

Abstract
TGF-β is a pleiotropic cytokine that accumulates during kidney injuries, resulting in various renal diseases. We have reported previously that TGF-β1 induces the selective up-regulation of mitochondrial Nox4, playing critical roles in podocyte apoptosis. Here we investigated the regulatory mechanism of Nox4 up-regulation by mTORC1 activation on TGF-β1-induced apoptosis in immortalized podocytes. TGF-β1 treatment markedly increased the phosphorylation of mammalian target of rapamycin (mTOR) and its downstream targets p70S6K and 4EBP1. Blocking TGF-β receptor I with SB431542 completely blunted the phosphorylation of mTOR, p70S6K, and 4EBP1. Transient adenoviral overexpression of mTOR-WT and constitutively active mTORΔ augmented TGF-β1-treated Nox4 expression, reactive oxygen species (ROS) generation, and apoptosis, whereas mTOR kinase-dead suppressed the above changes. In addition, knockdown of mTOR mimicked the effect of mTOR-KD. Inhibition of mTORC1 by low-dose rapamycin or knockdown of p70S6K protected podocytes through attenuation of Nox4 expression and subsequent oxidative stress-induced apoptosis by TGF-β1. Pharmacological inhibition of the MEK-ERK cascade, but not the PI3K-Akt-TSC2 pathway, abolished TGF-β1-induced mTOR activation. Inhibition of either ERK1/2 or mTORC1 did not reduce the TGF-β1-stimulated increase in Nox4 mRNA level but significantly inhibited total Nox4 expression, ROS generation, and apoptosis induced by TGF-β1. Moreover, double knockdown of Smad2 and 3 or only Smad4 completely suppressed TGF-β1-induced ERK1/2-mTORactivation. Our data suggest that TGF-β1 increases translation of Nox4 through the Smad-ERK1/2-mTORC1 axis, which is independent of transcriptional regulation. Activation of this pathway plays a crucial role in ROS generation and mitochondrial dysfunction, leading to podocyte apoptosis. Therefore, inhibition of the ERK1/2-mTORC1 pathway could be a potential therapeutic and preventive target in proteinuric and chronic kidney diseases.
Keywords: apoptosis, ERK, mammalian target of rapamycin (mTOR), NADPH oxidase, podocyte, reactive oxygen species (ROS), SMAD transcription factor, TGF-β
Introduction
Podocyte damage is one of the key determinants of the majority of diabetic and non-diabetic glomerular diseases, leading to chronic kidney diseases and end-stage renal disease (1). Leakage of plasma proteins in the urine indicates the onset of renal diseases that are associated with podocyte injury (2–4). Recent evidence has shown that mammalian target of rapamycin (mTOR)3 signaling activation is an important mediator of diabetic nephropathy in mice and humans (5, 6). Increased mTOR activity has been reported in different glomerular diseases, and mTORC1 inhibition by rapamycin and everolimus shows beneficial effects against diabetic nephropathy, focal segmental glomerulosclerosis, minimal change disease, membranous nephropathy, etc. (7–10). But contrasting evidence also indicates that mTORC1 inhibition by the immunosuppressive drug rapamycin leads proteinuria and podocyte apoptosis and develops primary focal segmental glomerulosclerosis in renal transplant recipients (11). Therefore, it might be important to investigate the detailed molecular mechanisms to answer this puzzle and identify therapeutic targets.
mTOR is an atypical serine/threonine kinase belonging to the phosphatidylinositol 3-kinase-related kinase and is conserved from yeast to humans (12, 13). It forms two functionally different multiprotein complexes: mTOR complex 1 (mTORC1), sensitive to rapamycin, and mTOR complex 2 (mTORC2), rapamycin-insensitive (14). Functional mTORC1 consists of mTOR, mLST8, and raptor, whereas mTORC2 includes mTOR, mLST8, mSIN1, and rictor. These signaling pathways regulate major physiological events, including ribosome biogenesis and protein translation (15), lipid homeostasis (16), and mitochondrial biogenesis (17). However, it is also implicated in various disease progressions, such as type 2 diabetes and obesity (14). mTORC1 activation results in phosphorylation of ribosomal protein S6 via p70S6K and eukaryotic translation initiation factor 4E binding protein (4EBP), which potentiates the initiation of protein translation (18, 19). However, mTORC2 regulates cell survival through phosphorylation of Akt (Ser473) and cytoskeleton organization through phosphorylation of PKCα (20, 21). Tuberous sclerosis 1 (TSC1)/hamartin forms a heterodimer with tuberous sclerosis 2 (TSC2)/tuberin to negatively regulate mTORC1 activity. Many growth factors, including insulin, insulin-like growth factor, and TGF-β and nutrients such as amino acids exert their cellular input through modulation of mTOR complexes (22, 23). Akt, ERK1/2, and AMP-activated protein kinase are well known intermediate signaling molecules that transmit extracellular signals to mTOR (15, 24).
TGF-β is a multifunctional cytokine involved in many cellular processes, including migration, growth inhibition, invasion, extracellular matrix remodeling, epithelial-mesenchymal transition, and fibrosis (25). Increased activity of TGF-β plays critical roles in the progression of renal diseases in animal models and human renal diseases where podocyte loss is highly evident (26, 27). Furthermore, accumulation of TGF-β and its pathogenic roles in primary focal segmental glomerulosclerosis, progressive glomerulosclerosis, and diabetic nephropathy have been reported (26, 28). In addition, adenovirus-mediated TGF-β1 gene transfer to kidney glomeruli leads to proteinuria in rats (29). It has been reported recently that podocyte-specific overexpression of TGF-β induces segmental glomerulosclerosis with dysfunction in endothelial cells and podocyte depletion (30). We have also published previously that TGF-β1 induces podocyte apoptosis through transcriptional up-regulation of NADPH oxidase 4 (Nox4) by activation of the Smad pathway (31). There is also evidence that high glucose-mediated inactivation of AMP-activated protein kinase activates mTOR in a TSC2-dependent manner, which accounts for Nox4-mediated podocyte depletion in diabetic mice (32). However, the possibility of mTOR activation by TGF-β1 in podocyte apoptosis has not been addressed so far. Moreover, the molecular mechanism of Nox4 regulation by the TGF-β1-mTOR axis has not yet been explored in detail.
In this study, we observed that Nox4 is up-regulated by TGF-β1 via rapamycin-sensitive mTORC1 activation because Nox4-induced oxidative stress and mitochondrial dysfunctions are ameliorated with rapamycin treatment or silencing of mTOR or its downstream target p70S6K. ERK1/2 plays an important role in TGF-β1-induced mTOR activation and translational regulation of Nox4 protein. Finally, Smad signaling regulates TGF-β1-mediated ERK1/2 phosphorylation in podocytes. Therefore, we suggest that inhibition of the ERK1/2-mTOR pathway could provide effective protection against podocyte depletion in glomerular kidney diseases.
Experimental Procedures
Cell Culture and Drugs
Immortalized mouse podocytes were obtained from the laboratory of Prof. Peter Mundel (Harvard Medical School) and cultured as described previously (31). Cells were grown on collagen-coated 100-mm dishes using low-glucose Dulbecco's modified Eagles medium supplemented with 5% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin. Cell proliferation was achieved at 33 °C in the presence of 20 units/ml mouse recombinant interferon γ, and, for differentiation, these cells were thermoshifted to 37 °C in the absence of interferonγ for 10∼14 days. Before experiments, cells were changed with media containing 0.2% FBS for 24 h.
Chemicals were purchased as follows: SB431542 (catalog no. S4317) and rapamycin (catalog no. R8781) from Sigma, U0126 (catalog no. 9903) and PD184352 (catalog no. 12147) from Cell Signaling Technology (Danvers, MA), and cycloheximide (catalog no. 14126) from Cayman Chemical (Ann Arbor, MI).
