS, Cody V, Galitsky N, Luft JR, Pangborn W, Chunduru SK, Spencer HT, Appleman JR, Blakley RL. Methotrexate-resistant variants of human dihydrofolate reductase with substitutions of leucine 22. Kinetics, crystallography, and potential as selectable markers. J Biol Chem. 1995;270:5057–5064. doi: 10.1074/jbc.270.10.5057. [DOI] [PubMed] [Google Scholar]
- 94.Allay JA, Persons DA, Galipeau J, Riberdy JM, Ashmun RA, Blakley RL, Sorrentino BP. In vivo selection of retrovirally transduced hematopoietic stem cells. Nat Med. 1998;4:1136–1143. doi: 10.1038/2632. [DOI] [PubMed] [Google Scholar]
- 95.Havenga M, Valerio D, Hoogerbrugge P, Es H. In vivo methotrexate selection of murine hemopoietic cells transduced with a retroviral vector for Gaucher disease. Gene Ther. 1999;6:1661–1669. doi: 10.1038/sj.gt.3301037. [DOI] [PubMed] [Google Scholar]
- 96.Persons DA, Allay JA, Bonifacino A, Lu T, Agricola B, Metzger ME, Donahue RE, Dunbar CE, Sorrentino BP. Transient in vivo selection of transduced peripheral blood cells using antifolate drug selection in rhesus macaques that received transplants with hematopoietic stem cells expressing dihydrofolate reductase vectors. Blood. 2004;103:796–803. doi: 10.1182/blood-2003-05-1572. [DOI] [PubMed] [Google Scholar]
- 97.Gori JL, McIvor RS, Kaufman DS. Methotrexate supports in vivo selection of human embryonic stem cell derived-hematopoietic cells expressing dihydrofolate reductase. Bioeng Bugs. 2010;1:434–436. doi: 10.4161/bbug.1.6.12390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stead RB, Kwok WW, Storb R, Miller AD. Canine model for gene therapy: inefficient gene expression in dogs reconstituted with autologous marrow infected with retroviral vectors. Blood. 1988;71:742–747. [PubMed] [Google Scholar]
- 99.de Miguel D, García-Suárez J, Martín Y, Gil-Fernández JJ, Burgaleta C. Severe acute renal failure following high-dose methotrexate therapy in adults with haematological malignancies: a significant number result from unrecognized co-administration of several drugs. Nephrol Dial Transplant. 2008;23:3762–3766. doi: 10.1093/ndt/gfn503. [DOI] [PubMed] [Google Scholar]
- 100.Grönroos MH, Jahnukainen T, Möttönen M, Perkkiö M, Irjala K, Salmi TT. Long-term follow-up of renal function after high-dose methotrexate treatment in children. Pediatr Blood Cancer. 2008;51:535–539. doi: 10.1002/pbc.21650. [DOI] [PubMed] [Google Scholar]
- 101.Vilay AM, Mueller BA, Haines H, Alten JA, Askenazi DJ. Treatment of methotrexate intoxication with various modalities of continuous extracorporeal therapy and glucarpidase. Pharmacotherapy. 2010;30:111. doi: 10.1592/phco.30.1.111. [DOI] [PubMed] [Google Scholar]
- 102.Vang SI, Schmiegelow K, Frandsen T, Rosthøj S, Nersting J. Mercaptopurine metabolite levels are predictors of bone marrow toxicity following high-dose methotrexate therapy of childhood acute lymphoblastic leukaemia. Cancer Chemother Pharmacol. 2015;75:1089–1093. doi: 10.1007/s00280-015-2717-8. [DOI] [PubMed] [Google Scholar]
- 103.Jonnalagadda M, Brown CE, Chang WC, Ostberg JR, Forman SJ, Jensen MC. Efficient selection of genetically modified human T cells using methotrexate-resistant human dihydrofolate reductase. Gene Ther. 2013;20:853–860. doi: 10.1038/gt.2012.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gori JL, Beard BC, Williams NP, Ironside C, Swanson D, Scott McIvor R, Kiem HP. In vivo protection of activated Tyr22-dihydrofolate reductase gene-modified canine T lymphocytes from methotrexate. J Gene Med. 2013;15:233–241. doi: 10.1002/jgm.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Allay JA, Dumenco LL, Koc ON, Liu L, Gerson SL. Retroviral transduction and expression of the human alkyltransferase cDNA provides nitrosourea resistance to hematopoietic cells. Blood. 1995;85:3342–3351. [PubMed] [Google Scholar]
- 106.Gerson SL. Clinical relevance of MGMT in the treatment of cancer. J Clin Oncol. 2002;20:2388–2399. doi: 10.1200/JCO.2002.06.110. [DOI] [PubMed] [Google Scholar]
- 107.Gerson SL, Miller K, Berger NA. O6 alkylguanine-DNA alkyltransferase activity in human myeloid cells. J Clin Invest. 1985;76:2106–2114. doi: 10.1172/JCI112215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gerson SL, Trey JE, Miller K, Berger NA. Comparison of O6-alkylguanine-DNA alkyltransferase activity based on cellular DNA content in human, rat and mouse tissues. Carcinogenesis. 1986;7:745–749. doi: 10.1093/carcin/7.5.745. [DOI] [PubMed] [Google Scholar]
