

Abstract
The tunicate Ciona intestinalis, an invertebrate chordate, has recently emerged as a powerful model organism for gene regulation analysis. However, few studies have been conducted to identify and characterize its transcription start sites (TSSs) and promoters at the genome-wide level. Here, using TSS-seq, we identified TSSs at the genome-wide scale and characterized promoters in C. intestinalis. Specifically, we identified TSS clusters (TSCs), high-density regions of TSS-seq tags, each of which appears to originate from an identical promoter. TSCs were found not only at known TSSs but also in other regions, suggesting the existence of many unknown transcription units in the genome. We also identified candidate promoters of 79 ribosomal protein (RP) genes, each of which had the major TSS in a polypyrimidine tract and showed a sharp TSS distribution like human RP gene promoters. Ciona RP gene promoters, however, did not appear to have typical TATA boxes, unlike human RP gene promoters. In Ciona non-RP promoters, two pyrimidine-purine dinucleotides, CA and TA, were frequently used as TSSs. Despite the absence of CpG islands, Ciona TATA-less promoters showed low expression specificity like CpG-associated human TATA-less promoters. By using TSS-seq, we also predicted trans-spliced gene TSSs and found that their downstream regions had higher G+T content than those of non-trans-spliced gene TSSs. Furthermore, we identified many putative alternative promoters, some of which were regulated in a tissue-specific manner. Our results provide valuable information about TSSs and promoter characteristics in C. intestinalis and will be helpful in future analysis of transcriptional regulation in chordates.
To understand how transcription occurs and how gene expression is regulated, it is necessary to discover and characterize promoter regions. The high-throughput sequencing in combination with oligo-capping (Maruyama and Sugano 1994; Suzuki et al. 1997) or the cap trapper (Carninci et al. 1996) method, which generates millions of 5′ sequences derived from 5′ capped mRNAs transcribed by RNA polymerase II, have enabled us to identify transcription start sites (TSSs) on a genome-wide scale and have contributed to the identification and characterization of core promoters (Suzuki et al. 2001; Carninci et al. 2005; Kawaji et al. 2006; van Heeringen et al. 2011). Several studies using the high-throughput sequencing method have characterized core promoters in terms of TSS distribution and associated motifs and have shown that there are several promoter classes (Carninci et al. 2006; Yamamoto et al. 2009; Zhao et al. 2011).
A genome-wide study of human promoters has defined two distinct promoter classes based on the shape of TSS distribution: sharp-type promoters in which transcription occurs within a narrow genomic region and broad-type promoters in which TSSs are dispersed over a larger genomic region. Sharp- and broad-type promoters are more likely to be associated with TATA boxes and CpG islands, respectively (Carninci et al. 2006). These two promoter classes have also been found in Drosophila melanogaster (Rach et al. 2009; Ni et al. 2010; Hoskins et al. 2011), but there is a difference in the associated motifs between human and Drosophila; the broad-type promoters in Drosophila are more likely to be associated with nonpositionally fixed motifs such as a DNA replication-related element (DRE) (Ni et al. 2010). In human and Drosophila, it has been shown that broad-type promoters exhibit more precise nucleosome positioning than sharp-type promoters (Nozaki et al. 2011; Rach et al. 2011), and human broad-type promoters have been shown to exhibit a 10.5-bp periodic distribution of WW (where W is A or T) motifs at the region corresponding to the position of the +1 nucleosome downstream from the dominant TSS (Forrest et al. 2014). As for ribosomal protein (RP) gene promoters, in human and Drosophila, it has been shown that they possess a polypyrimidine initiator motif, where transcription starts with a cytosine nucleotide, and exhibit a sharp-type TSS distribution (Yoshihama et al. 2002; Perry 2005; Yamashita et al. 2008; Parry et al. 2010).
The tunicate Ciona intestinalis, which is an invertebrate chordate, has recently emerged as a powerful model organism for biological research (Sasakura et al. 2012; Stolfi and Christiaen 2012). The draft genome sequence of C. intestinalis was published in 2002 (Dehal et al. 2002), and the improved genome assembly, called the Kyoto Hoya (KH) assembly, was released in 2008 (Satou et al. 2008). The genome is ∼160 Mbp in size (Dehal et al. 2002) and has approximately 15,000 genes according to the KH gene model available in the Ghost database (Satou et al. 2005), indicating that C. intestinalis has a considerably smaller genome and more compactly organized genes than vertebrates. This compactness allows us to search a smaller space to discover cis-regulatory elements (Johnson et al. 2005; Kusakabe 2005; Irvine 2013). In addition, it has been shown that tunicates are the closest invertebrate relatives of human (Delsuc et al. 2006; Putnam et al. 2008), emphasizing their importance as model organisms for elucidating regulatory programs in chordates and their evolution after the divergence of the tunicate and vertebrate lineages. Moreover, reporter genes can be easily introduced into fertilized eggs of C. intestinalis using electroporation techniques, facilitating the identification of cis-regulatory elements (Corbo et al. 1997; Takahashi et al. 1999; Di Gregorio and Levine 2002; Harafuji et al. 2002; Johnson et al. 2004; Kusakabe et al. 2004). These potential advantages make C. intestinalis a very useful organism to analyze chordate gene regulation. However, few studies have been conducted to identify and characterize TSSs and promoters at the genome-wide level in C. intestinalis.
In C. intestinalis, a different type of splicing, called spliced leader (SL) trans-splicing, occurs (Vandenberghe et al. 2001; Ganot et al. 2004; Hastings 2005). Approximately half of its genes undergo SL trans-splicing (Satou et al. 2006). In SL trans-splicing, the original 5′ end sequence of a pre-mRNA, called an outron, is replaced by a short noncoding RNA, known as an SL, with a spliceosomal mechanism (Vandenberghe et al. 2001). Thus, the 5′ ends of SL trans-spliced mature mRNAs do not represent original TSSs. Currently, the KH gene model does not provide the TSS of a given SL trans-spliced gene, making it difficult to analyze cis-regulatory regions of SL trans-spliced genes. Matsumoto et al. (2010) showed that there are many genes that give rise to both trans-spliced and non-trans-spliced mRNAs over a very wide range of fractional trans-splicing rates; they can be grouped into frequently trans-spliced genes and infrequently trans-spliced genes. This suggests that, for infrequently trans-spliced genes and frequently trans-spliced but highly expressed genes, TSS-seq possibly captures 5′ ends of not only mRNAs that have undergone trans-splicing but also mRNAs that have not undergone trans-splicing and have retained regions corresponding to outrons. Indeed, Khare et al. (2011) identified and validated the TSS of a trans-spliced gene (the Troponin I gene) by using TSS-seq and another approach. TSS-seq therefore would provide a way to identify TSSs of some trans-spliced genes.
Here, we identified TSSs at the genome-wide level using multiple samples derived from various tissues and developmental stages in C. intestinalis. We then identified TSS clusters (TSCs), which are high-density regions of TSSs, by clustering TSSs to determine promoter regions on the genome. TSCs are not equal to promoters. Every TSC, however, should accompany a promoter region on or near it for ensuring the transcription start from it. Thus, in this study, we do not distinguish between the words “TSC” and “promoter” for brevity. The identified TSCs were used to characterize promoters in C. intestinalis. Because the studies of human promoters have been performed by using cap analysis gene expression (CAGE) data, we also reanalyzed human TSSs obtained by using TSS-seq to compare the general characteristics of TSSs and promoters between C. intestinalis and human. In addition, we predicted candidate promoters of SL trans-spliced genes based on our data, and identified putative alternative promoters in C. intestinalis.
Results
Identification of TSSs and trans-splice acceptor sites
To identify TSSs and trans-splice acceptor sites (TASs) in C. intestinalis, we mapped TSS-seq reads, which were obtained from four different tissue samples (ovary, heart, body wall muscle, neural complex) and larva (see Methods). The raw reads were preprocessed and classified into SL(−) reads and SL(+) reads, which were derived from the 5′ end sequence of non-trans-spliced mRNAs and trans-spliced mRNAs after removing SL sequences, respectively (Supplemental Table 1; see Supplemental Methods). These two types of reads were mapped to the reference genome separately (see Supplemental Methods). The 5′ end positions of uniquely mapped SL(−) reads and SL(+) reads were considered candidate TSSs and TASs, respectively (Supplemental Table 2). For human candidate TSSs, we used the data of 15 adult tissues in the database of TSSs (DBTSS) (see Supplemental Methods; Yamashita et al. 2012).