Quantitative Real-time PCR
To isolate total RNA, podocytes were grown on 100-mm dishes. Cells were washed twice with PBS before trypsinization. RNA was isolated according to the RNeasy kit (catalog no. 74134, Qiagen GmbH, Hilden, Germany). cDNA was synthesized from 0.5–1 μg of RNA with a reverse transcription kit (Applied Bioscience, Foster City, CA) using oligo(dT) in a reaction volume of 20–40 μl according to instructions of the company. cDNA was subjected to real-time PCR to evaluate mRNA expression of Nox4 with the help of a sequence-specific primer (forward, 5′-CCACAGACCTGGATTTGGAT-3′; reverse, 5′-TGGTGACAGGTTTGTTGCTC-3′). The β-actin (forward, 5′-AAGAGCTATGAGCTGCCTGA-3′; reverse, 5′-CACAGGATTCCATACCCAAG-3′) primer was considered a reference control. Experiments were performed in triplicate in a real-time PCR machine (7900HT, Applied Bioscience) using SYBR Green PCR Master Mix (catalog no. 204143, Qiagen GmbH). Data analysis was performed according to the ΔΔCT method (33).
Western Blot Analysis
For isolation of total protein, podocytes were differentiated in 100-mm dishes or on 6-well plates. The cells were washed carefully twice with ice-cold PBS and lysed with cold radioimmune precipitation assay buffer (Thermo Scientific, Rockford, IL) containing protease and phosphatase inhibitor mixture (Thermo Scientific, catalog nos. 88665 and 88667, respectively). The crude lysates were centrifuged in a tabletop centrifuge at 13,200 rpm for 20 min at 4 °C, and clear supernatants were carefully transferred to a new Eppendorf tube. A BCA protein assay kit (Thermo Scientific) was used to determine the concentration of total protein. Equal amounts of protein were loaded to 6%∼10% SDS-PAGE and transferred to a PVDF membrane (Millipore Corp., Billerica, MA). The membrane was blocked with either with 6% skim milk or 5% BSA, followed by primary antibody incubation at 4 °C overnight. Dilutions and catalog information for primary antibodies is as follows: total and phospho-mTOR (1:5000 and 1:2000, respectively; catalog nos. 2983 and 5536, respectively), total and phospho-p70S6K (1:2000 and 1:5000, respectively; catalog nos. 9202 and 9204, respectively), total and phospho-Smad2 (1:2500 and 1:1000, respectively; catalog nos. 5339 and 3108, respectively), total and phospho-ERK1/2 (1:10,000 each; catalog nos. 9102 and 9101, respectively), phospho-4EBP1 (1:5000, catalog no. 2855), phospho-Akt (Thr308, 1:2500, catalog no. 9275), cleaved caspase-3 (1:1000, catalog no. 9661), phospho-TSC2 (Ser939, 1:2000, catalog no. 3615), phospho-TSC2 (Thr1462, 1:2000, catalog no. 3617), and total (t-)TSC2 (1:2500, catalog no. 4308) from Cell Signaling Technology; AU1 (1:1000, catalog no. PA1-26547) from Thermo Scientific; Nox4 (1:200, catalog no. sc-30141) from Santa Cruz Biotechnology; and Nox4 (1 μg/ml, catalog no. ab60940) and β-actin (1:10,000, catalog no. ab6246) from Abcam (Cambridge, UK). HRP-conjugated goat anti-rabbit secondary antibody (catalog no. 31460) was purchased from Thermo Scientific. All antibodies were prepared in 0.1% Tris-buffered saline and Tween 20 (TBST) containing 5% BSA. Secondary antibody was prepared in 6% skim milk and incubated for 1 h at room temperature. Membranes were developed using ECL solution (Luminata Forte, Millipore Corp.), and the images were captured with a UVP Biospectrum-600 imaging system.
Measurement of Reactive Oxygen Species (ROS)
Measurement of reactive oxygen species in podocytes was performed as described previously (31). Intracellular ROS generation was detected by the fluorescent dye chloromethyl-H2-dichlorofluorescein diacetate (catalog no. C6827, Molecular Probes). This dye rapidly oxidizes to fluorescent 2,7-dichlorofluorescein in the presence of intracellular ROS and produces a bright fluorescent light. For ROS measurement, 5000 podocytes were seeded on 12-mm coverslips and differentiated for 10 days before application of TGF-β1 and other inhibitors. Cells were washed out using Krebs-Ringer bicarbonate solution (135 mm NaCl, 3.6 mm KCl, 2 mm NaHCO3, 0.5 mm NaH2PO4, 0.5 mm MgSO4, 1.5 mm CaCl2, and 10 mm HEPES (pH 7.4)) twice and loaded with 5 μm chloromethyl-H2-dichlorofluorescein diacetate for 20 min at 37 °C. Images (excitation/emission, 490/535 nm) were captured using an inverted microscope (IX81, Olympus, Tokyo, Japan) with a confocal spinning disk (CSU10, Yokogawa Electric Corp., Tokyo, Japan), and images were analyzed using Metamorph 6.1 software (Molecular Devices, Sunnyvale, CA).
Detection of Mitochondrial Membrane Potential
The lipophilic cationic dye 5,5′,6,6′-tetrachloro-1,19,3,39-tetraethylbenzimidazolyl-carbocyanine iodide (catalog no. T3168, Molecular Probes) was used to determine the mitochondrial membrane potential in podocytes. 5,5′,6,6′-tetrachloro-1,19,3,39-tetraethylbenzimidazolyl-carbocyanine iodide monomers is a plasma membrane-permeable dye entering into mitochondria depending on the mitochondrial membrane potential. Inside mitochondria, monomers aggregate to form J aggregates and transmit red fluorescence (excitation/emission, 540/590 nm), whereas the free monomers outside of mitochondria transmit green fluorescence (excitation/emission, 490/535 nm). The ratio of red/green fluorescence is used as an indicator to measure the mitochondrial membrane potential. This experiment was performed in 96-well microplates (catalog no. 3603, Corning Inc., Corning, NY). Podocytes, grown on collagen-coated 96-well plates, were washed twice with warm Krebs-Ringer bicarbonate solution, and 300 nm 5,5′,6,6′-tetrachloro-1,19,3,39-tetraethylbenzimidazolyl-carbocyanine iodide was loaded for 30 min. Fluorescence intensities were recorded by a fluorescence microplate reader (Flexstation II, Molecular Devices), and data were analyzed as described previously (34).
Measurement of total Nox Activity
Total NADPH oxidase activity from podocyte lysate was measured as described previously (31). Podocytes were differentiated on 100-mm culture dishes for 13 days. Cells were kept on ice for 15 min, washed three times with ice-cold PBS, and collected from the dishes by scrapers in PBS containing protease inhibitor. Cells were centrifuged at 2000 rpm for 3 min at 4 °C. The supernatant was discarded, and cell pellets were dissolved in lysis buffer (20 mm KH2PO4, 1 mm EGTA, and protease inhibitor mixture (pH 7.0)). Using a Dounce homogenizer, cell suspensions were homogenized with 100 strokes, and the homogenates were used freshly to measure total Nox activity. 50 μg of homogenates was added to 50 mm phosphate buffer (pH 7.0) containing 1 mm EGTA, 150 mm sucrose, 5 μm lucigenin, and 100 μm NADPH. Photoparticles generated during the reaction were detected by a luminometer (Synergy 2, BioTek Instruments, Winooski, VT) for 10 min continuously and expressed as relative light units. Superoxide generation was expressed as relative light units per minute per microgram of protein.