- 109.Dumenco LL, Allay E, Norton K, Gerson SL. The prevention of thymic lymphomas in transgenic mice by human O6-alkylguanine-DNA alkyltransferase. Science. 1993;259:219–222. doi: 10.1126/science.8421782. [DOI] [PubMed] [Google Scholar]
- 110.Maze R, Kapur R, Kelley MR, Hansen WK, Oh SY, Williams DA. Reversal of 1,3-bis(2-chloroethyl)-1-nitrosourea-induced severe immunodeficiency by transduction of murine long-lived hemopoietic progenitor cells using O6-methylguanine DNA methyltransferase complementary DNA. J Immunol. 1997;158:1006–1013. [PubMed] [Google Scholar]
- 111.Allay JA, Koç ON, Davis BM, Gerson SL. Retroviral-mediated gene transduction of human alkyltransferase complementary DNA confers nitrosourea resistance to human hematopoietic progenitors. Clin Cancer Res. 1996;2:1353–1359. [PubMed] [Google Scholar]
- 112.Clemons M, Kelly J, Watson AJ, Howell A, McElhinney RS, McMurry TB, Margison GP. O6-(4-bromothenyl)guanine reverses temozolomide resistance in human breast tumour MCF-7 cells and xenografts. Br J Cancer. 2005;93:1152–1156. doi: 10.1038/sj.bjc.6602833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Crone TM, Kanugula S, Pegg AE. Mutations in the Ada O6-alkylguanine-DNA alkyltransferase conferring sensitivity to inactivation by O6-benzylguanine and 2,4-diamino-6-benzyloxy-5-nitrosopyrimidine. Carcinogenesis. 1995;16:1687–1692. doi: 10.1093/carcin/16.8.1687. [DOI] [PubMed] [Google Scholar]
- 114.Roth JC, Ismail M, Reese JS, Lingas KT, Ferrari G, Gerson SL. Cotransduction with MGMT and Ubiquitous or Erythroid-Specific GFP Lentiviruses Allows Enrichment of Dual-Positive Hematopoietic Progenitor Cells In Vivo. ISRN Hematol. 2012;2012:212586. doi: 10.5402/2012/212586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Neff T, Horn PA, Peterson LJ, Thomasson BM, Thompson J, Williams DA, Schmidt M, Georges GE, von Kalle C, Kiem HP. Methylguanine methyltransferase-mediated in vivo selection and chemoprotection of allogeneic stem cells in a large-animal model. J Clin Invest. 2003;112:1581–1588. doi: 10.1172/JCI18782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Neff T, Beard BC, Peterson LJ, Anandakumar P, Thompson J, Kiem HP. Polyclonal chemoprotection against temozolomide in a large-animal model of drug resistance gene therapy. Blood. 2005;105:997–1002. doi: 10.1182/blood-2004-08-3169. [DOI] [PubMed] [Google Scholar]
- 117.Gerull S, Beard BC, Peterson LJ, Neff T, Kiem HP. In vivo selection and chemoprotection after drug resistance gene therapy in a nonmyeloablative allogeneic transplantation setting in dogs. Hum Gene Ther. 2007;18:451–456. doi: 10.1089/hum.2006.039. [DOI] [PubMed] [Google Scholar]
- 118.Pollok KE, Hartwell JR, Braber A, Cooper RJ, Jansen M, Ragg S, Bailey BJ, Erickson LC, Kreklau EL, Williams DA. In vivo selection of human hematopoietic cells in a xenograft model using combined pharmacologic and genetic manipulations. Hum Gene Ther. 2003;14:1703–1714. doi: 10.1089/104303403322611728. [DOI] [PubMed] [Google Scholar]
- 119.Zielske SP, Reese JS, Lingas KT, Donze JR, Gerson SL. In vivo selection of MGMT(P140K) lentivirus-transduced human NOD/SCID repopulating cells without pretransplant irradiation conditioning. J Clin Invest. 2003;112:1561–1570. doi: 10.1172/JCI17922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Persons DA, Allay ER, Sawai N, Hargrove PW, Brent TP, Hanawa H, Nienhuis AW, Sorrentino BP. Successful treatment of murine beta-thalassemia using in vivo selection of genetically modified, drug-resistant hematopoietic stem cells. Blood. 2003;102:506–513. doi: 10.1182/blood-2003-03-0677. [DOI] [PubMed] [Google Scholar]