Identification of TSCs and TAS clusters
In both C. intestinalis and human, we identified TSCs, which are high-density regions of candidate TSSs, by clustering candidate TSSs to estimate the promoters from which the TSSs originate (see Methods). The TSCs with a small number of tags (fewer than 100 tags) were not used in subsequent analysis because they might be derived from erroneously oligo-capped truncated transcripts or cryptic transcripts, which are thought to be inherent to the basic transcriptional machinery (Yamashita et al. 2011). This clustering and selection yielded 9792 and 15,498 TSCs in C. intestinalis and human, respectively. These TSCs were used as an initial set of TSCs.
In C. intestinalis, it has been reported that there are many nearest-neighbor pairs of TASs that are ≤50 nucleotides (nt) apart. The distribution of the distance between the major and minor alternative sites of the pair shows strong maxima at the +3 position, where the zero position represents major sites (Matsumoto et al. 2010). In Matsumoto et al. (2010), it was also suggested that the alternate use of short-interval acceptor sites reflects a stochastic aspect of the splicing mechanism and would have no impact on the structure of encoded proteins. This result and suggestion led us to cluster candidate TASs that are close to each other and consider each cluster as a TAS. In C. intestinalis, we identified clusters of TASs, which we call TACs, by clustering candidate TASs using a 4-bp sliding window. TACs with a small number of tags (fewer than 100 tags) were removed because they might be derived from TSS-seq reads caused by sequencing errors. In addition, TACs with AG immediately upstream of the representative position (the most frequent TAS in each TAC) were selected and used in subsequent analysis because the AG motif is a feature of TASs (Agabian 1990; Nilsen 1993). This clustering and selection yielded 5373 TACs (Supplemental Table 3). Most (88%) of them were located at known TASs. The other TACs, which were not located at known TASs, were considered to represent new TASs.
Identification of RP gene TSSs
We first searched for TSSs of RP genes in C. intestinalis. In human and Drosophila, it has been known that TSSs of RP genes have special characteristics; transcription of RP genes preferentially starts from a polypyrimidine initiator motif and each RP promoter shows a sharp TSS distribution (Yoshihama et al. 2002; Perry 2005; Yamashita et al. 2008; Parry et al. 2010). It is therefore expected that TSSs of RP genes can be determined more clearly than those of other genes. First, to determine the gene loci of all 79 RP genes in the KH assembly, we identified orthologous genes of all 79 human RPs in C. intestinalis. Although 78 of 79 RP orthologous genes were identified by BLAST and KH gene annotations (for BLAST, see Supplemental Methods), we could not identify the orthologous gene of human RPL41 due to the lack of annotation of Ciona Rpl41 in the KH model. To identify the gene locus of Rpl41, we mapped the cDNA sequence of Rpl41 obtained from the Ribosomal Protein Gene database (RPG) (Nakao et al. 2004) by the BLAST-like alignment tool (BLAT) (Kent 2002) and found that it was located within the KH.C9.469 gene locus. We then manually inspected all the 79 RP genes and successfully identified their candidate promoters (79 TSCs) in the initial set of TSCs, each of which had the representative TSS (the most frequent TSS) in a polypyrimidine sequence and showed a sharp TSS distribution.
Most (72/78) of the representative TSSs identified for annotated RP genes were exactly located at their annotated TSSs, strongly suggesting that the 72 representative TSSs, which start from polypyrimidine tracts, represent true major RP gene TSSs. On the other hand, those of six RP genes (Rps2, Rps5, Rps21, Rpl21, Rpl29, and Rpl37) were ≥15 nt apart from their annotated TSSs. However, we regarded the representative TSSs as the strongest candidates of major TSSs of the six RP genes, because (1) unlike the annotated TSSs of the six RP genes, they were found in polypyrimidine tracts as well as the major TSSs of the 72 RP genes and (2) they had a substantial number of mapped TSS-seq tags compared with the annotated TSSs, including nearby regions (Supplemental Figs. 1–6). Notably, the representative TSS of Rpl21 was found ∼1700 bp upstream of its annotated TSS. This large discrepancy is because Rpl21 is the second gene in an operon (KHOP.805). The Rpl21 promoter (TSC) was not located near the 5′ end of the Rpl21 gene; rather, it was near the 5′ end of the operon, and a TAC was located at the 5′ end of the Rpl21 (Supplemental Fig. 7), suggesting that the Rpl21 is transcribed as a polycistronic form and is resolved by SL trans-splicing into a monocistronic transcript that encodes the RP.
Based on these results, we considered that the 79 TSCs are derived from RP gene promoters and that their representative TSSs are the major TSSs of RP genes. For this reason, the 79 TSCs will be used to characterize RP gene promoters in this study (for the complete list of the major TSSs of 79 RP genes, see Supplemental File 1). Also, they will not be subject to any filtering of TSCs performed in subsequent sections.
One-base pair-width TSCs
In the initial set of TSCs, we found many TSCs in intergenic regions in both C. intestinalis and human (Supplemental Tables 4, 5). Remarkably, they contained many 1-bp-width TSCs (Supplemental Fig. 8A), and the 1-bp-width TSCs in intergenic regions exhibited a clear CTGG motif (Supplemental Fig. 8B). However, these TSCs with CTGG motif were likely to be generated by mishybridization (nonspecific hybridization) of 5′ oligos in TSS-seq (Supplemental Fig. 9), and therefore were removed from further analysis (see Supplemental Methods; Supplemental Tables 6, 7).
After removing the CTGG TSCs, we found that there were still many 1-bp-width TSCs, especially in introns and intergenic regions in both C. intestinalis and human (Supplemental Fig. 10). Notably, these 1-bp-width TSCs showed A+T-rich downstream regions (Supplemental Fig. 8C), and many 1-bp-width TSCs with A+T-rich downstream regions were found in both C. intestinalis and human (Supplemental Fig. 11). In addition to the A+T-rich TSCs, another type of 1-bp-width TSC was found near splice donor sites on the reverse strand or their vicinity in C. intestinalis (Supplemental Fig. 8D). These two types of 1-bp-width TSCs did not exhibit a pyrimidine-purine (PyPu) motif (Supplemental Fig. 12), a known signature of human and C. intestinalis promoters (Carninci et al. 2006; Okamura et al. 2011), at the −1,0 position, where the zero position represents the representative TSS. Although it is unclear whether these two types of 1-bp-width TSCs are noise or atypical promoters, we removed them in both C. intestinalis and human as well to increase the reliability of our data set (see Supplemental Methods; Supplemental Tables 6, 7). Other 1-bp-width TSCs were included in subsequent analysis.
TSCs in CDSs and 3′ UTRs
We also found many TSCs in coding DNA sequences (CDSs) and 3′ untranslated regions (UTRs) in both C. intestinalis and human (Supplemental Tables 4, 5). For each CDS, we calculated the proportion of the region covered by TSCs, and found that >50% was covered by TSCs in most (80%) of the CDSs (Supplemental Figs. 13A, 14A), indicating that TSS-seq tags are broadly distributed over the CDSs. In addition, there were many transcript models in which a wide range of the entire coding region was covered by TSCs, while the entire intron was almost not (Supplemental Figs. 13B, 14B). It seems that the TSS-seq tags were specifically distributed in coding regions in many transcripts. The TSCs in CDSs and 3′ UTRs had an initiator motif that was quite distinct from the PyPu initiator motif (Supplemental Figs. 13C, 14C). In order to increase the reliability of our data set, we removed TSCs in CDSs and 3′ UTRs from subsequent analysis in both C. intestinalis and human (see Supplemental Methods; Supplemental Tables 6, 7).
Core promoter elements in C. intestinalis promoters
After removing the three types of 1-bp-width TSCs and TSCs in CDSs and 3′ UTRs, we obtained 1844 TSCs at known (annotated) TSSs besides the 79 RP TSCs in C. intestinalis (Supplemental Table 8). Because of the overlap between these 1844 TSCs and known TSSs, they were considered to be reliable TSCs and represent known promoters. We therefore used a total of 1923 TSCs, including the 79 RP TSCs, to investigate the characteristics of promoters. Similarly, we obtained 5073 TSCs at known TSSs in human (Supplemental Table 9) and used them to compare promoter characteristics between Ciona and human. The width of most of the TSCs was <100 bp in both C. intestinalis and human (Supplemental Fig. 15), which is consistent with a previous study of mammalian promoters (Forrest et al. 2014).
We examined the positions of the best-known core promoter element, TATA box, in C. intestinalis promoters. The positions of TATA boxes were predicted using TRANSFAC's MATCH program (see Supplemental Methods). TATA boxes were preferentially positioned at −32 to −29 in C. intestinalis, as well as in human (Fig. 1). Based on this observation, promoters with predicted TATA boxes within −32 to −29 were defined as TATA-containing promoters.