TUNEL Assay
Podocytes were grown on 12-mm coverglasses coated with rat collagen and were differentiated for 10 days at 37 °C without interferon γ. Cells were exchanged with 0.2% FBS-containing medium 24 h before addition of TGF-β1 and inhibitors. In the case of siRNA treatment, 0.2% FBS-containing medium was replaced 24 h after siRNA treatment. TUNEL was performed according to the instructions of the manufacturer using an ApopTag fluorescein in situ apoptosis detection kit (catalog no. S7110, Millipore). Cells were counterstained with DAPI. Signals from the nuclei of apoptotic cells merged with DAPI (blue) and fluorescein (green) were considered TUNEL-positive.
siRNA Transfection
Cells were grown on 6-well cell culture dishes and allowed to differentiate for 10 days. The culture medium was changed to antibiotic-free medium 1 day before siRNA transfection. Transfection was carried out using DharmaFECT 1 siRNA transfection reagent (catalog no. T-2001-03, Thermo Fisher Scientific) according to the directions of the manufacturer. Opti-MEM-reduced serum medium was used to prepare the siRNA-transfection reagent complex. The medium was changed 6 h after the addition of transfection reagent. Protein was isolated 72 h after siRNA treatment. SiGENOME SMARTpool siRNA duplexes for Smad2, Smad3, Smad4, and p70S6K were purchased from Dharmacon (Thermo Fisher Scientific Inc.). TSC2 siRNA was purchased from Bioneer Inc. (Daejeon, Korea). siRNAs for mTOR and non-targeting sequences (siControl) were synthesized from Bioneer Inc. The sequences for mTOR, TSC2, and non-targeting siRNA were 5′-CAUUCGCAUUCAGUCCAUA-3′, 5′-CAUCAUUGAACGACUACUU-3′, and 5′-CCUACGCCACCAAUUUCGU-3′, respectively. The sequence for mTOR siRNA was obtained from a published article where the efficiency of siRNA had already been tested (35).
Adenoviral Infection
Adenoviral mTOR constructs of wild-type (mTOR-WT), constitutively active (mTORΔ), kinase-dead (mTOR-KD), and control virus expressing firefly luciferase (Fluc) were gifts from Prof. C. Rhodes (University of Chicago, Chicago, IL). mTORΔ was generated by deleting amino acid sequences 2430–2450 with 5-fold higher activity compared with wild-type mTOR assayed under in vitro conditions. mTOR-KD carried an Asp2338-to-Ala mutation rendering catalytic inactivation (36). Each mTOR construct was attached to an N-terminal AU1 tag, facilitating identification of exogenous mTOR expression. All four adenoviruses were amplified in HEK293 cells and purified by an ibiPureTMAdeno adenovirus purification kit (catalog no. 60681) according to the instructions of the manufacturer. For all experiments, infections with a multiplicity of infection of 100 were used to infect podocytes as described previously (36).
Statistical Analysis
Experimental data are expressed as mean ± S.E. All data shown here represent three or more independent experiments where n indicates the number of independent experiments. Statistical comparisons were made by Student's t test or one-way analysis if not stated otherwise, and p < 0.05 was considered to be significant.
Results
TGF-β1 Phosphorylates and Activates the mTOR Signaling Cascade in Podocytes
Immortalized podocyte cell line expressing the differentiation marker synaptopodin at 37 °C in the absence of interferon γ has been checked previously (31). Under similar culture conditions, podocytes were treated with TGF-β1 (5 ng/ml) in a time-dependent manner to check the activation of mTOR signaling. Phosphorylation of mTOR at Ser2448 was evident 5 min after TGF-β1 treatment and reached its peak at 6 h. At the same time, p70S6K and 4EBP1, which are the downstream targets of mTORC1, were phosphorylated at Thr421/Ser424 and Thr37/46, respectively, reached a maximum level at 6 h, and were reduced slightly at the 24-h time point. No statistically significant changes in expression of t-mTOR and t-p70S6K protein level were observed during this time interval. Additionally, Akt, which is a well known upstream regulator of the mTOR pathway, was also checked. Phosphorylation of Akt (Thr308 and Ser473) was increased by TGF-β1, with a maximum peak at 6 h (Fig. 1, A and B). The TGF-β receptor I (TGF-βRI)-specific inhibitor SB431542 (10 μm) completely blocked the activation of mTOR signaling. TGF-β1-mediated phosphorylations of mTOR, p70S6K, and 4EBP1 was prevented completely in the presence of SB431542. Inhibition of p-Smad2 and p-Smad3 by SB431542 was used as a positive control for TGF-β signaling activation in this experiment (Fig. 1, C–E).
FIGURE 1.

TGF-β1 activates mTOR signaling in podocytes. A, activation of mTOR was checked by TGF-β1 treatment in podocytes. Fully differentiated cells were treated with 0.2% FBS for 24 h before treatment with 5 ng/ml TGF-β1 at different time points. p-mTOR (Ser2448), t-mTOR, p-p70S6K (Thr421/Ser424), t-p70S6K, p-4EBP1 (Thr37/46), p-Akt (Thr308 and Ser473), and t-Akt and β-actin were detected by Western blot analysis. B, densitometry analysis of p-mTOR, p-p70S6K, and p-4EBP1 from Fig. 1A (n = 3). C, inhibition of TGF-β1 and mTOR signaling using SB431542 (SB) was assessed. SB431542 (10 μm) was applied 1 h before addition of TGF-β1, and protein was isolated after 6 h of TGF-β1 stimulation. Western blots for p-mTOR (Ser2448), t-mTOR, p-p70S6K (Thr421/Ser424), t-p70S6K, p-4EBP1 (Thr37/46), p-Smad2, t-Smad2, p-Smad3, t-Smad3, and β-actin were performed. For Western blot analysis, 15 μg of protein was loaded for each sample except β-actin (5 μg). Experiments were performed three times or more. D and E, quantitative analyses of p-mTOR and p-p70S6K from C. Values are mean ± S.E. *, p < 0.05; **, p < 0.01.
mTOR Regulates Nox4 Protein Synthesis in Podocytes
We have described previously that TGF-β1-induced Nox4 is responsible for TGF-β1-mediated podocyte apoptosis (31). Different growth factors are known to regulate mTOR activity to increase protein synthesis in a cell type-dependent manner. Therefore, the role of mTOR on TGF-β1-mediated Nox4 protein synthesis was evaluated here in podocytes. To confirm the role of mTOR in TGF-β1-mediated oxidative stress by Nox4, mTOR was knocked down using siRNA (100 nm). Application of simTOR significantly reduced the mTOR protein level, as validated by Western blotting (Fig. 2A). Treatment with simTOR suppressed TGF-β1-induced Nox4 protein up-regulation and completely recovered TGF-β1-stimulated ROS generation in podocytes (Fig. 2, B and C). The protein translation inhibitor cycloheximide was applied to confirm the role of translational regulation as one of the possible mechanisms for increased Nox4 protein synthesis by TGF-β1 application. TGF-β1-mediated Nox4 protein synthesis was abolished completely by cycloheximide treatment (Fig. 2D). Using an adenoviral delivery system, we further evaluated the effect of mTOR on Nox4 protein expression through loss-of-function and gain-of-function mutations in mTOR. Overexpression of mTOR constructs was confirmed because t-mTOR expression was increased substantially and AU1 expression was clearly detected in all groups except the control group (Fluc). p70S6K phosphorylation was increased at the basal level as well as by TGF-β1 treatment in podocytes expressing mTOR-WT and mTORΔ, whereas p-p70S6K was reduced in the mTOR-KD group in comparison with the control (Fig. 2E). Overexpression of mTOR-WT increased TGF-β1-stimulated Nox4 expression, ROS generation, and TUNEL-positive apoptotic nuclei compared with the control vector. Expression of mTORΔ further augmented the Nox4 level, ROS generation, and podocyte apoptosis. However, mTOR-KD completely abrogated the above changes under a TGF-β1 stimulus (Fig. 2, F–H).
FIGURE 2.