- 121.Cai S, Ernstberger A, Wang H, Bailey BJ, Hartwell JR, Sinn AL, Eckermann O, Linka Y, Goebel WS, Hanenberg H, et al. In vivo selection of hematopoietic stem cells transduced at a low multiplicity-of-infection with a foamy viral MGMT(P140K) vector. Exp Hematol. 2008;36:283–292. doi: 10.1016/j.exphem.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Larochelle A, Choi U, Shou Y, Naumann N, Loktionova NA, Clevenger JR, Krouse A, Metzger M, Donahue RE, Kang E, et al. In vivo selection of hematopoietic progenitor cells and temozolomide dose intensification in rhesus macaques through lentiviral transduction with a drug resistance gene. J Clin Invest. 2009;119:1952–1963. doi: 10.1172/JCI37506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhao H, Pestina TI, Nasimuzzaman M, Mehta P, Hargrove PW, Persons DA. Amelioration of murine beta-thalassemia through drug selection of hematopoietic stem cells transduced with a lentiviral vector encoding both gamma-globin and the MGMT drug-resistance gene. Blood. 2009;113:5747–5756. doi: 10.1182/blood-2008-10-186684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Schroeder JA, Chen Y, Fang J, Wilcox DA, Shi Q. In vivo enrichment of genetically manipulated platelets corrects the murine hemophilic phenotype and induces immune tolerance even using a low multiplicity of infection. J Thromb Haemost. 2014;12:1283–1293. doi: 10.1111/jth.12633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gori JL, Beard BC, Ironside C, Karponi G, Kiem HP. In vivo selection of autologous MGMT gene-modified cells following reduced-intensity conditioning with BCNU and temozolomide in the dog model. Cancer Gene Ther. 2012;19:523–529. doi: 10.1038/cgt.2012.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Beard BC, Trobridge GD, Ironside C, McCune JS, Adair JE, Kiem HP. Efficient and stable MGMT-mediated selection of long-term repopulating stem cells in nonhuman primates. J Clin Invest. 2010;120:2345–2354. doi: 10.1172/JCI40767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chang AH, Stephan MT, Lisowski L, Sadelain M. Erythroid-specific human factor IX delivery from in vivo selected hematopoietic stem cells following nonmyeloablative conditioning in hemophilia B mice. Mol Ther. 2008;16:1745–1752. doi: 10.1038/mt.2008.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Roth JC, Alberti MO, Ismail M, Lingas KT, Reese JS, Gerson SL. MGMT enrichment and second gene co-expression in hematopoietic progenitor cells using separate or dual-gene lentiviral vectors. Virus Res. 2015;196:170–180. doi: 10.1016/j.virusres.2014.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shepherd BE, Kiem HP, Lansdorp PM, Dunbar CE, Aubert G, LaRochelle A, Seggewiss R, Guttorp P, Abkowitz JL. Hematopoietic stem-cell behavior in nonhuman primates. Blood. 2007;110:1806–1813. doi: 10.1182/blood-2007-02-075382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Adair JE, Beard BC, Trobridge GD, Neff T, Rockhill JK, Silbergeld DL, Mrugala MM, Kiem HP. Extended survival of glioblastoma patients after chemoprotective HSC gene therapy. Sci Transl Med. 2012;4:133ra57. doi: 10.1126/scitranslmed.3003425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Stout JT, Caskey CT. HPRT: gene structure, expression, and mutation. Annu Rev Genet. 1985;19:127–148. doi: 10.1146/annurev.ge.19.120185.001015. [DOI] [PubMed] [Google Scholar]
- 132.Aubrecht J, Goad ME, Schiestl RH. Tissue specific toxicities of the anticancer drug 6-thioguanine is dependent on the Hprt status in transgenic mice. J Pharmacol Exp Ther. 1997;282:1102–1108. [PubMed] [Google Scholar]
- 133.Hacke K, Szakmary A, Cuddihy AR, Rozengurt N, Lemp NA, Aubrecht J, Lawson GW, Rao NP, Crooks GM, Schiestl RH, et al. Combined preconditioning and in vivo chemoselection with 6-thioguanine alone achieves highly efficient reconstitution of normal hematopoiesis with HPRT-deficient bone marrow. Exp Hematol. 2012;40:3–13.e3. doi: 10.1016/j.exphem.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hacke K, Treger JA, Bogan BT, Schiestl RH, Kasahara N. Genetic modification of mouse bone marrow by lentiviral vector-mediated delivery of hypoxanthine-Guanine phosphoribosyltransferase short hairpin RNA confers chemoprotection against 6-thioguanine cytotoxicity. Transplant Proc. 2013;45:2040–2044. doi: 10.1016/j.transproceed.2013.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Porter CC, DeGregori J. Interfering RNA-mediated purine analog resistance for in vitro and in vivo cell selection. Blood. 2008;112:4466–4474. doi: 10.1182/blood-2008-03-146571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Choudhary R, Baturin D, Fosmire S, Freed B, Porter CC. Knockdown of HPRT for selection of genetically modified human hematopoietic progenitor cells. PLoS One. 2013;8:e59594. doi: 10.1371/journal.pone.0059594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Elion GB. The purine path to chemotherapy. Science. 1989;244:41–47. doi: 10.1126/science.2649979. [DOI] [PubMed] [Google Scholar]