Figure 1.
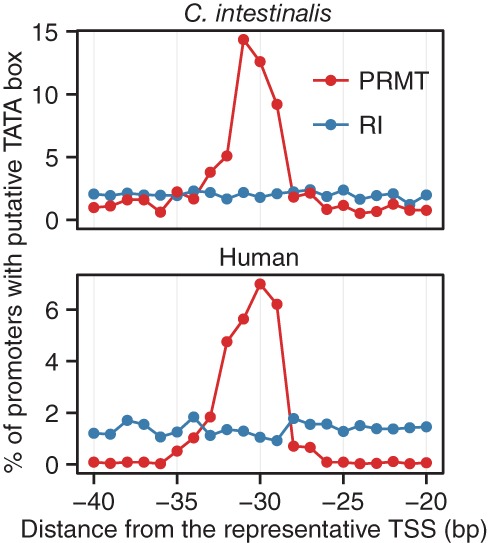
Distribution of TATA boxes. The positions of putative TATA boxes were examined in core promoter regions. The x- and y-axes represent the distance from the representative TSS and the percentage of promoters with the putative TATA box at a given position, respectively. In addition to promoter sequences, the positions of putative TATA boxes in random intergenic regions are also shown (blue line). Intergenic regions (3000 and 6000) were randomly extracted from the C. intestinalis and human reference genomes, respectively. (PRMT) Promoter regions, (RI) random intergenic regions.
Unlike TATA boxes, other known core promoter elements (BRE, XCPE1, DCE, MTE, DPE) (Juven-Gershon et al. 2008) and four Drosophila core promoter elements (DRE, motifs 1, 6, and 7) (Ohler et al. 2002) were predicted in a very small percentage of promoters and did not show positional preference at their known locations in C. intestinalis promoters (Supplemental Fig. 16). In addition, the distribution of them in promoter sequences was not clearly distinguished from that in random intergenic regions, suggesting that they are not overrepresented in Ciona promoters.
Characterization of RP gene promoters
We characterized Ciona RP gene promoters using the 79 RP TSCs. Because TSSs of 79 human RP genes have been already analyzed in a previous study (Perry 2005), we used them as the representative TSSs of human RP gene promoters.
As described in a previous section, we identified all 79 RP gene promoters (TSCs), each of which has a sharp TSS distribution and a polypyrimidine tract (Fig. 2A; Supplemental Figs. 17, 18). Only the Rplp1 promoter showed a somewhat different sharp TSS distribution from the other RP gene promoters. This promoter had two polypyrimidine tracts ∼30 bp apart from each other, and transcription preferentially started from C in both polypyrimidine tracts (Fig. 2A). This type has also been observed in human (e.g., RPL39), where transcription begins at different cytosines within separate polypyrimidine tracts (Yoshihama et al. 2002).
Figure 2.
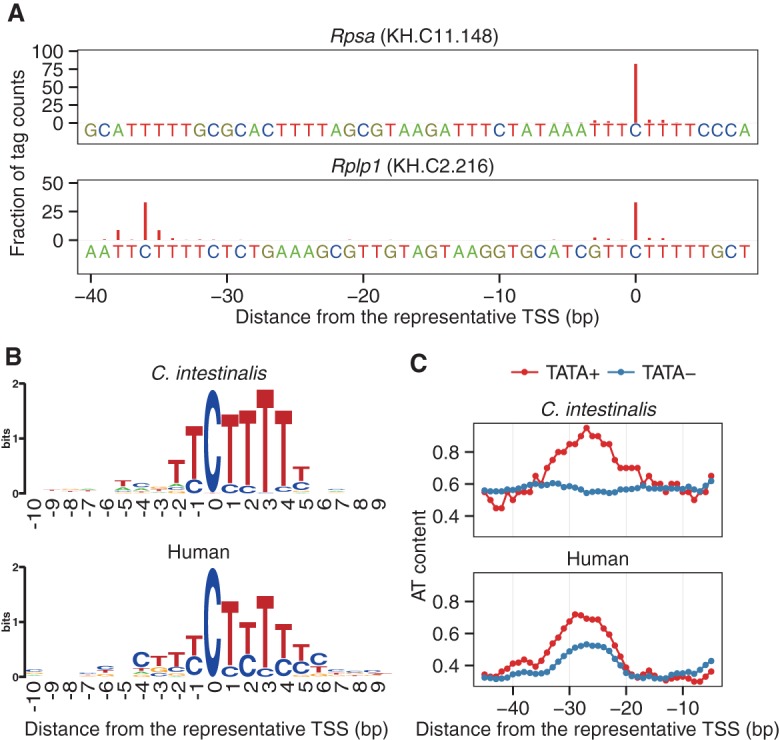
Characterization of ribosomal protein (RP) gene promoters. (A) TSS distributions and polypyrimidine tracts of RP gene promoters. The TSS distributions of the Rpsa and Rplp1 promoters are shown as examples. The nucleotides at the corresponding positions, including polypyrimidine tracts, are also shown. (B) Polypyrimidine initiator of RP gene promoters. The sequences from −10 to +9 of the 79 RP gene promoters were aligned relative to the representative TSS. The TSSs reported by Perry (2005) were used as the representative TSSs of human RP gene promoters. (C) Distribution of the A+T content of RP gene promoters. The A+T content was calculated by using a 10-bp sliding window. TATA+ and TATA− represent TATA-containing and TATA-less RP gene promoters, respectively.
The representative TSSs of RP genes corresponded to C in all the 79 RP genes except Rpl22. In the Rpl22 promoter, it was located at thymine, which was 2 nt downstream from C in a polypyrimidine tract (Supplemental Fig. 19). We aligned the core promoter sequences relative to the representative TSS and found a well-conserved polypyrimidine initiator motif in C. intestinalis, as well as in human (Fig. 2B). The central 6-nt polypyrimidine sequence from −1 to +4 seems to be more strictly conserved in C. intestinalis than in human.
The presence of TATA boxes in RP gene promoters was examined based on our definition of TATA-containing promoters. Of the 79 RP gene promoters, only two were predicted to have TATA boxes in C. intestinalis, whereas there are 16 RP gene promoters in human. We also investigated the A+T-richness of TATA-less RP gene promoters around the −30 position. In human, TATA-less RP gene promoters exhibited A+T-richness around the −30 position like TATA-containing RP gene promoters (Fig. 2C), suggesting that human RP gene promoters often have TATA boxes or TATA-like sequences as previously reported in (Yoshihama et al. 2002; Perry 2005). On the other hand, in C. intestinalis, unlike the two TATA-containing RP gene promoters, TATA-less RP gene promoters did not exhibit A+T-richness around the −30 position (Fig. 2C).
Characterization of nonribosomal protein gene promoters
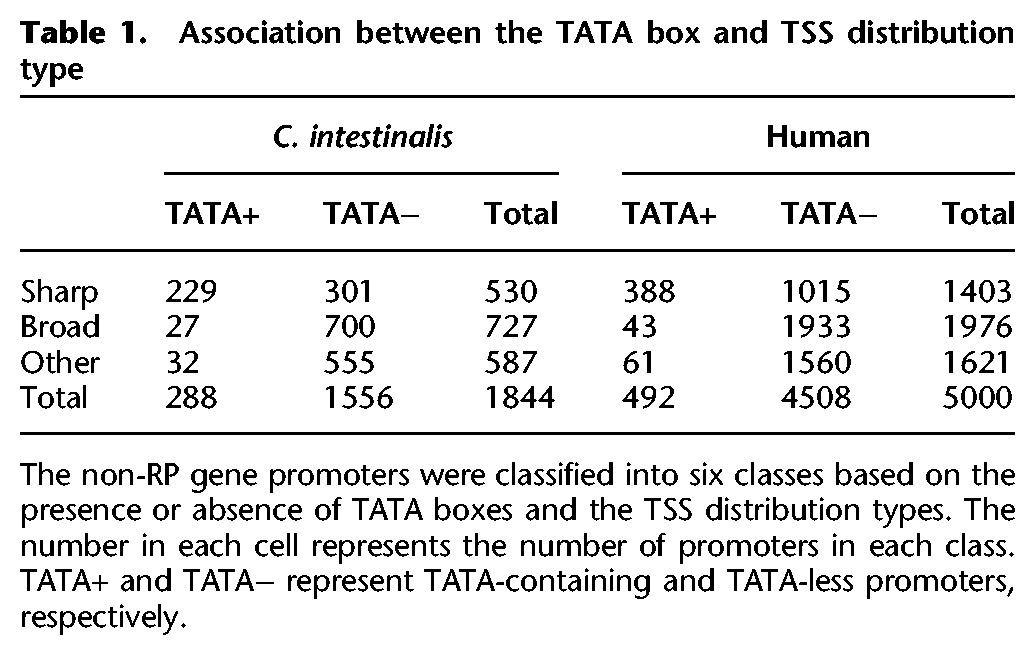
We characterized nonribosomal protein (non-RP) gene promoters in terms of core promoter elements and their TSS distribution. Of the TSCs at known TSSs, 1844 and 5000 TSCs at non-RP gene TSSs were used for the characterization of non-RP gene promoters in C. intestinalis and human, respectively. We first classified the non-RP gene promoters based on TSS distribution types and the presence or absence of TATA boxes to examine the association between these characteristics. The non-RP gene promoters were divided into three classes based on their TSS distribution types: “sharp,” “broad,” and “other” (see Supplemental Methods; Supplemental Fig. 20). The “other” group represents promoters that were not classified into either the “sharp” or “broad” category because of the ambiguity of the TSS distribution shape. They were further classified into TATA-containing and TATA-less promoters based on the presence or absence of TATA boxes (Table 1). In both C. intestinalis and human, most TATA-containing promoters showed a sharp TSS distribution, and almost all broad-type promoters were TATA-less promoters. These results were consistent with the fact that TATA-binding protein is responsible for specifying the exact position of transcription initiation (Baumann et al. 2010). However, in spite of the absence of TATA boxes, TATA-less promoters did not always show a broad TSS distribution; there were many TATA-less promoters with a sharp TSS distribution. In this study, we examined the characteristics of the three main promoter classes: TATA-containing sharp-type promoters, TATA-less sharp-type promoters, and TATA-less broad-type promoters. The “other” promoters were not considered in further analysis because their TSS distribution shapes seemed ambiguous.