mTOR regulates Nox4 expression and oxidative stress in podocytes. A, representative blot for t-mTOR and densitometry analysis showing the effect of simTOR (100 nm)-mediated knockdown in podocytes (n = 3). B, Western blot for Nox4 in siControl- and simTOR-treated podocytes in the presence or absence of TGF-β1 treatment for 24 h. C, t-mTOR was silenced using simTOR, and ROS generation was checked after 72 h of siRNA treatment (n = 3). D, the protein level for Nox4 was detected after treatment with or without TGF-β1 and cycloheximide (2 and 10 μm, respectively) (n = 3). E, podocytes were infected with adenovirus constructs (multiplicity of infection, 100) carrying Fluc, mTOR-WT, mTORΔ, and mTOR-KD and treated with TGF-β1 for 6 h. Western blot analysis was performed to check the expression of AU1, t-mTOR, and phospho- and total p70S6K. F, Western blot for Nox4 after adenoviral delivery of Fluc, mTOR-WT, mTORΔ, and mTOR-KD in podocytes treated with TGF-β1 for 24 h. G, ROS measurement using 2,7-dichlorofluorescein (DCF) after adenoviral infection of Fluc, mTOR-WT, mTORΔ, and mTOR-KD in podocytes treated with TGF-β1 for 24 h (n = 3). H, apoptosis was detected by TUNEL assay after TGF-β1 treatment in podocytes expressing Fluc, mTOR-WT, mTORΔ, and mTOR-KD. All values are mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Activation of mTORC1 Mediates TGF-β1-induced Oxidative Stress and Apoptosis in Podocytes
To deduce the involvement of mTORC1 in TGF-β1-mediated oxidative stress by Nox4, podocytes were treated with rapamycin, a well known pharmacological inhibitor for mTORC1. Low-dose (10 nm) rapamycin treatment (1-h pretreatment) clearly reduced TGF-β1-induced augmentation of the Nox4 protein level at 24 h (Fig. 3A). Interestingly, when the Nox4 mRNA level was evaluated by real-time PCR, no significant reduction of Nox4 mRNA was observed by rapamycin pretreatment along with TGF-β1 stimulation (Fig. 3B). In addition, 10 nm rapamycin treatment completely prevented total Nox activity and ROS elevation by TGF-β1 treatment (Fig. 3, C and D). TGF-β1-mediated mitochondrial dysfunction was also recovered significantly by rapamycin treatment (Fig. 3E). Podocyte apoptosis was evaluated by expression of cleaved caspase-3 and TUNEL staining. TGF-β1 significantly increased cleaved aspase-3 expression and TUNEL-positive nuclei, which were inhibited by rapamycin treatment (Fig. 3, F and G). To further confirm the role of mTORC1, p70S6K was knocked down by siRNA (50 nm) treatment. sip70S6K significantly reduced the total p70S6K protein level and protected TGF-β1-induced Nox4 expression, ROS generation, and apoptosis in podocytes (Fig. 3, H–K).
FIGURE 3.

Inhibition of mTORC1 protects podocytes from TGF-β1-induced oxidative stress and apoptosis. Total protein and RNA were extracted after 24 h of TGF-β1 treatment with or without rapamycin (10 nm) treatment. A, 30 μg of total lysate was loaded to detect Nox4 by Western blot analysis. B, the mRNA level of Nox4 was detected by real-time PCR (n = 4). C, quantitative analysis of total Nox activity was performed with cultured podocytes after 24 h of TGF-β1 treatment (n = 3). RLU, relative light units. D, analysis of 2,7-dichlorofluorescein (DCF) intensity indicating the inhibitory effect of rapamycin on cellular ROS generation after 24 h of TGF-β1 stimulation (n = 4). E, using the fluorescence dye 5,5′,6,6′-tetrachloro-1,19,3,39-tetraethylbenzimidazolyl-carbocyanine iodide (JC-1), the mitochondrial membrane potential was assessed (n = 5). F, 40∼50 μg of protein was loaded to check the expression of cleaved caspase-3 after 48 h of TGF-β1 and rapamycin treatment (n = 3). G, TUNEL assay indicating the protecting effect of rapamycin against TGF-β1-mediated podocyte cell death (n = 3). H, densitometry analysis showing the reduction in p70S6K protein level after siRNA-mediated knockdown of p70S6K (n = 3). I, Western blot analysis was performed with si-p70S6K-treated podocytes, and the Nox4 level was assayed. J, ROS measurements in p70S6K-silenced podocytes with TGF-β1 treatment for 24 h. K, TUNEL assay indicating TGF-β1-stimulated podocyte apoptosis in the control and si-p70S6K-treated groups (n = 3). All values are mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Inhibition of ERK1/2 Phosphorylation Protects TGF-β1-induced Nox4 Up-regulation and Apoptosis in Podocytes
We also observed that TGF-β1 stimulation strongly phosphorylated ERK1/2 within 5 min and remained activated until 24 h, with a maximum phosphorylation peak at 6 h (Fig. 4A). The TGF-βRI kinase inhibitor SB431542 abrogated ERK1/2 phosphorylation (Fig. 4, B and C). We examined the effects of the ERK inhibitors U0126 and PD184352 against TGF-β1-stimulated Nox4 protein synthesis, ROS generation, loss of mitochondrial membrane potential, and caspase-3 activation. Application of U0126 (5 μm) and PD184352 (1 μm) completely abrogated ERK1/2 phosphorylation (Fig. 4D). Both ERK inhibitors efficiently prevented Nox4 protein up-regulation by TGF-β1 treatment for 24 h (Fig. 4E). Similar to the effect of rapamycin, inhibition of the ERK1/2 pathway by U0126 (5 and 10 μm) did not reverse the mRNA level of Nox4 up-regulated by TGF-β1 (Fig. 4F). Preincubation with U0126 for 1 h completely blocked TGF-β1-mediated total Nox activity (Fig. 4G). Application of PD184352 significantly inhibited TGF-β1-mediated total reactive oxygen species generation by podocytes (Fig. 4H). The protective effect of U0126 was also observed against the loss of mitochondrial membrane potential mediated by TGF-β1 at the 24-h time point (Fig. 4I). Apoptosis marker caspase-3 activation and increase of TUNEL-positive apoptotic nuclei by TGF-β1 treatment were diminished by U0126 or PD184352 treatment, respectively (Fig. 4, J and K).
FIGURE 4.

Inhibition of the ERK1/2 pathway prevents podocyte apoptosis by TGF-β1. Inhibition of ERK1/2 was carried out by 1-h pretreatment with U0126 (5 μm) and PD184352 (1 μm), which prevented TGF-β1-induced podocyte apoptosis. A, time-dependent ERK1/2 phosphorylation was checked by Western blotting (n = 4). B and C, TGF-β1-mediated ERK1/2 phosphorylation was blocked by 1-h pretreatment with the TGF-βRI inhibitor SB431542 (SB) (n = 3). D, the specificities of ERK inhibitors were confirmed by Western blot analysis showing a marked reduction of p-ERK1/2 by U0126 and PD184352 treatment (n = 2). E, Western blot analysis for Nox4 in the presence of U0126 and PD184352 under TGF-β1 treatment (n = 3). F–I, real-time PCR data showing no reduction in TGF-β1-induced Nox4 mRNA by U0126 (5 and 10 μm) treatment (F, n = 4). Cells were stimulated with TFG-β1 for 24 h, and total Nox activity (G, n = 4), ROS generation (H, n = 4), and mitochondrial membrane potential (I, n = 4) were assessed after ERK inhibition. RLU, relative light units; DCF, 2,7-dichlorofluorescein; JC-1, 5,5′,6,6′-tetrachloro-1,19,3,39-tetraethylbenzimidazolyl-carbocyanine iodide. J, Western blot analysis showing inhibition of cleaved caspase-3 activation by U0126 treatment (n = 3). K, podocyte apoptosis detected by TUNEL assay in the presence and/or absence of TGF-β1 and PD184352. All values are mean ± S.E. *, p < 0.05; **, p < 0.01.