- 138.Torres RJ, Puig JG. Hypoxanthine-guanine phosophoribosyltransferase (HPRT) deficiency: Lesch-Nyhan syndrome. Orphanet J Rare Dis. 2007;2:48. doi: 10.1186/1750-1172-2-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Spencer DM, Wandless TJ, Schreiber SL, Crabtree GR. Controlling signal transduction with synthetic ligands. Science. 1993;262:1019–1024. doi: 10.1126/science.7694365. [DOI] [PubMed] [Google Scholar]
- 140.Blau CA, Peterson KR, Drachman JG, Spencer DM. A proliferation switch for genetically modified cells. Proc Natl Acad Sci USA. 1997;94:3076–3081. doi: 10.1073/pnas.94.7.3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Spencer DM, Belshaw PJ, Chen L, Ho SN, Randazzo F, Crabtree GR, Schreiber SL. Functional analysis of Fas signaling in vivo using synthetic inducers of dimerization. Curr Biol. 1996;6:839–847. doi: 10.1016/s0960-9822(02)00607-3. [DOI] [PubMed] [Google Scholar]
- 142.Cameron AM, Steiner JP, Roskams AJ, Ali SM, Ronnett GV, Snyder SH. Calcineurin associated with the inositol 1,4,5-trisphosphate receptor-FKBP12 complex modulates Ca2+ flux. Cell. 1995;83:463–472. doi: 10.1016/0092-8674(95)90124-8. [DOI] [PubMed] [Google Scholar]
- 143.Walensky LD, Dawson TM, Steiner JP, Sabatini DM, Suarez JD, Klinefelter GR, Snyder SH. The 12 kD FK 506 binding protein FKBP12 is released in the male reproductive tract and stimulates sperm motility. Mol Med. 1998;4:502–514. [PMC free article] [PubMed] [Google Scholar]
- 144.Shou W, Aghdasi B, Armstrong DL, Guo Q, Bao S, Charng MJ, Mathews LM, Schneider MD, Hamilton SL, Matzuk MM. Cardiac defects and altered ryanodine receptor function in mice lacking FKBP12. Nature. 1998;391:489–492. doi: 10.1038/35146. [DOI] [PubMed] [Google Scholar]
- 145.Neff T, Blau CA. Pharmacologically regulated cell therapy. Blood. 2001;97:2535–2540. doi: 10.1182/blood.v97.9.2535. [DOI] [PubMed] [Google Scholar]
- 146.Carlson JC, Kanter A, Thuduppathy GR, Cody V, Pineda PE, McIvor RS, Wagner CR. Designing protein dimerizers: the importance of ligand conformational equilibria. J Am Chem Soc. 2003;125:1501–1507. doi: 10.1021/ja026264y. [DOI] [PubMed] [Google Scholar]
- 147.Clackson T, Yang W, Rozamus LW, Hatada M, Amara JF, Rollins CT, Stevenson LF, Magari SR, Wood SA, Courage NL, et al. Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Proc Natl Acad Sci USA. 1998;95:10437–10442. doi: 10.1073/pnas.95.18.10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Burnett SH, Kershen EJ, Zhang J, Zeng L, Straley SC, Kaplan AM, Cohen DA. Conditional macrophage ablation in transgenic mice expressing a Fas-based suicide gene. J Leukoc Biol. 2004;75:612–623. doi: 10.1189/jlb.0903442. [DOI] [PubMed] [Google Scholar]
- 149.Emery DW, Tubb J, Nishino Y, Nishino T, Otto KG, Stamatoyannopoulos G, Blau CA. Selection with a regulated cell growth switch increases the likelihood of expression for a linked gamma-globin gene. Blood Cells Mol Dis. 2005;34:235–247. doi: 10.1016/j.bcmd.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 150.Siatskas C, Underwood J, Ramezani A, Hawley RG, Medin JA. Specific pharmacological dimerization of KDR in lentivirally transduced human hematopoietic cells activates anti-apoptotic and proliferative mechanisms. FASEB J. 2005;19:1752–1754. doi: 10.1096/fj.05-4006fje. [DOI] [PubMed] [Google Scholar]
- 151.Abdel-Azim H, Zhu Y, Hollis R, Wang X, Ge S, Hao QL, Smbatyan G, Kohn DB, Rosol M, Crooks GM. Expansion of multipotent and lymphoid-committed human progenitors through intracellular dimerization of Mpl. Blood. 2008;111:4064–4074. doi: 10.1182/blood-2007-08-107466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Okazuka K, Beard BC, Emery DW, Schwarzwaelder K, Spector MR, Sale GE, von Kalle C, Torok-Storb B, Kiem HP, Blau CA. Long-term regulation of genetically modified primary hematopoietic cells in dogs. Mol Ther. 2011;19:1287–1294. doi: 10.1038/mt.2011.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bade LK, Goldberg JE, Dehut HA, Hall MK, Schwertfeger KL. Mammary tumorigenesis induced by fibroblast growth factor receptor 1 requires activation of the epidermal growth factor receptor. J Cell Sci. 2011;124:3106–3117. doi: 10.1242/jcs.082651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Fischer-Posovszky P, Wang QA, Asterholm IW, Rutkowski JM, Scherer PE. Targeted deletion of adipocytes by apoptosis leads to adipose tissue recruitment of alternatively activated M2 macrophages. Endocrinology. 2011;152:3074–3081. doi: 10.1210/en.2011-1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bashamboo A, Taylor AH, Samuel K, Panthier JJ, Whetton AD, Forrester LM. The survival of differentiating embryonic stem cells is dependent on the SCF-KIT pathway. J Cell Sci. 2006;119:3039–3046. doi: 10.1242/jcs.03038. [DOI] [PubMed] [Google Scholar]