Table 1.
Association between the TATA box and TSS distribution type
We investigated which dinucleotides were preferentially used as TSSs. In C. intestinalis, three PyPu dinucleotides (CA, TA, and TG) were significantly used as TSSs in all the three classes compared with their background frequencies in the genome (Bonferroni-corrected P < 0.01; binomial test) (Fig. 3). This characteristic was conserved between C. intestinalis and human, although CG was also often used as TSSs in human. Of the four PyPu dinucleotides, CA and TA were more frequently used as TSSs in C. intestinalis than the others, whereas only CA was dominant in human. We also found differences in TSS usage between sharp-type and broad-type promoters. The sharp-type promoters showed higher TSS usage of CA than the broad-type promoters. Instead, the other PyPu dinucleotides (TA, TG, and CG) were more frequently used in the broad-type promoters than the sharp-type promoters. This difference seems to be conserved between C. intestinalis and human (Supplemental Fig. 21).
Figure 3.

Frequency of dinucleotides used as TSSs. The frequencies of 16 dinucleotides at the −1,0 position, where the zero position represents the representative TSS, were examined in non-RP gene promoters. Each bar represents the percentage of each dinucleotide. The percentage of each dinucleotide in the genome sequence is also shown. To examine which dinucleotides were used as TSSs in each promoter class, we compared the percentage of each dinucleotide in each promoter class and the genome, and we evaluated the difference using the binomial test. (*) P < 0.05 and (**) P < 0.01, Bonferroni-corrected.
In human, it is known that broad-type and TATA-less promoters are associated with CpG islands and ubiquitously expressed genes (Yamashita et al. 2005; Carninci et al. 2006; Yang et al. 2007). Indeed, our study showed that human TATA-less promoters had a significantly higher CpG content and lower expression specificity than TATA-containing promoters (Bonferroni-corrected P < 0.01; Mann-Whitney U test) (Fig. 4). On the other hand, it is known that C. intestinalis does not seem to have CpG islands (Okamura et al. 2011). In this study, we examined whether there are differences in the CpG content of the promoter classes. We did not find significant differences in the CpG content of the three promoter classes, suggesting that neither TATA boxes nor TSS distribution types are associated with CpG dinucleotides in C. intestinalis. Nevertheless, like human TATA-less promoters, Ciona TATA-less promoters showed significantly lower expression specificity than TATA-containing promoters (Bonferroni-corrected P < 0.01; Mann-Whitney U test) (Fig. 4).
Figure 4.
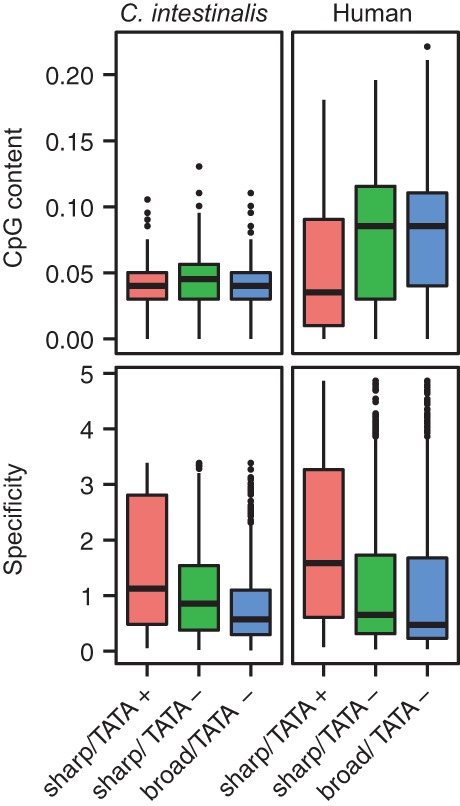
CpG content and expression specificity. The CpG content was calculated in each core promoter sequence (−100 to +99) in each promoter class. The CpG content was defined as the number of CpG sites divided by (N − 1), where N is the length of the core promoter sequence (200 bp). The expression specificity was evaluated by relative entropy (see Supplemental Methods).
A recent analysis of human promoters using CAGE data with greater sequencing depth showed that broad-type promoters exhibit more precise nucleosome positioning than sharp-type promoters and a 10.5-bp periodic distribution of WW motifs at +1 nucleosome position (Forrest et al. 2014). Also in this study, we observed the periodic distribution of WW motifs in human broad-type promoters especially in the +50 to +90 region, although the periodicity was not clearer than that in the previous study due to the lower sequencing depth of the TSS-seq data (Supplemental Fig. 22). The periodic distribution of WW motifs was also found in C. intestinalis broad-type promoters, and its periodicity in the +120 to +210 region seemed to be clearer than sharp-type promoters (Supplemental Fig. 22), suggesting that broad-type promoters show more precise nucleosome positioning than sharp-type promoters in C. intestinalis.
Identification of putative promoters in C. intestinalis
In addition to the TSCs at known TSSs that were used for promoter characterization analysis above, many TSCs were found in 5′ UTRs, introns, and intergenic regions in C. intestinalis (Supplemental Table 8). Because these TSCs were not supported by annotated TSSs, their reliability was considered to be lower than that of the TSCs at known TSSs. To check the quality of them, we examined their initiator motifs and found that they had relatively conserved PyPu initiator motifs (Supplemental Fig. 23), suggesting that many of them are true TSCs. To increase their reliability as much as possible for subsequent analysis, we selected the TSCs with the dinucleotides TA, CA, or TG at their peak TSSs because most (88%) of non-RP promoters used these three PyPu dinucleotides as TSSs in C. intestinalis, as shown in Figure 3. The peak TSSs of a TSC were defined as all TSSs with a frequency that was more than half of that of the most frequent TSS. These selected TSCs were considered to be putative promoters, which are newly identified promoters and have not been annotated in the KH model (version 2013). Many putative promoters were found in 5′ UTRs, introns, and intergenic regions. Some putative promoters (32 TSCs) overlapped with known TASs (Supplemental Fig. 24). In the subsequent sections, a set of the selected TSCs and the TSCs at known TSSs, including the 79 RP TSCs, is referred to as a final set of TSCs (Supplemental Table 6).