ERK1/2 Regulates TGF-β1-induced mTOR Activation in a TSC2-independent Manner in Podocytes
Previous literature has shown that phosphorylation of ERK1/2 regulates mTOR activation in different cell types (24, 37, 38). To elucidate the role of ERK1/2 in mTOR activation by TGF-β1, ERK1/2 activation was inhibited by U0126 and PD184352, and 1 h of preincubation with these inhibitors significantly reduced mTOR and p70S6K phosphorylation by TGF-β1 (Fig. 5, A–C). Next we wanted to identify the mechanism of mTOR phosphorylation by ERK1/2. PMA-induced TSC2 phosphorylation at position Ser664 via ERK has been described by Ma et al. (38). Stimulation with TGF-β1 significantly phosphorylated TSC2 (Ser664) at 6 h, and this phosphorylation was inhibited completely by U0126 and PD184352. As a positive control, HeLa cells were treated with nocodazole (10 μm) for 1 h to stimulate phosphorylation of TSC2 at Ser664 (Fig. 5D). Another potential target of mTOR activation by TGF-β1 is protein kinase B or Akt. Akt (Thr308) phosphorylation was clearly detected after 1 h of TGF-β1 treatment, and the downstream target TSC2 was phosphorylated simultaneously (Ser939 and Thr1462) by TGF-β1. These phosphorylations were clearly inhibited by the PI3K inhibitor wortmannin in a dose-dependent manner (Fig. 5E). However, wortmannin did not show any significant inhibition of p70S6K phosphorylation mediated by TGF-β1 stimulation in podocytes (Fig. 5F). To confirm the role of ERK1/2-p-TSC2 (Ser664) in the activation of mTOR by TGF-β1, TSC2 was knocked down by specific siRNA oligos (100 nm). The TSC2 protein level was reduced significantly by siRNA-mediated TSC2 knockdown. However, TGF-β1-induced phosphorylation of p70S6K was not changed by silencing of TSC2 in podocytes. When U0126 and rapamycin were added to siTSC2-treated groups, phosphorylation of p70S6K by TGF-β1 was diminished completely. Moreover, insulin-induced phosphorylation of p70S6K was also not inhibited by TSC2 knockdown in podocytes (Fig. 5G).
FIGURE 5.

ERK1/2 phosphorylation mediates mTOR activation in a tuberin-independent manner. A and B, phosphorylation of mTOR and p70S6K was checked by Western blot analysis after TGF-β1 treatment for 6 h. Pretreatment with U0126 and PD184352 reduced mTOR and p70S6K activation (n = 3). C, densitometry analysis of B. D, Western blot analysis result showing that treatment of TGF-β1 for 6 h increased phosphorylation of TSC2 (Ser664), which was blocked by U0126. The total cell lysate from HeLa cell treated with nocodazole (10 μm) for 1 h was loaded as a positive control (n = 4). E, Western blot analysis for p-Akt (Ser308), p-TSC2 (Ser939 and Thr1462), and t-TSC2. Wortmannin (50 and 100 nm) inhibited TGF-β1-induced Akt and TSC2 phosphorylation. F, Western blot analysis showing that 100 nm wortmannin blocked insulin-induced p70S6K phosphorylation but not TGF-β1-induced p70S6K phosphorylation (n = 3). G, Western blot analysis showing that siTSC2 did not reverse TGF-β1- and insulin-induced p70S6K phosphorylation and that addition of U0126 and rapamycin completely prevented TGF-β1-induced p-p70S6K in siTSC2-treated groups (n = 3). Values are mean ± S.E. *, p < 0.05.
The Smad Pathway Stimulates TGF-β1-induced ERK1/2-mTOR Activation in Podocytes
We have published previously that Nox4 mRNA was transcriptionally up-regulated by Smad2 or Smad3 upon TGF-β1 addition. Here we observed that mTOR activation facilitated translation of the Nox4 protein level (Fig. 2B). Therefore, we were interested to verify the possibility of ERK1/2 activation by Smad2 and Smad3. siRNA-mediated combined knockdown of Smad2 and 3 (50 nm each) completely inhibited phosphorylation of ERK1/2 and p70S6K (Fig. 6, A–C). Similar events were observed when Smad4 was knocked down. siSmad4 (50 nm) completely reversed TGF-β1-induced ERK1/2, mTOR, and p70S6K phosphorylation (Fig. 6, D–F). Interestingly, none of the individual knockdowns of Smad2 or Smad3 was effective to suppress TGF-β1-stimulated ERK1/2 and p70S6K phosphorylation (Fig. 6, G–I).
FIGURE 6.

Smad regulates phosphorylation of ERK1/2 and mTOR activation in podocytes. A, podocytes were transfected with a mixture of siSmad2 (50 nm) and siSmad3 (50 nm), and expression levels of p-ERK1/2, t-ERK1/2, p-p70S6K, t-p70S6K, t-Smad2, and t-Smad3 were checked by Western blot analysis after TGF-β1 treatment for 6 h. β-actin was used as a loading control. B and C, densitometry analyses of p-ERK1/2 and p-p70S6K from A, respectively (n = 3). D, cells were treated with 50 nm of siSmad4 and TGF-β1 for 6 h. Western blot analysis was performed for p-ERK1/2, t-ERK1/2, p-mTOR, t-mTOR, p-p70S6K, t-p70S6K, Smad4, and β-actin. E and F, densitometry analyses of p-ERK1/2 and p-p70S6K from D, respectively (n = 3). G, Western blot analysis showing that siRNA-mediated individual knockdown of Smad2 or Smad3 (100 nm each) did not recover TGF-β1-induced p-ERK1/2 and p-p70S6K phosphorylation (n = 3). H and I, densitometry analyses of p-ERK1/2 and p-p70S6K from G, respectively. All values are mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s., not significant.
Discussion
Here we report on TGF-β1-stimulated Nox4 up-regulation through ERK1/2 and mTOR signaling, which accelerates oxidative stress-induced mitochondrial dysfunction and apoptosis in immortalized podocytes. Inhibition of either mTORC1 by low-dose rapamycin and p70S6K siRNA treatment or of ERK1/2 by U0126 and PD184352 protected podocytes from activation of caspase-3 and apoptosis. Previously, we have reported that TGF-β1 selectively up-regulates the transcription of Nox4 mRNA by Smad2/3, resulting in an elevated mitochondrial Nox4 protein level, oxidative stress, mitochondrial dysfunction, and apoptosis in podocytes (31). This study identifies a distinct molecular mechanism of translational regulation of Nox4 protein that is considered another crucial step in TGF-β1 signaling to augment oxidative stress in podocytes. Activation of ERK1/2-mTORC1 signaling by TGF-β1 occurs in a Smad-dependent pathway, leading to an increased Nox4 protein level. We also show that mTOR activation is independent of ERK1/2 or Akt-targeted TSC2 phosphorylation. Taken together, we suggest that TGF-β1-induced transcriptional regulation of Nox4 mRNA by the Smad-dependent pathway converges with translational regulation by the Smad-ERK1/2-mTORC1 pathway, accounting for oxidative stress-induced apoptosis in cultured podocytes.
Although the main function of mTORC1 is in growth control, recent findings suggest its overactivity in the progression of different glomerular diseases in human and animal models. In contrast, inhibition of mTORC1 also leads to pathogenic alterations such as podocyte apoptosis and proteinuria (11). Several pieces of evidence suggest that TGF-β1 can stimulate mTORC1 and mTORC2 activation in a cell type-dependent manner (39–42). We observed rapid mTOR phosphorylation in podocytes after 5 min of TGF-β1 treatment. Within the same time window, Smad2, Smad3 (31), and mTOR signaling are activated when podocytes are challenged with exogenous TGF-β1 (Fig. 1A). This observation led us to identify a possible cross-talk between Smad signaling and mTOR activation in podocytes. Upon binding of TGF-β1 to TGF-βRII on the extracellular surface, it transmits a signal by phosphorylating TGF-βRI, which activates either Smad-dependent or -independent signaling. TGF-βRI kinase activity is a prerequisite for phosphorylation of Smad2 and 3, which can be inhibited completely by SB431542, a potent inhibitor of TGF-βRI. However, Smad-independent activation of p38 MAPK in mesangial cells does not rely on TGF-βRI kinase activity (43, 44). In our results, phosphorylation of mTOR and p70S6K was inhibited completely by SB431542, implying that TGF-βRI kinase activity is a mandatory to initiate mTORC1 (Fig. 1, C–E). It has been observed previously that TGF-β1 stimulation induces apoptosis in undifferentiated podocytes cultured at 33 °C (27). We also observed strong phosphorylation of p70S6K by TGF-β1 treatment to undifferentiated podocytes and inhibition of caspase-3 activation by rapamycin, indicating conservation of the mTORC1-mediated apoptotic signal even in undifferentiated podocytes (data not shown).