- 156.Richard RE, Blau CA. Small-molecule-directed mpl signaling can complement growth factors to selectively expand genetically modified cord blood cells. Stem Cells. 2003;21:71–78. doi: 10.1634/stemcells.21-1-71. [DOI] [PubMed] [Google Scholar]
- 157.Iuliucci JD, Oliver SD, Morley S, Ward C, Ward J, Dalgarno D, Clackson T, Berger HJ. Intravenous safety and pharmacokinetics of a novel dimerizer drug, AP1903, in healthy volunteers. J Clin Pharmacol. 2001;41:870–879. doi: 10.1177/00912700122010771. [DOI] [PubMed] [Google Scholar]
- 158.Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhou X, Dotti G, Krance RA, Martinez CA, Naik S, Kamble RT, Durett AG, Dakhova O, Savoldo B, Di Stasi A, et al. Inducible caspase-9 suicide gene controls adverse effects from alloreplete T cells after haploidentical stem cell transplantation. Blood. 2015;125:4103–4113. doi: 10.1182/blood-2015-02-628354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zhou X, Di Stasi A, Tey SK, Krance RA, Martinez C, Leung KS, Durett AG, Wu MF, Liu H, Leen AM, et al. Long-term outcome after haploidentical stem cell transplant and infusion of T cells expressing the inducible caspase 9 safety transgene. Blood. 2014;123:3895–3905. doi: 10.1182/blood-2014-01-551671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Nagasawa Y, Wood BL, Wang L, Lintmaer I, Guo W, Papayannopoulou T, Harkey MA, Nourigat C, Blau CA. Anatomical compartments modify the response of human hematopoietic cells to a mitogenic signal. Stem Cells. 2006;24:908–917. doi: 10.1634/stemcells.2005-0484. [DOI] [PubMed] [Google Scholar]
- 162.Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, Kanemaki M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods. 2009;6:917–922. doi: 10.1038/nmeth.1401. [DOI] [PubMed] [Google Scholar]
- 163.Austin TW, Solar GP, Ziegler FC, Liem L, Matthews W. A role for the Wnt gene family in hematopoiesis: expansion of multilineage progenitor cells. Blood. 1997;89:3624–3635. [PubMed] [Google Scholar]
- 164.Morshed M, Shamm M, Hossain M, Anwar Habib M, Moinuddin Ahmed M, Ibrahim M, Umar Ali M, Azizul Islam M. The effect of plant hormone indoleacetic acid (IAA) on hematological and biochemical parameters in mice. Bangladesh J Physiol Pharmacol. 2006;22:5–8. [Google Scholar]
- 165.Urban DJ, Roth BL. DREADDs (designer receptors exclusively activated by designer drugs): chemogenetic tools with therapeutic utility. Annu Rev Pharmacol Toxicol. 2015;55:399–417. doi: 10.1146/annurev-pharmtox-010814-124803. [DOI] [PubMed] [Google Scholar]
- 166.Park JS, Rhau B, Hermann A, McNally KA, Zhou C, Gong D, Weiner OD, Conklin BR, Onuffer J, Lim WA. Synthetic control of mammalian-cell motility by engineering chemotaxis to an orthogonal bioinert chemical signal. Proc Natl Acad Sci U S A. 2014;111:5896–5901. doi: 10.1073/pnas.1402087111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci USA. 2007;104:5163–5168. doi: 10.1073/pnas.0700293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lo Celso C, Scadden DT. The haematopoietic stem cell niche at a glance. J Cell Sci. 2011;124:3529–3535. doi: 10.1242/jcs.074112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Notta F, Doulatov S, Laurenti E, Poeppl A, Jurisica I, Dick JE. Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science. 2011;333:218–221. doi: 10.1126/science.1201219. [DOI] [PubMed] [Google Scholar]
- 170.Cashen AF, Nervi B, DiPersio J. AMD3100: CXCR4 antagonist and rapid stem cell-mobilizing agent. Future Oncol. 2007;3:19–27. doi: 10.2217/14796694.3.1.19. [DOI] [PubMed] [Google Scholar]