Candidate promoters of SL trans-spliced genes
We predicted candidate promoters of some SL trans-spliced genes by selecting pairs of TSCs and TACs. Given the fact that there are frequently and infrequently trans-spliced genes (Matsumoto et al. 2010) and the possibility that TSS-seq captures the 5′ ends of transcripts with and without the original 5′ regions (Khare et al. 2011), we can expect that, for infrequently trans-spliced genes and frequently trans-spliced but highly expressed genes, there are not only TACs at the 5′ ends of their SL-type transcript models but also TSCs upstream of them. In these cases, both of the clusters (the TAC and the upstream TSC) may show the same or similar expression specificity because they are derived from the same trans-spliced gene. We therefore searched for pairs of clusters that were significantly highly expressed in the same sample. The significance of expression was evaluated by a relative entropy and hypergeometric test (see Supplemental Methods). The TACs (Supplemental Table 3) and the final set of TSCs were used in this search. The minimum and maximum distances between the TAC and the upstream TSC, that is, the minimum and maximum length of outron, were set to 51 and 2000 bp, respectively. The 51-bp lower limit was chosen based on the report showing that synthetic AU-rich RNA, ≥51 nt, placed upstream of a 3′ splice site results in efficient trans-splicing in Caenorhabditis elegans (Conrad et al. 1995). The 2000-bp upper limit was derived from the report showing that most (>90%) of outrons are <2000 bp in C. elegans (Kruesi et al. 2013). In this study, the outron means discarded 5′ end regions of “non-operon-type” trans-spliced genes. The pairs of clusters that correspond to annotated “operon-type” trans-spliced genes were excluded in this search.
We found 264 pairs of clusters with the same expression specificity. The predicted pairs were classified into two unannotated-operon-type pairs and 262 non-operon-type pairs (for the definition of these two types, see Supplemental Methods). The unannotated-operon-type pairs may indicate operons that have not been annotated yet, and their TSCs may represent operon gene promoters. The mean distance between the TACs and the upstream TSCs of the non-operon-type pairs, which is the mean length of putative outrons, was 438 bp (Supplemental Fig. 25). The peak of the outron length distribution was found to be ∼100 bp, and many outrons were <500 bp. These characteristics of outron length were similar to those in C. elegans (Kruesi et al. 2013). The putative outrons had similar A, T, and C content as introns but had significantly higher G content than introns (Bonferroni-corrected P < 0.01; Mann-Whitney U test) (Supplemental Fig. 26).
The 262 non-operon-type pairs consisted of 262 TACs and 233 TSCs. Some TACs can pair with multiple TSCs. The 233 TSCs corresponded to 161 putative promoters and 72 known promoters, including seven RP gene promoters. The 161 putative promoters were considered novel candidate promoters of non-operon-type trans-spliced genes. Figure 5 shows the novel candidate promoter of the trans-spliced transcript (KH.S455.4.v1.A.SL1-1) encoding Heat shock protein beta-1. The TSC 489 bp upstream of TASs was significantly highly expressed in body wall muscle and heart, as well as the TAC at the TASs, and may represent TSSs of the Heat shock protein beta-1 gene. The full list of candidate promoters can be found in Supplemental File 2.
Figure 5.

Candidate promoter of the Heat shock protein beta-1 gene. The TSC and TAC are marked by red and green dotted rectangles, respectively. The arrows indicate annotated TASs. The region of the putative outron is represented by a double-headed arrow. (BWM) Body wall muscle, (NC) neural complex.
Characterization of trans-spliced gene promoters
To examine the difference between trans-spliced gene promoters and non-trans-spliced gene promoters, the 1844 non-RP promoters were classified into five classes: predicted trans-spliced gene promoters, non-trans-spliced gene promoters, annotated operon gene promoters, predicted operon gene promoters, and not-determined (ND) promoters. We examined N1 + N2 content around TSSs, where N1 and N2 are different nucleotides, and found that trans-spliced gene promoters clearly showed higher G+T content than non-trans-spliced gene promoters at regions (+1 to +20) immediately downstream from TSSs (Supplemental Fig. 27). We therefore focused on G+T content and examined whether the high G+T content of trans-spliced gene promoters is conserved regardless of outron length. We found that trans-spliced gene promoters exhibited high G+T content around 10–20 bp downstream from TSSs regardless of their putative outron length (Fig. 6; Supplemental Table 10), and the downstream regions (+1 to +20) of non-trans-spliced gene promoters had significantly low G+T content compared with all the other classes (FDR < 0.05; Mann-Whitney U test). We also applied the same analysis to the 79 RP gene promoters and examined G+T content. Although the number of trans-spliced RP gene promoters was very small and a statistically significant difference was not observed, they showed higher G+T content ∼30 bp downstream from TSSs than non-trans-spliced RP gene promoters (Fig. 6; Supplemental Table 10).
Figure 6.

Distribution of G+T content. Average G+T content was calculated using a 20-bp sliding window in each promoter class. The class of predicted operon gene promoters was not shown because the number of them was very small. The number in the parentheses represents the outron length of trans-spliced genes. (RI) Random intergenic regions.
Identification of putative alternative promoters
We examined alternative promoters in C. intestinalis by assigning the final set of TSCs to the genes to which they belong as follows. First, all of the TSCs except intergenic TSCs were assigned to the genes on which they were located. Second, intergenic TSCs were assigned to the nearest downstream genes if they were located within ≤500 bp of the 5′ ends. In addition, the intergenic TSCs that had been predicted to be candidate promoters of SL trans-spliced genes were assigned to the corresponding genes.
Out of the final set of TSCs (3206 TSCs), 2703 were assigned to 2581 genes. About 4.5% (115/2581) of the genes had two or more alternative promoters (TSCs) (Supplemental Table 11). Supplemental Figure 28 shows the putative alternative promoters of the Betagamma crystallin gene (KH.S605.3) (Shimeld et al. 2005). Interestingly, this gene had two alternative promoters with different expression specificity: One had neural complex–specific expression, and the other had larva-specific expression, suggesting that this gene has two distinct transcriptional mechanisms in larva and the adult neural complex. The larva-specific promoter had a canonical TATA box ∼30 bp upstream, whereas the neural complex–specific promoter did not. Our results provided information about putative alternative promoters and their expression specificity (for details, see Supplemental File 3). These data will be useful for deciphering transcriptional regulatory codes controlling tissue-specific expression in chordates.
Discussion
In this study, we identified candidate TSSs at the genome-wide level in C. intestinalis by applying TSS-seq to a total of five samples. The candidate TSSs were clustered into TSCs to detect arrays of TSSs that appear to originate from the same promoter. As a result, many TSCs were found not only at known TSSs but also in other regions, such as CDSs, 3′ UTRs, and intergenic regions. However, some of the identified TSCs do not appear to be derived from promoters. For instance, we found three types of 1-bp-width TSCs: CTGG, A+T-rich TSCs, and TSCs near splice donor sites on the reverse strand. Zhao et al. (2011) also reported a different type of 1-bp-width TSCs, called ultra-dense TSS distributions, in mouse and human promoter analysis using CAGE data, and they showed that some of them are likely due to mapping errors in the CAGE protocol. The CTGG TSCs, which have a CTGG motif immediately upstream of the representative position, are probably technical artifacts generated by the mishybridization of 5′ oligos in the TSS-seq protocol. It is therefore important to remove the CTGG TSCs when using TSS-seq data. Although we do not know the generation mechanism of the other two types of 1-bp-width TSCs, the transient pausing of RNA polymerase II within A+T-rich regions and the following backtracking and cleavage (Nechaev et al. 2010) might be involved in the generation of the A+T-rich TSCs. In addition to the 1-bp-width TSCs, TSCs in exons, such as CDSs and 3′ UTRs, may be derived from 5′ ends of truncated mRNAs, including recapped RNAs (Schoenberg and Maquat 2009; Yamashita et al. 2011). Considering that TSCs in exons often had their peaks near splice acceptor sites (Supplemental Fig. 29), truncations of mRNAs might occur not after they become mature mRNAs but during splicing events.
Major TSSs of 79 Ciona RP genes were identified for the first time in this study. In the KH model, there are multiple transcript models with different 5′ ends for each RP gene; there are multiple annotated TSSs for each RP gene, and it is unclear which TSSs are major (most frequently used) TSSs. Our results thus provide important and useful information for TSSs of RP gene promoters. Somewhat surprisingly, the major TSSs of six RP genes were not located at any of their annotated TSSs and were considered newly identified TSSs. Interestingly, the Ciona Rpl21 was suggested to undergo operon-type trans-splicing, although RP genes are thought to be infrequently and/or undetectably trans-spliced genes due to the presence of a terminal oligo-pyrimidine (TOP) sequence at their 5′ ends (Matsumoto et al. 2010). This result may suggest that the Ciona SL sequence can function as the TOP, as suggested in Oikopleura dioica (Danks et al. 2015).