The roles of Nox4 in the effacement and apoptosis of podocytes have been studied in an experimental diabetic mice model and in a puromycin aminonucleoside-induced rat model of focal segmental glomerulosclerosis (32, 45–47). We also have reported, in a previous publication, that inhibition of Nox4 by the pharmacological inhibitor diphenyleneiodonium or RNA interference ameliorates TGF-β1-mediated oxidative stress, mitochondrial dysfunction, and apoptosis in podocytes (31). Here we observed that change in mTOR protein expression modulates the Nox4 level in podocytes. Gain-of-function (using mTOR-WT and mTORΔ) and loss-of-function (using mTORΔ and simTOR) studies indicate that mTOR kinase activity is crucial for Nox4 protein expression, determining the ROS level and apoptosis (Fig. 2, B, C, and E–H). Additionally, pharmacological inhibition of mTORC1 by rapamycin suppressed TGF-β1-stimulated Nox4 protein up-regulation and total Nox activity, dissipated ROS generation, and recovered from loss of mitochondrial membrane potential, caspase-3 activation, and apoptosis (Fig. 3, A and C–G). Moreover, silencing of p70S6K showed a reduction in Nox4 and ROS generation to the level similar to rapamycin treatment, confirming the role of mTORC1 in the regulation of Nox4 in podocytes (Fig. 3, H–K). However, the up-regulated Nox4 mRNA level was not changed at all when rapamycin was applied (Fig. 3B). This implies that mTOR may up-regulate translation mechanism or increase protein stability by inhibiting the proteasomal degradation of Nox4 under TGF-β1 treatment in podocytes. To confirm this finding, cycloheximide, a protein translation inhibitor, was employed, and, indeed, pretreatment with cycloheximide blocked up-regulation of Nox4 protein, confirming that mTOR activation by TGF-β1 regulates the translation of the Nox4 protein level in podocytes (Fig. 2D).
Evidence shows that activation of the ERK MAPK pathway induces pathogenic changes in podocytes. Gain-of-function mutation in the transient receptor potential C6 (TRPC6) channel facilitates a Ca2+ influx that activates ERK signaling (48). Yu et al. (49) have shown that TGF-β1-induced Smad3-ERK1/2-NF-κB activation and Fyn-dependent TRPC6 phosphorylation lead to podocyte apoptosis. Interestingly, we observed that TGF-β1 mediates biphasic phosphorylation of ERK1/2 in podocytes (Fig. 4A). Rapid phosphorylation of ERK1/2 at 5 min may be due to Smad-independent direct phosphorylation of ShcA by TGF-βRI, which associates with Grb2 and Sos, leading to Ras-Raf-MEK-ERK pathway activation (50). The later peak at 6 h might result from a Smad-dependent increase in mRNA transcription and translation of ERK1/2 (51). However, we did not further investigate this mechanism in this study. We observed that Nox4 protein up-regulation depends on ERK phosphorylation because U0126 and PD184352 completely inhibited the increase in Nox4 protein level and total Nox activity in podocytes (Fig. 4, E–G). Similar to rapamycin treatment, ERK inhibition did not show any reduction in Nox4 mRNA level increased by TGF-β1 (Fig. 4F), implying regulation of the Nox4 translation process by the ERK pathway. Moreover, ERK inhibition also partially recovered loss of the mitochondrial membrane potential, reduced ROS generation, and prevented caspase-3 activation and apoptosis (Fig. 4, H–K). These results clearly suggest that podocyte apoptosis is mediated via ERK1/2 activation and that a probable cross-talk exists between ERK and mTOR signaling in podocytes.
We considered three possible intermediate pathways that might regulate TGF-β1-induced mTOR activation. These are the Akt, ERK, and Smad pathways, and they were explored to find the potential upstream regulator of mTOR in podocytes. Lamouille et al. (41) observed that translational regulation of epithelial-mesenchymal transition genes is mediated by TGF-β-induced mTOR activation through PI3K and Akt. Akt is considered a prosurvival pathway, and TGF-β1 is known to activate Akt through CD2AP in podocytes (52). Similar to the observation by Abe et al. (53), we detected TSC2 phosphorylation at Thr1462. In addition, p-TSC2 at Ser939 by TGF-β1 was also detected (Fig. 5E). Phosphorylation of these two positions on TSC2 by TGF-β1 indicates the possible involvement of Akt in mTOR activation (15). Surprisingly, inhibition of PI3K-Akt signaling using wortmannin did not significantly suppress mTORC1 activation, as assessed by phosphorylation of p70S6K (Fig. 5F). A similar result was obtained when Akt was blocked directly by triciribine, an Akt inhibitor (data not shown). The significance of in vitro phosphorylation of TSC2 at Ser939 and Thr1462 by TGF-β1 was hardly conceivable to us until now. However, these data clearly indicate that the PI3K-Akt pathway is not involved in TGF-β1-mediated mTOR activation in podocytes. The role of Akt in mTOR phosphorylation in vivo may also be excluded by the observation that there is no increase but, rather, a decrease in p-Akt in the isolated glomeruli from db/db mice compared with db/+ mice (54). Notably, TGF-β1 expression is increased in the kidney cortex of db/db mice (55).
There is evidence of Smad pathways regulating ERK1/2 and mTOR signaling. Phosphorylation of Smad3 stimulates mTOR activation to increase the synthesis of collagen in mesangial cells (42). In another study, sustained activation of Smad3 by TGF-β1 suppresses deptor expression to activate mTORC1 and mTORC2 in a similar cell type (39). In renal fibroblasts, Smad3-Nox4-induced ROS generation phosphorylates ERK1/2,leading to myofibroblastic transformation (56). In podocytes, Smad3 phosphorylation has been reported previously to regulate ERK1/2 activation (49). However, their study has limitations because no inhibitory evidence of Smad3 for ERK1/2 activation is presented. Combined silencing of Smad2 and 3 or only Smad4 completely suppressed TGF-β1-stimulated ERK1/2, mTOR, and p70S6K phosphorylation, indicating that Smad activation is upstream of the ERK-mTOR cascade (Fig. 6, A–F). Surprisingly, knockdown of either Smad2 or Smad3 did not prevent TGF-β1-induced ERK1/2 and p70S6K phosphorylation (Fig. 6, G–I). This observation shows the redundancy of Smad2 and 3 in ERK1/2-mTOR activation and differs from the transcriptional regulation of Nox4 mRNA (31). Therefore, Smad signaling simultaneously initiates two independent pathways. One pathway regulates transcription in the nucleus and increases the concentration of Nox4 mRNA, whereas the other one activates translation machineries in the cytoplasm to accelerate Nox4 protein synthesis in podocytes. Our data also indicate that ERK1/2 activation by TGF-β1 varies between podocytes and mesangial cells and, therefore, should be considered carefully when developing new therapeutic strategies.