- 171.DiPersio JF, Micallef IN, Stiff PJ, Bolwell BJ, Maziarz RT, Jacobsen E, Nademanee A, McCarty J, Bridger G, Calandra G. Phase III prospective randomized double-blind placebo-controlled trial of plerixafor plus granulocyte colony-stimulating factor compared with placebo plus granulocyte colony-stimulating factor for autologous stem-cell mobilization and transplantation for patients with non-Hodgkin’s lymphoma. J Clin Oncol. 2009;27:4767–4773. doi: 10.1200/JCO.2008.20.7209. [DOI] [PubMed] [Google Scholar]
- 172.DiPersio JF, Stadtmauer EA, Nademanee A, Micallef IN, Stiff PJ, Kaufman JL, Maziarz RT, Hosing C, Früehauf S, Horwitz M, et al. Plerixafor and G-CSF versus placebo and G-CSF to mobilize hematopoietic stem cells for autologous stem cell transplantation in patients with multiple myeloma. Blood. 2009;113:5720–5726. doi: 10.1182/blood-2008-08-174946. [DOI] [PubMed] [Google Scholar]
- 173.Ghobadi A, Rettig MP, Cooper ML, Holt MS, Ritchey JK, Eissenberg L, DiPersio JF. Bortezomib is a rapid mobilizer of hematopoietic stem cells in mice via modulation of the VCAM-1/VLA-4 axis. Blood. 2014;124:2752–2754. doi: 10.1182/blood-2014-08-595967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lapidot T. Mechanism of human stem cell migration and repopulation of NOD/SCID and B2mnull NOD/SCID mice. The role of SDF-1/CXCR4 interactions. Ann N Y Acad Sci. 2001;938:83–95. doi: 10.1111/j.1749-6632.2001.tb03577.x. [DOI] [PubMed] [Google Scholar]
- 175.Lai CY, Yamazaki S, Okabe M, Suzuki S, Maeyama Y, Iimura Y, Onodera M, Kakuta S, Iwakura Y, Nojima M, et al. Stage-specific roles for CXCR4 signaling in murine hematopoietic stem/progenitor cells in the process of bone marrow repopulation. Stem Cells. 2014;32:1929–1942. doi: 10.1002/stem.1670. [DOI] [PubMed] [Google Scholar]
- 176.Kahn J, Byk T, Jansson-Sjostrand L, Petit I, Shivtiel S, Nagler A, Hardan I, Deutsch V, Gazit Z, Gazit D, et al. Overexpression of CXCR4 on human CD34+ progenitors increases their proliferation, migration, and NOD/SCID repopulation. Blood. 2004;103:2942–2949. doi: 10.1182/blood-2003-07-2607. [DOI] [PubMed] [Google Scholar]
- 177.Brenner S, Whiting-Theobald N, Kawai T, Linton GF, Rudikoff AG, Choi U, Ryser MF, Murphy PM, Sechler JM, Malech HL. CXCR4-transgene expression significantly improves marrow engraftment of cultured hematopoietic stem cells. Stem Cells. 2004;22:1128–1133. doi: 10.1634/stemcells.2003-0196. [DOI] [PubMed] [Google Scholar]
- 178.Spoo AC, Lübbert M, Wierda WG, Burger JA. CXCR4 is a prognostic marker in acute myelogenous leukemia. Blood. 2007;109:786–791. doi: 10.1182/blood-2006-05-024844. [DOI] [PubMed] [Google Scholar]
- 179.Burger JA, Kipps TJ. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood. 2006;107:1761–1767. doi: 10.1182/blood-2005-08-3182. [DOI] [PubMed] [Google Scholar]
- 180.Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K, Ito K, Koh GY, Suda T. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118:149–161. doi: 10.1016/j.cell.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 181.Lewandowski D, Barroca V, Ducongé F, Bayer J, Van Nhieu JT, Pestourie C, Fouchet P, Tavitian B, Roméo PH. In vivo cellular imaging pinpoints the role of reactive oxygen species in the early steps of adult hematopoietic reconstitution. Blood. 2010;115:443–452. doi: 10.1182/blood-2009-05-222711. [DOI] [PubMed] [Google Scholar]
- 182.Thorén LA, Liuba K, Bryder D, Nygren JM, Jensen CT, Qian H, Antonchuk J, Jacobsen SE. Kit regulates maintenance of quiescent hematopoietic stem cells. J Immunol. 2008;180:2045–2053. doi: 10.4049/jimmunol.180.4.2045. [DOI] [PubMed] [Google Scholar]
- 183.Sawai N, Persons DA, Zhou S, Lu T, Sorrentino BP. Reduction in hematopoietic stem cell numbers with in vivo drug selection can be partially abrogated by HOXB4 gene expression. Mol Ther. 2003;8:376–384. doi: 10.1016/s1525-0016(03)00205-3. [DOI] [PubMed] [Google Scholar]
- 184.Gori JL, Korashon LW, Chandrasekaran D, Sauvageau G, Kiem HP. Effective Expansion and Engraftment Of Nonhuman Primate CD34 Hematopoietic Stem Cells After Co-Culture With The Small Molecule UM171. Blood. 2013;122:1656–1656. [Google Scholar]
- 185.Carrancio S, Romo C, Ramos T, Lopez-Holgado N, Muntion S, Prins HJ, Martens AC, Briñón JG, San Miguel JF, Del Cañizo MC, et al. Effects of MSC coadministration and route of delivery on cord blood hematopoietic stem cell engraftment. Cell Transplant. 2013;22:1171–1183. doi: 10.3727/096368912X657431. [DOI] [PubMed] [Google Scholar]