Ciona RP gene promoters possessed a well-conserved polypyrimidine initiator motif with a C start site like human RP gene promoters. Especially, the central 6-nt polypyrimidine sequences from −1 to +4 were highly conserved in C. intestinalis. This conserved polypyrimidine initiator has also been found in 52 Drosophila RP gene promoters, and mutational analysis has demonstrated that the central 6-nt sequences are most important for the transcription of RP genes (Parry et al. 2010). The 6-nt pyrimidine region with a C start site probably plays an important role in transcription also in C. intestinalis. However, in the Ciona Rpl22 promoter, transcription did not preferentially start from cytosine but started from thymine 2 nt downstream from the cytosine, suggesting that cytosine is not an absolute start site of RP gene promoters. Although it is unknown why transcription does not start from cytosine, the presence of cytosine nucleotides in all the RP gene polypyrimidine initiators may suggest their importance in the initiators; a poly-thymine initiator without cytosine may not be sufficient for the transcription of Rpl22. Taken together, the cytosine or thymine in polypyrimidine tracts appears to be a dominant start site of Ciona RP genes. However, it seems that transcription sometimes starts from purine nucleotides (A or G) of PyPu sites closely downstream from polypyrimidine tracts at a relatively low frequency (Supplemental Fig. 30). These start sites are considered minor TSSs of RP genes, but the mRNAs transcribed from these minor TSSs do not possess TOP sequences at their 5′ ends, which are thought to serve as a cis-regulatory element that inhibits the binding of translational regulatory proteins or the translational machinery itself (Yamashita et al. 2008). Therefore the post-transcriptional regulation of them may be different from that of mRNAs with TOP sequences.
Most of Ciona RP gene promoters did not appear to have typical TATA boxes around the −30 position, unlike human RP gene promoters. Because the general features of RP gene promoter architecture are usually well conserved in human, chicken, amphibians, and fish (Perry 2005), the absence of TATA boxes in C. intestinalis suggests that RP gene promoters acquired them during an early stage of vertebrate evolution. As a result, we wonder whether RP gene promoters have TATA boxes in primitive vertebrates, such as lamprey. Recent analysis showed that TBP (TATA box- binding protein)-related factor TRF2, but not TBP, is required for transcription of RP genes with polypyrimidine initiator motifs in Drosophila (Wang et al. 2014). Ciona TATA-less RP genes therefore also may use a TRF2-based transcription system. Extensive analysis of overrepresented motifs in RP gene promoters have been done in many species, such as human, Drosophila, yeast, C. elegans, and basal metazoans (Tanay et al. 2005; Roepcke et al. 2006; Ma et al. 2009; Perina et al. 2011; Sleumer et al. 2012). Our preliminary study of C. intestinalis RP gene promoters focused only on a polypyrimidine initiator motif and the best-characterized promoter motif, TATA box. Further detailed analysis of RP gene promoter architecture is required to elucidate the transcriptional mechanism of RP genes in C. intestinalis and to understand the evolution of RP gene promoters in chordates.
In Ciona non-RP promoters, two PyPu dinucleotides (CA and TA) were the most frequently used as TSSs, while only CA was dominantly used in human. This difference probably arises from the A+T-richness of the C. intestinalis genome. Indeed, the T content is higher around the TSS than the A, C, and G content in C. intestinalis promoters (Supplemental Fig. 31). On the other hand, CG was used more often in TSSs in human than in C. intestinalis, reflecting the high GC content of human promoters.
In human, TATA-less promoters had a higher CpG content and lower expression specificity than TATA-containing promoters, suggesting that they are associated with CpG islands and housekeeping genes (Yamashita et al. 2005; Carninci et al. 2006; Yang et al. 2007). In C. intestinalis, TATA-less promoters did not exhibit a high CpG content, supporting the hypothesis that C. intestinalis does not have CpG islands (Okamura et al. 2011). Nevertheless, C. intestinalis TATA-less promoters showed lower expression specificity than TATA-containing promoters, like human TATA-less promoters. These results raise a new question: Do Ciona promoters have a substitute for CpG islands that is associated with housekeeping genes? It is presently unknown what elements play an important role in recruitment of RNA polymerase II into Ciona TATA-less promoters. In human broad-type TATA-less promoters, CpG islands can provide multiple binding sites of the transcription factor SP1 that binds to GC box motifs and recruits TBP to promoters (Butler and Kadonaga 2002; Deaton and Bird 2011). Unknown elements in C. intestinalis TATA-less promoters may be present at multiple sites in promoters as do GC boxes in CpG islands and function as binding sites of unknown factors that recruit general transcription factors. To answer whether such elements exist or not, we need more comprehensive and detailed analyses focusing on TATA-less promoters. At least, known polymerase II promoter elements in the JASPAR database (Sandelin et al. 2004) and Drosophila promoter elements (DRE, motif 1, 6, and 7) (Ohler et al. 2002) were not enriched in either sharp-type or broad-type TATA-less promoters (Supplemental Fig. 32).
In this study, 15.6% (288/1844) of Ciona non-RP promoters were estimated to have TATA boxes. This proportion was significantly higher than that of human non-RP promoters (9.8%). Some of this difference may be explained by a limitation of TATA box prediction; we used PWMs of vertebrate TATA boxes for prediction of Ciona TATA boxes because there are presently no PWMs of TATA boxes for tunicates, including Ciona in the TRANSFAC and the JASPAR database. In this prediction, the sites with a score above a defined threshold, which is predefined by experts for each vertebrate PWM, were predicted to be TATA boxes. However, because the regions around TSSs have higher A+T content in Ciona, the threshold used may be lenient, producing higher false positives; therefore, the real proportion of TATA-containing promoters may be <15% in Ciona. Another possibility is that the number of Ciona promoters used is smaller than that of human promoters. Future studies using more samples would improve this estimation.
Many putative promoters (TSCs that were not located at known TSSs) were found in intergenic regions, suggesting that there are many transcription units that have not been annotated yet in the KH model. Some of these unannotated transcripts may code for noncoding RNAs because a number of candidate noncoding RNAs, including microRNAs, have been identified in C. intestinalis (Sasakura et al. 2012). Interestingly, some putative promoters (TSCs) overlapped with TASs. This result may suggest that the regions near TASs can function as alternative promoters of trans-spliced genes. These trans-spliced genes might not require trans-splicing to be expressed. It has been speculated that one of the functions of trans-splicing is to remove 5′ UTRs that include elements compromising mRNA transport, translation, or stability (Hastings 2005). The transcription from alternative promoters near TASs might be an alternative way to make pre-mRNAs not include deleterious elements at their 5′ UTRs.
In this study, to obtain the putative promoters, we selected only the TSCs with PyPu motifs at frequent TSSs. However, this selection may not be enough to completely remove false TSCs because we did not use additional NGS data, such as ChIP-seq data for histone modifications, which can be used for evidence of active promoters (Lenhard et al. 2012). In addition, it does not consider true TSCs without PyPu motifs; they are not included in a final set of putative promoters, and therefore, the analysis is limited to promoters with PyPu motifs. These are thought to be limitations of our study. However, at least, we confirmed that the selected TSCs exhibited an initiator motif very similar to that of non-RP promoters (Supplemental Fig. 33), suggesting that most of them are true TSCs. They were therefore considered to represent putative promoters.
TSS-seq allowed us to predict TSSs of some trans-spliced genes. A previous report that identified the TSS of the trans-spliced troponin I gene by using the same method as TSS-seq (Khare et al. 2011) and the fact that our TSS-seq tags were found at the reported TSS (Supplemental Fig. 34) indicate that our data are also useful for identification of TSSs of trans-spliced genes. By comparing the promoters of predicted trans-spliced genes and non-trans-spliced genes, we found that the trans-spliced gene promoters exhibited higher G+T content at <20 bp downstream from TSSs than non-trans-spliced gene promoters. The preferential association of G+T-rich regions with trans-spliced gene promoters was observed, not only for genes in general but also within the RP gene subset. The G+T-rich regions downstream from TSSs therefore might play some role in trans-splicing.
The predictions of trans-spliced gene TSSs were performed based on the assumption that pairs of TSCs and TACs have the same expression pattern, but there is a possibility that the rate of trans-splicing of a given gene is controlled differently across tissues. In this case, pairs of TSCs and TACs will not have the same expression pattern, and therefore, our method can miss such pairs. However, whereas the distribution of predicted outron length showed the same properties as that reported in C. elegans (Kruesi et al. 2013), the distribution of distance between pairs of TSCs and TACs that have different expression patterns did not show them (Supplemental Fig. 35). It seems that the assumption is useful for the prediction, at least in this study. Although predicted TSSs are not experimentally validated by additional approaches, these data will contribute to future analysis of cis-regulatory regions of the trans-spliced genes.