Activation of mTOR by ERK1/2 plays a critical role in cancer progression, and it occurs through phosphorylation of TSC2 at Ser664. Inactivation of TSC2 by ERK1/2-mediated phosphorylation accelerates mTOR activation (37, 38). ERK1/2 can also phosphorylate raptor through phosphorylation of the p90 ribosomal S6 kinase and, thereby, phosphorylates mTOR (57). Direct phosphorylation of raptor by ERK1/2 is also known to activate mTOR by phosphorylation (58). We identified that ERK1/2 is a potential upstream regulator of TGF-β1-induced mTOR activation in podocytes because U0126 and PD184352 clearly inhibited mTOR and p70S6K phosphorylation (Fig. 5, A–C). We also detected Ser664 phosphorylation on TSC2 after TGF-β1 treatment, which was completely inhibited by U0126 (Fig. 5D). Surprisingly, siRNA-mediated knockdown of TSC2 did not suppress TGF-β1-stimulated p70S6K phosphorylation, indicating that TSC2 phosphorylation does not play any role in TGF-β1-ERK1/2-mediated mTOR activation (Fig. 5G). However, when the ERK1/2 inhibitor U0126 was applied to siTSC2-treated groups, p-p70S6K disappeared completely. These observations clearly point out that TSC2-independent activation of mTOR occurs through ERK1/2 in podocytes. Studies have also shown that TSC2 is not a critical target of Akt during Drosophila development (59) and that Akt can activate mTOR by regulating ATP and AMP-activated protein kinase activity (60). Therefore, the role of TSC2 phosphorylation by ERK1/2 in mTOR activation is questionable in podocytes. The detailed mechanism of mTOR activation remains to be elucidated, and we are working in that direction at present. However, in this manuscript, we clearly demonstrate the role of ERK1/2 in TGF-β1-mediated mTOR activation, which occurs in a TSC2-independent pathway in cultured podocytes. It should also be noted that insulin-induced mTOR activation was not prevented by TSC2 knockdown, suggesting the existence of some other signaling routes transmitting a physiologic and pathophysiologic response across the cell membrane. A report has shown that insulin can bypass TSC2 phosphorylation in Akt-dependent mTOR activation through PRAS40 (proline-rich Akt/PKB substrate, 40 kDa) (61). A recent study has also shown that regulation of TSC2 by TGF-β1 is cell type-specific. Reduction of TSC2 in renal tubular cells mediates EMT and tubulointerstitial fibrosis by TGF-β1 (62). Therefore, although TSC2-mediated mTOR activation has been documented as a mediator of diabetic nephropathy (6, 32), TSC2-independented mTOR activation may participate in diabetes and/or other different types of nephritic diseases.
In summary, using a differentiated podocyte cell line, we demonstrated the role of the ERK1/2-mTORC1 signaling axis in TGF-β-induced podocyte apoptosis. Activation of Nox4 by mTOR produces oxidative stress-induced mitochondrial dysfunction and apoptosis in podocytes. The TGF-β1-induced ERK1/2-mTORC1 pathway accelerates the translation of Nox4 protein without participating in the transcription of Nox4 mRNA, which is primarily mediated by Smad2 and 3 (Fig. 7). Moreover, this mechanism of disease progression may not be restricted to Nox4 regulation and may be applicable to other TGF-β target genes in podocytes and other cell types. We also propose the beneficial effect of rapamycin against proteinuric kidney diseases caused by elevated TGF-β1. Moreover, the prosurvival effect of the ERK pathway inhibitor could be another promising therapy against podocyte depletion, especially PD184352, which may need more attention because it is used frequently in cancer research and is already being tested in clinical trials (63).
FIGURE 7.
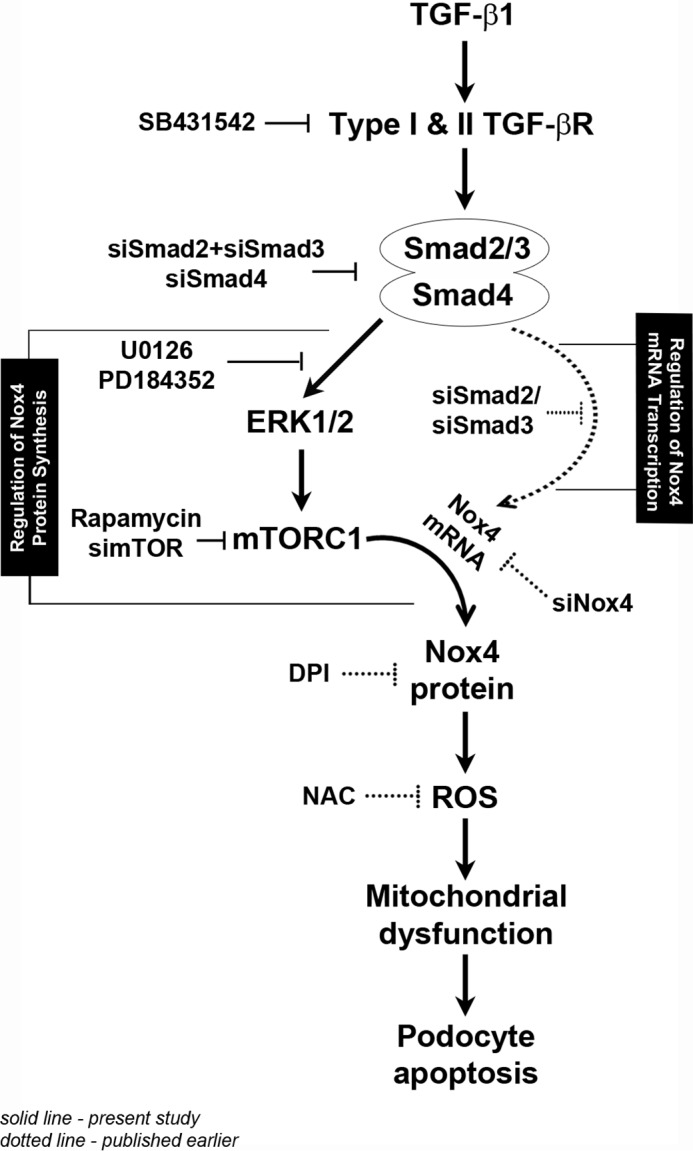
The proposed mechanism of TGF-β1-stimulated podocyte apoptosis. This schematic illustrates the mechanism of Nox4 up-regulation and oxidative stress-induced apoptosis by TGF-β1 in podocytes. We have reported previously that activation of the transcription factors Smad2 and Smad3 increases the Nox4 mRNA level (dotted lines). Here we show that simultaneous Smad-dependent activation of the ERK1/2-mTORC1 axis augments translation of the elevated Nox4 mRNA pool to elevate Nox4 protein to a pathogenic level, resulting in oxidative stress and podocyte apoptosis (solid lines). A detailed explanation of this figure is provided under “Discussion.” DPI, diphenyleneiodonium; NAC, N-acetylcysteine.
Author Contributions
K. S. P. and R. D. conceived and coordinated the study and wrote the paper. R. D. prepared most of the figures. S. X., T. T. N., X. Q., S. K. C., and S. J. K. performed some experiments and provided technical assistance. E. Y. L. and S. K. C. contributed to data analysis and discussions. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Prof. C. Rhodes (University of Chicago, Chicago) for adenoviral mTOR constructs.
This work was supported by National Research Foundation (NRF-2013R1A1A4A01010780) and Yonsei University Future-leading Research Initiative of 2014 (2014-22-0127). The authors declare that they have no conflicts of interest with the contents of this article.
- mToR
- mammalian target of rapamycin
- ROS
- reactive oxygen species
- KD
- kinase-dead
- Fluc
- firefly luciferase
- t-
- total
- TGF-βRI
- TGF-β receptor I
- simTOR
- mTOR siRNA
- sip70S6K
- p70S6K siRNA.