- 186.Stopp S, Bornhäuser M, Ugarte F, Wobus M, Kuhn M, Brenner S, Thieme S. Expression of the melanoma cell adhesion molecule in human mesenchymal stromal cells regulates proliferation, differentiation, and maintenance of hematopoietic stem and progenitor cells. Haematologica. 2013;98:505–513. doi: 10.3324/haematol.2012.065201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zimmermann AG, Spychala J, Mitchell BS. Characterization of the human inosine-5’-monophosphate dehydrogenase type II gene. J Biol Chem. 1995;270:6808–6814. doi: 10.1074/jbc.270.12.6808. [DOI] [PubMed] [Google Scholar]
- 188.Zimmermann AG, Gu JJ, Laliberté J, Mitchell BS. Inosine-5’-monophosphate dehydrogenase: regulation of expression and role in cellular proliferation and T lymphocyte activation. Prog Nucleic Acid Res Mol Biol. 1998;61:181–209. doi: 10.1016/s0079-6603(08)60827-2. [DOI] [PubMed] [Google Scholar]
- 189.Jonsson CA, Carlsten H. Mycophenolic acid inhibits inosine 5’-monophosphate dehydrogenase and suppresses immunoglobulin and cytokine production of B cells. Int Immunopharmacol. 2003;3:31–37. doi: 10.1016/s1567-5769(02)00210-2. [DOI] [PubMed] [Google Scholar]
- 190.Franklin TJ, Cook JM. The inhibition of nucleic acid synthesis by mycophenolic acid. Biochem J. 1969;113:515–524. doi: 10.1042/bj1130515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ransom JT. Mechanism of action of mycophenolate mofetil. Ther Drug Monit. 1995;17:681–684. doi: 10.1097/00007691-199512000-00023. [DOI] [PubMed] [Google Scholar]
- 192.Sintchak MD, Fleming MA, Futer O, Raybuck SA, Chambers SP, Caron PR, Murcko MA, Wilson KP. Structure and mechanism of inosine monophosphate dehydrogenase in complex with the immunosuppressant mycophenolic acid. Cell. 1996;85:921–930. doi: 10.1016/s0092-8674(00)81275-1. [DOI] [PubMed] [Google Scholar]
- 193.Kharfan-Dabaja M, Mhaskar R, Reljic T, Pidala J, Perkins JB, Djulbegovic B, Kumar A. Mycophenolate mofetil versus methotrexate for prevention of graft-versus-host disease in people receiving allogeneic hematopoietic stem cell transplantation. Cochrane Database Syst Rev. 2014;7:CD010280. doi: 10.1002/14651858.CD010280.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Yam P, Jensen M, Akkina R, Anderson J, Villacres MC, Wu J, Zaia JA, Yee JK. Ex vivo selection and expansion of cells based on expression of a mutated inosine monophosphate dehydrogenase 2 after HIV vector transduction: effects on lymphocytes, monocytes, and CD34+ stem cells. Mol Ther. 2006;14:236–244. doi: 10.1016/j.ymthe.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 195.Farazi T, Leichman J, Harris T, Cahoon M, Hedstrom L. Isolation and characterization of mycophenolic acid-resistant mutants of inosine-5’-monophosphate dehydrogenase. J Biol Chem. 1997;272:961–965. doi: 10.1074/jbc.272.2.961. [DOI] [PubMed] [Google Scholar]
- 196.Sangiolo D, Lesnikova M, Nash RA, Jensen MC, Nikitine A, Kiem HP, Georges GE. Lentiviral vector conferring resistance to mycophenolate mofetil and sensitivity to ganciclovir for in vivo T-cell selection. Gene Ther. 2007;14:1549–1554. doi: 10.1038/sj.gt.3303018. [DOI] [PubMed] [Google Scholar]
- 197.Jonnalagadda M, Brown CE, Chang WC, Ostberg JR, Forman SJ, Jensen MC. Engineering human T cells for resistance to methotrexate and mycophenolate mofetil as an in vivo cell selection strategy. PLoS One. 2013;8:e65519. doi: 10.1371/journal.pone.0065519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Suzuki N, Mukai HY, Yamamoto M. In vivo regulation of erythropoiesis by chemically inducible dimerization of the erythropoietin receptor intracellular domain. PLoS One. 2015;10:e0119442. doi: 10.1371/journal.pone.0119442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Almarza D, Bussadori G, Navarro M, Mavilio F, Larcher F, Murillas R. Risk assessment in skin gene therapy: viral-cellular fusion transcripts generated by proviral transcriptional read-through in keratinocytes transduced with self-inactivating lentiviral vectors. Gene Ther. 2011;18:674–681. doi: 10.1038/gt.2011.12. [DOI] [PubMed] [Google Scholar]
- 200.Moiani A, Mavilio F. Alternative splicing caused by lentiviral integration in the human genome. Methods Enzymol. 2012;507:155–169. doi: 10.1016/B978-0-12-386509-0.00008-9. [DOI] [PubMed] [Google Scholar]