Methods
Data sources
In order to determine TSSs in C. intestinalis, we applied TSS-seq (Yamashita et al. 2011) to four different adult tissues (ovary, heart, body wall muscle, and neural complex) and one developmental stage (larva) to obtain five sets of reads data. Adult tissues were obtained from mature adults of C. intestinalis that were collected from harbors in Murotsu, Hyogo, Japan, or were obtained from the National BioResource Project. Larvae were obtained as described previously (Yoshida et al. 2007). The reads data are 36-nt single-end reads.
Identification of TSCs and TACs
TSCs and TACs were identified by clustering candidate TSSs and TASs, respectively. Details of the clustering method are described in Supplemental Methods. Each cluster consisted of up to five and 15 clusters derived from each sample in C. intestinalis and human, respectively. The representative position of each cluster was determined by a majority vote of the most frequent positions obtained from clusters with 100 tags or more derived from each sample. If there were two or more representative positions, the most upstream position was used. Also, of the clusters derived from each sample with the most frequent position as the representative position, the cluster with the greatest number of tags was selected as the representative cluster. The clusters with more than 100 tags in the representative cluster were not used in this study. Unless otherwise noted, the TSS distribution of a TSC indicates that of the representative TSC. The width of a TSC was defined as the distance from the fifth percentile to the 95th percentile of the TSS distribution.
Data access
TSS-seq data from this study have been deposited in the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra/) under accession number SRP063032.
Supplementary Material
Acknowledgments
This study was supported in part by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS; 22310120 and 25290067). Computational resources were provided by the supercomputer system at Human Genome Center, Institute of Medical Science, the University of Tokyo.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.184648.114.
References
- Agabian N. 1990. Trans splicing of nuclear pre-mRNAs. Cell 61: 1157–1160. [DOI] [PubMed] [Google Scholar]
- Baumann M, Pontiller J, Ernst W. 2010. Structure and basal transcription complex of RNA polymerase II core promoters in the mammalian genome: an overview. Mol Biotechnol 45: 241–247. [DOI] [PubMed] [Google Scholar]
- Butler JE, Kadonaga JT. 2002. The RNA polymerase II core promoter: a key component in the regulation of gene expression. Genes Dev 16: 2583–2592. [DOI] [PubMed] [Google Scholar]
- Carninci P, Kvam C, Kitamura A, Ohsumi T, Okazaki Y, Itoh M, Kamiya M, Shibata K, Sasaki N, Izawa M, et al. 1996. High-efficiency full-length cDNA cloning by biotinylated CAP trapper. Genomics 37: 327–336. [DOI] [PubMed] [Google Scholar]
- Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, et al. 2005. The transcriptional landscape of the mammalian genome. Science 309: 1559–1563. [DOI] [PubMed] [Google Scholar]
- Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, Semple CAM, Taylor MS, Engstrom PG, Frith MC, et al. 2006. Genome-wide analysis of mammalian promoter architecture and evolution. Nat Genet 38: 626–635. [DOI] [PubMed] [Google Scholar]
- Conrad R, Lea K, Blumenthal T. 1995. SL1 trans-splicing specified by AU-rich synthetic RNA inserted at the 5′ end of Caenorhabditis elegans pre-mRNA. RNA 1: 164–170. [PMC free article] [PubMed] [Google Scholar]
- Corbo JC, Levine M, Zeller RW. 1997. Characterization of a notochord-specific enhancer from the Brachyury promoter region of the ascidian, Ciona intestinalis. Development 124: 589–602. [DOI] [PubMed] [Google Scholar]
- Danks GB, Raasholm M, Campsteijn C, Long AM, Manak JR, Lenhard B, Thompson EM. 2015. Trans-splicing and operons in metazoans: translational control in maternally regulated development and recovery from growth arrest. Mol Biol Evol 32: 585–599. [DOI] [PubMed] [Google Scholar]
- Deaton AM, Bird A. 2011. CpG islands and the regulation of transcription. Genes Dev 25: 1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehal P, Satou Y, Campbell RK, Chapman J, Degnan B, De Tomaso A, Davidson B, Di Gregorio A, Gelpke M, Goodstein DM, et al. 2002. The draft genome of Ciona intestinalis: insights into chordate and vertebrate origins. Science 298: 2157–2167. [DOI] [PubMed] [Google Scholar]
- Delsuc F, Brinkmann H, Chourrout D, Philippe H. 2006. Tunicates and not cephalochordates are the closest living relatives of vertebrates. Nature 439: 965–968. [DOI] [PubMed] [Google Scholar]
- Di Gregorio A, Levine M. 2002. Analyzing gene regulation in ascidian embryos: new tools for new perspectives. Differentiation 70: 132–139. [DOI] [PubMed] [Google Scholar]
- Forrest AR, Kawaji H, Rehli M, Baillie JK, de Hoon MJ, Haberle V, Lassman T, Kulakovskiy IV, Lizio M, Itoh M, et al. 2014. A promoter-level mammalian expression atlas. Nature 507: 462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganot P, Kallesøe T, Reinhardt R, Chourrout D, Thompson EM. 2004. Spliced-leader RNA trans splicing in a chordate, Oikopleura dioica, with a compact genome. Mol Cell Biol 24: 7795–7805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harafuji N, Keys DN, Levine M. 2002. Genome-wide identification of tissue-specific enhancers in the Ciona tadpole. Proc Natl Acad Sci 99: 6802–6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings KE. 2005. SL trans-splicing: easy come or easy go? Trends Genet 21: 240–247. [DOI] [PubMed] [Google Scholar]
- Hoskins RA, Landolin JM, Brown JB, Sandler JE, Takahashi H, Lassmann T, Yu C, Booth BW, Zhang D, Wan KH, et al. 2011. Genome-wide analysis of promoter architecture in Drosophila melanogaster. Genome Res 21: 182–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine SQ. 2013. Study of cis-regulatory elements in the ascidian Ciona intestinalis. Curr Genomics 14: 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Davidson B, Brown CD, Smith WC, Sidow A. 2004. Noncoding regulatory sequences of Ciona exhibit strong correspondence between evolutionary constraint and functional importance. Genome Res 14: 2448–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Zhou Q, Yagi K, Satoh N, Wong W, Sidow A. 2005. De novo discovery of a tissue-specific gene regulatory module in a chordate. Genome Res 15: 1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juven-Gershon T, Hsu J-Y, Theisen JWM, Kadonaga JT. 2008. The RNA polymerase II core promoter: the gateway to transcription. Curr Opin Cell Biol 20: 253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaji H, Frith MC, Katayama S, Sandelin A, Kai C, Kawai J, Carninci P, Hayashizaki Y. 2006. Dynamic usage of transcription start sites within core promoters. Genome Biol 7: R118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. 2002. BLAT—the BLAST-like alignment tool. Genome Res 12: 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khare P, Mortimer SI, Cleto CL, Okamura K, Suzuki Y, Kusakabe T, Nakai K, Meedel TH, Hastings KE. 2011. Cross-validated methods for promoter/transcription start site mapping in SL trans-spliced genes, established using the Ciona intestinalis troponin I gene. Nucleic Acids Res 39: 2638–2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruesi WS, Core LJ, Waters CT, Lis JT, Meyer BJ. 2013. Condensin controls recruitment of RNA polymerase II to achieve nematode X-chromosome dosage compensation. eLife 2: e00808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusakabe T. 2005. Decoding cis-regulatory systems in ascidians. Zoolog Sci 22: 129–146. [DOI] [PubMed] [Google Scholar]
- Kusakabe T, Yoshida R, Ikeda Y, Tsuda M. 2004. Computational discovery of DNA motifs associated with cell type-specific gene expression in Ciona. Dev Biol 276: 563–580. [DOI] [PubMed] [Google Scholar]
- Lenhard B, Sandelin A, Carninci P. 2012. Metazoan promoters: emerging characteristics and insights into transcriptional regulation. Nat Rev Genet 13: 233–245. [DOI] [PubMed] [Google Scholar]
- Ma X, Zhang K, Li X. 2009. Evolution of Drosophila ribosomal protein gene core promoters. Gene 432: 54–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama K, Sugano S. 1994. Oligo-capping: a simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides. Gene 138: 171–174. [DOI] [PubMed] [Google Scholar]
- Matsumoto J, Dewar K, Wasserscheid J, Wiley GB, Macmil SL, Roe BA, Zeller RW, Satou Y, Hastings KE. 2010. High-throughput sequence analysis of Ciona intestinalis SL trans-spliced mRNAs: alternative expression modes and gene function correlates. Genome Res 20: 636–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao A, Yoshihama M, Kenmochi N. 2004. RPG: the Ribosomal Protein Gene database. Nucleic Acids Res 32: D168–D170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechaev S, Fargo DC, dos Santos G, Liu L, Gao Y, Adelman K. 2010. Global analysis of short RNAs reveals widespread promoter-proximal stalling and arrest of Pol II in Drosophila. Science 327: 335–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni T, Corcoran DL, Rach EA, Song S, Spana EP, Gao Y, Ohler U, Zhu J. 2010. A paired-end sequencing strategy to map the complex landscape of transcription initiation. Nat Methods 7: 521–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen TW. 1993. Trans-splicing of nematode premessenger RNA. Annu Rev Microbiol 47: 413–440. [DOI] [PubMed] [Google Scholar]
- Nozaki T, Yachie N, Ogawa R, Kratz A, Saito R, Tomita M. 2011. Tight associations between transcription promoter type and epigenetic variation in histone positioning and modification. BMC Genomics 12: 416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohler U, Liao GC, Niemann H, Rubin GM. 2002. Computational analysis of core promoters in the Drosophila genome. Genome Biol 3: RESEARCH0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K, Yamashita R, Takimoto N, Nishitsuji K, Suzuki Y, Kusakabe T, Nakai K. 2011. Profiling ascidian promoters as the primordial type of vertebrate promoter. BMC Genomics 12Suppl 3: S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry TJ, Theisen JW, Hsu JY, Wang YL, Corcoran DL, Eustice M, Ohler U, Kadonaga JT. 2010. The TCT motif, a key component of an RNA polymerase II transcription system for the translational machinery. Genes Dev 24: 2013–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perina D, Korolija M, Roller M, Harcet M, Jeličić B, Mikoč A, Ćetković H. 2011. Over-represented localized sequence motifs in ribosomal protein gene promoters of basal metazoans. Genomics 98: 56–63. [DOI] [PubMed] [Google Scholar]
- Perry RP. 2005. The architecture of mammalian ribosomal protein promoters. BMC Evol Biol 5: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam NH, Butts T, Ferrier DE, Furlong RF, Hellsten U, Kawashima T, Robinson-Rechavi M, Shoguchi E, Terry A, Yu JK, et al. 2008. The amphioxus genome and the evolution of the chordate karyotype. Nature 453: 1064–1071. [DOI] [PubMed] [Google Scholar]
- Rach EA, Yuan H-Y, Majoros WH, Tomancak P, Ohler U. 2009. Motif composition, conservation and condition-specificity of single and alternative transcription start sites in the Drosophila genome. Genome Biol 10: R73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rach EA, Winter DR, Benjamin AM, Corcoran DL, Ni T, Zhu J, Ohler U. 2011. Transcription initiation patterns indicate divergent strategies for gene regulation at the chromatin level. PLoS Genet 7: e1001274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roepcke S, Zhi D, Vingron M, Arndt PF. 2006. Identification of highly specific localized sequence motifs in human ribosomal protein gene promoters. Gene 365: 48–56. [DOI] [PubMed] [Google Scholar]
- Sandelin A, Alkema W, Engström P, Wasserman WW, Lenhard B. 2004. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res 32: D91–D94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasakura Y, Sierro N, Nakai K, Inaba K, Kusakabe T. 2012. Genome structure, functional genomics, and proteomics in ascidians. In Genome mapping and genomics in laboratory animals (ed. Denny P, Kole C), Vol. 4, pp. 87–132. Springer, Berlin Heidelberg. [Google Scholar]
- Satou Y, Kawashima T, Shoguchi E, Nakayama A, Satoh N. 2005. An integrated database of the ascidian, Ciona intestinalis: towards functional genomics. Zoolog Sci 22: 837–843. [DOI] [PubMed] [Google Scholar]
- Satou Y, Hamaguchi M, Takeuchi K, Hastings KE, Satoh N. 2006. Genomic overview of mRNA 5′-leader trans-splicing in the ascidian Ciona intestinalis. Nucleic Acids Res 34: 3378–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satou Y, Mineta K, Ogasawara M, Sasakura Y, Shoguchi E, Ueno K, Yamada L, Matsumoto J, Wasserscheid J, Dewar K, et al. 2008. Improved genome assembly and evidence-based global gene model set for the chordate Ciona intestinalis: new insight into intron and operon populations. Genome Biol 9: R51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenberg DR, Maquat LE. 2009. Re-capping the message. Trends Biochem Sci 34: 435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimeld SM, Purkiss AG, Dirks RP, Bateman OA, Slingsby C, Lubsen NH. 2005. Urochordate βγ-crystallin and the evolutionary origin of the vertebrate eye lens. Curr Biol 15: 1684–1689. [DOI] [PubMed] [Google Scholar]
- Sleumer MC, Wei G, Wang Y, Chang H, Xu T, Chen R, Zhang MQ. 2012. Regulatory elements of Caenorhabditis elegans ribosomal protein genes. BMC Genomics 13: 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolfi A, Christiaen L. 2012. Genetic and genomic toolbox of the chordate Ciona intestinalis. Genetics 192: 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Yoshitomo-Nakagawa K, Maruyama K, Suyama A, Sugano S. 1997. Construction and characterization of a full length-enriched and a 5′-end-enriched cDNA library. Gene 200: 149–156. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Tsunoda T, Sese J, Taira H, Mizushima-Sugano J, Hata H, Ota T, Isogai T, Tanaka T, Nakamura Y, et al. 2001. Identification and characterization of the potential promoter regions of 1031 kinds of human genes. Genome Res 11: 677–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Mitani Y, Satoh G, Satoh N. 1999. Evolutionary alterations of the minimal promoter for notochord-specific Brachyury expression in ascidian embryos. Development 126: 3725–3734. [DOI] [PubMed] [Google Scholar]
- Tanay A, Regev A, Shamir R. 2005. Conservation and evolvability in regulatory networks: the evolution of ribosomal regulation in yeast. Proc Natl Acad Sci 102: 7203–7208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heeringen SJ, Akhtar W, Jacobi UG, Akkers RC, Suzuki Y, Veenstra GJC. 2011. Nucleotide composition-linked divergence of vertebrate core promoter architecture. Genome Res 21: 410–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe AE, Meedel TH, Hastings KEM. 2001. mRNA 5′-leader trans-splicing in the chordates. Genes Dev 15: 294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YL, Duttke SH, Chen K, Johnston J, Kassavetis GA, Zeitlinger J, Kadonaga JT. 2014. TRF2, but not TBP, mediates the transcription of ribosomal protein genes. Genes Dev 28: 1550–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto YY, Yoshitsugu T, Sakurai T, Seki M, Shinozaki K, Obokata J. 2009. Heterogeneity of Arabidopsis core promoters revealed by high-density TSS analysis. Plant J 60: 350–362. [DOI] [PubMed] [Google Scholar]
- Yamashita R, Suzuki Y, Sugano S, Nakai K. 2005. Genome-wide analysis reveals strong correlation between CpG islands with nearby transcription start sites of genes and their tissue specificity. Gene 350: 129–136. [DOI] [PubMed] [Google Scholar]
- Yamashita R, Suzuki Y, Takeuchi N, Wakaguri H, Ueda T, Sugano S, Nakai K. 2008. Comprehensive detection of human terminal oligo-pyrimidine (TOP) genes and analysis of their characteristics. Nucleic Acids Res 36: 3707–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita R, Sathira NP, Kanai A, Tanimoto K, Arauchi T, Tanaka Y, Hashimoto S, Sugano S, Nakai K, Suzuki Y. 2011. Genome-wide characterization of transcriptional start sites in humans by integrative transcriptome analysis. Genome Res 21: 775–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita R, Sugano S, Suzuki Y, Nakai K. 2012. DBTSS: DataBase of Transcriptional Start Sites progress report in 2012. Nucleic Acids Res 40: D150–D154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Bolotin E, Jiang T, Sladek FM, Martinez E. 2007. Prevalence of the initiator over the TATA box in human and yeast genes and identification of DNA motifs enriched in human TATA-less core promoters. Gene 389: 52–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida R, Horie T, Tsuda M, Kusakabe TG. 2007. Comparative genomics identifies a cis-regulatory module that activates transcription in specific subsets of neurons in Ciona intestinalis larvae. Dev Growth Differ 49: 657–667. [DOI] [PubMed] [Google Scholar]
- Yoshihama M, Uechi T, Asakawa S, Kawasaki K, Kato S, Higa S, Maeda N, Minoshima S, Tanaka T, Shimizu N, et al. 2002. The human ribosomal protein genes: sequencing and comparative analysis of 73 genes. Genome Res 12: 379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Valen E, Parker BJ, Sandelin A. 2011. Systematic clustering of transcription start site landscapes. PLoS One 6: e23409. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.