- 201.Belshaw PJ, Spencer DM, Crabtree GR, Schreiber SL. Controlling programmed cell death with a cyclophilin-cyclosporin-based chemical inducer of dimerization. Chem Biol. 1996;3:731–738. doi: 10.1016/s1074-5521(96)90249-5. [DOI] [PubMed] [Google Scholar]
- 202.Wang J, Chun HJ, Wong W, Spencer DM, Lenardo MJ. Caspase-10 is an initiator caspase in death receptor signaling. Proc Natl Acad Sci USA. 2001;98:13884–13888. doi: 10.1073/pnas.241358198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Ramos CA, Asgari Z, Liu E, Yvon E, Heslop HE, Rooney CM, Brenner MK, Dotti G. An inducible caspase 9 suicide gene to improve the safety of mesenchymal stromal cell therapies. Stem Cells. 2010;28:1107–1115. doi: 10.1002/stem.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Barese CN, Felizardo TC, Sellers SE, Keyvanfar K, Di Stasi A, Metzger ME, Krouse AE, Donahue RE, Spencer DM, Dunbar CE. Regulated apoptosis of genetically modified hematopoietic stem and progenitor cells via an inducible caspase-9 suicide gene in rhesus macaques. Stem Cells. 2015;33:91–100. doi: 10.1002/stem.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Dey D, Evans GRD. Suicide Gene Therapy by Herpes Simplex Virus-1 Thymidine Kinase (HSV-TK) Vol InTech. 2011;2:65–76. [Google Scholar]
- 206.Lupo-Stanghellini MT, Provasi E, Bondanza A, Ciceri F, Bordignon C, Bonini C. Clinical impact of suicide gene therapy in allogeneic hematopoietic stem cell transplantation. Hum Gene Ther. 2010;21:241–250. doi: 10.1089/hum.2010.014. [DOI] [PubMed] [Google Scholar]
- 207.Tiberghien P, Ferrand C, Lioure B, Milpied N, Angonin R, Deconinck E, Certoux JM, Robinet E, Saas P, Petracca B, et al. Administration of herpes simplex-thymidine kinase-expressing donor T cells with a T-cell-depleted allogeneic marrow graft. Blood. 2001;97:63–72. doi: 10.1182/blood.v97.1.63. [DOI] [PubMed] [Google Scholar]
- 208.Nasu Y, Saika T, Ebara S, Kusaka N, Kaku H, Abarzua F, Manabe D, Thompson TC, Kumon H. Suicide gene therapy with adenoviral delivery of HSV-tK gene for patients with local recurrence of prostate cancer after hormonal therapy. Mol Ther. 2007;15:834–840. doi: 10.1038/sj.mt.6300096. [DOI] [PubMed] [Google Scholar]
- 209.Trask TW, Trask RP, Aguilar-Cordova E, Shine HD, Wyde PR, Goodman JC, Hamilton WJ, Rojas-Martinez A, Chen SH, Woo SL, et al. Phase I study of adenoviral delivery of the HSV-tk gene and ganciclovir administration in patients with current malignant brain tumors. Mol Ther. 2000;1:195–203. doi: 10.1006/mthe.2000.0030. [DOI] [PubMed] [Google Scholar]
- 210.Goodrich JM, Bowden RA, Fisher L, Keller C, Schoch G, Meyers JD. Ganciclovir prophylaxis to prevent cytomegalovirus disease after allogeneic marrow transplant. Ann Intern Med. 1993;118:173–178. doi: 10.7326/0003-4819-118-3-199302010-00003. [DOI] [PubMed] [Google Scholar]
- 211.Scaife M, Pacienza N, Au BC, Wang JC, Devine S, Scheid E, Lee CJ, Lopez-Perez O, Neschadim A, Fowler DH, et al. Engineered human Tmpk fused with truncated cell-surface markers: versatile cell-fate control safety cassettes. Gene Ther. 2013;20:24–34. doi: 10.1038/gt.2011.210. [DOI] [PubMed] [Google Scholar]
- 212.Ball CR, Pilz IH, Schmidt M, Fessler S, Williams DA, von Kalle C, Glimm H. Stable differentiation and clonality of murine long-term hematopoiesis after extended reduced-intensity selection for MGMT P140K transgene expression. Blood. 2007;110:1779–1787. doi: 10.1182/blood-2006-11-053710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Beard BC, Sud R, Keyser KA, Ironside C, Neff T, Gerull S, Trobridge GD, Kiem HP. Long-term polyclonal and multilineage engraftment of methylguanine methyltransferase P140K gene-modified dog hematopoietic cells in primary and secondary recipients. Blood. 2009;113:5094–5103. doi: 10.1182/blood-2008-09-176412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Olszko ME, Adair JE, Linde I, Rae DT, Trobridge P, Hocum JD, Rawlings DJ, Kiem HP, Trobridge GD. Foamy viral vector integration sites in SCID-repopulating cells after MGMTP140K-mediated in vivo selection. Gene Ther. 2015;22:591–595. doi: 10.1038/gt.2015.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Zychlinski D, Schambach A, Modlich U, Maetzig T, Meyer J, Grassman E, Mishra A, Baum C. Physiological promoters reduce the genotoxic risk of integrating gene vectors. Mol Ther. 2008;16:718–725. doi: 10.1038/mt.2008.5. [DOI] [PubMed] [Google Scholar]