References
- 1. Shankland S. J. (2006) The podocyte's response to injury: role in proteinuria and glomerulosclerosis. Kidney Int. 69, 2131–2147 [DOI] [PubMed] [Google Scholar]
- 2. Jefferson J. A., Alpers C. E., and Shankland S. J. (2011) Podocyte biology for the bedside. Am. J. Kidney Dis. 58, 835–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kriz W., Shirato I., Nagata M., LeHir M., and Lemley K. V. (2013) The podocyte's response to stress: the enigma of foot process effacement. Am. J. Physiol. Renal Physiol. 304, F333–347 [DOI] [PubMed] [Google Scholar]
- 4. Mathieson P. W. (2012) The podocyte as a target for therapies: new and old. Nat. Rev. Nephrol. 8, 52–56 [DOI] [PubMed] [Google Scholar]
- 5. Inoki K., Mori H., Wang J., Suzuki T., Hong S., Yoshida S., Blattner S. M., Ikenoue T., Rüegg M. A., Hall M. N., Kwiatkowski D. J., Rastaldi M. P., Huber T. B., Kretzler M., Holzman L. B., Wiggins R. C., and Guan K. L. (2011) mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J. Clin. Invest. 121, 2181–2196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gödel M., Hartleben B., Herbach N., Liu S., Zschiedrich S., Lu S., Debreczeni-Mór A., Lindenmeyer M. T., Rastaldi M. P., Hartleben G., Wiech T., Fornoni A., Nelson R. G., Kretzler M., Wanke R., Pavenstädt H., Kerjaschki D., Cohen C. D., Hall M. N., Rüegg M. A., Inoki K., Walz G., and Huber T. B. (2011) Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J. Clin. Invest. 121, 2197–2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ito N., Nishibori Y., Ito Y., Takagi H., Akimoto Y., Kudo A., Asanuma K., Sai Y., Miyamoto K., Takenaka H., and Yan K. (2011) mTORC1 activation triggers the unfolded protein response in podocytes and leads to nephrotic syndrome. Lab. Invest. 91, 1584–1595 [DOI] [PubMed] [Google Scholar]
- 8. Rangan G. K., and Coombes J. D. (2007) Renoprotective effects of sirolimus in non-immune initiated focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 22, 2175–2182 [DOI] [PubMed] [Google Scholar]
- 9. Yang Y., Wang J., Qin L., Shou Z., Zhao J., Wang H., Chen Y., and Chen J. (2007) Rapamycin prevents early steps of the development of diabetic nephropathy in rats. Am. J. Nephrol. 27, 495–502 [DOI] [PubMed] [Google Scholar]
- 10. Bonegio R. G., Fuhro R., Wang Z., Valeri C. R., Andry C., Salant D. J., and Lieberthal W. (2005) Rapamycin ameliorates proteinuria-associated tubulointerstitial inflammation and fibrosis in experimental membranous nephropathy. J. Am. Soc. Nephrol. 16, 2063–2072 [DOI] [PubMed] [Google Scholar]
- 11. Letavernier E., Bruneval P., Mandet C., Duong Van Huyen J. P., Péraldi M. N., Helal I., Noël L. H., and Legendre C. (2007) High sirolimus levels may induce focal segmental glomerulosclerosis de novo. Clin. J. Am. Soc. Nephrol. 2, 326–333 [DOI] [PubMed] [Google Scholar]
- 12. Cutler N. S., Heitman J., and Cardenas M. E. (1999) TOR kinase homologs function in a signal transduction pathway that is conserved from yeast to mammals. Mol. Cell. Endocrinol. 155, 135–142 [DOI] [PubMed] [Google Scholar]
- 13. Martin K. A., and Blenis J. (2002) Coordinate regulation of translation by the PI 3-kinase and mTOR pathways. Adv. Cancer Res. 86, 1–39 [DOI] [PubMed] [Google Scholar]
- 14. Laplante M., and Sabatini D. M. (2012) mTOR signaling in growth control and disease. Cell 149, 274–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ma X. M., and Blenis J. (2009) Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 10, 307–318 [DOI] [PubMed] [Google Scholar]
- 16. Lamming D. W., and Sabatini D. M. (2013) A central role for mTOR in lipid homeostasis. Cell Metab. 18, 465–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Morita M., Gravel S. P., Chénard V., Sikström K., Zheng L., Alain T., Gandin V., Avizonis D., Arguello M., Zakaria C., McLaughlan S., Nouet Y., Pause A., Pollak M., Gottlieb E., Larsson O., St-Pierre J., Topisirovic I., and Sonenberg N. (2013) mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 18, 698–711 [DOI] [PubMed] [Google Scholar]
- 18. Holz M. K., Ballif B. A., Gygi S. P., and Blenis J. (2005) mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 123, 569–580 [DOI] [PubMed] [Google Scholar]
- 19. Shahbazian D., Roux P. P., Mieulet V., Cohen M. S., Raught B., Taunton J., Hershey J. W., Blenis J., Pende M., and Sonenberg N. (2006) The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J. 25, 2781–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sarbassov D. D., Ali S. M., Kim D. H., Guertin D. A., Latek R. R., Erdjument-Bromage H., Tempst P., and Sabatini D. M. (2004) Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 14, 1296–1302 [DOI] [PubMed] [Google Scholar]
- 21. Moore S. F., Hunter R. W., and Hers I. (2011) mTORC2 protein complex-mediated Akt (protein kinase B) serine 473 phosphorylation is not required for Akt1 activity in human platelets [corrected]. J. Biol. Chem. 286, 24553–24560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Efeyan A., and Sabatini D. M. (2013) Nutrients and growth factors in mTORC1 activation. Biochem. Soc. Trans. 41, 902–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bar-Peled L., and Sabatini D. M. (2014) Regulation of mTORC1 by amino acids. Trends Cell Biol. 24, 400–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vollenbröker B., George B., Wolfgart M., Saleem M. A., Pavenstädt H., and Weide T. (2009) mTOR regulates expression of slit diaphragm proteins and cytoskeleton structure in podocytes. Am. J. Physiol. Renal Physiol. 296, F418–426 [DOI] [PubMed] [Google Scholar]
- 25. Akhurst R. J., and Hata A. (2012) Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 11, 790–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Border W. A., and Noble N. A. (1994) Transforming growth factor β in tissue fibrosis. N. Engl. J. Med. 331, 1286–1292 [DOI] [PubMed] [Google Scholar]
- 27. Schiffer M., Bitzer M., Roberts I. S., Kopp J. B., ten Dijke P., Mundel P., and Böttinger E. P. (2001) Apoptosis in podocytes induced by TGF-β and Smad7. J. Clin. Invest. 108, 807–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim J. H., Kim B. K., Moon K. C., Hong H. K., and Lee H. S. (2003) Activation of the TGF-β/Smad signaling pathway in focal segmental glomerulosclerosis. Kidney Int. 64, 1715–1721 [DOI] [PubMed] [Google Scholar]
- 29. Ghayur A., Liu L., Kolb M., Chawla A., Lambe S., Kapoor A., and Margetts P. J. (2012) Adenovirus-mediated gene transfer of TGF-β1 to the renal glomeruli leads to proteinuria. Am. J. Pathol. 180, 940–951 [DOI] [PubMed] [Google Scholar]
- 30. Daehn I., Casalena G., Zhang T., Shi S., Fenninger F., Barasch N., Yu L., D'Agati V., Schlondorff D., Kriz W., Haraldsson B., and Bottinger E. P. (2014) Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J. Clin. Invest. 124, 1608–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Das R., Xu S., Quan X., Nguyen T. T., Kong I. D., Chung C. H., Lee E. Y., Cha S. K., and Park K. S. (2014) Upregulation of mitochondrial Nox4 mediates TGF-β-induced apoptosis in cultured mouse podocytes. Am. J. Physiol. Renal Physiol. 306, F155–167 [DOI] [PubMed] [Google Scholar]
- 32. Eid A. A., Ford B. M., Bhandary B., de Cassia Cavaglieri R., Block K., Barnes J. L., Gorin Y., Choudhury G. G., and Abboud H. E. (2013) Mammalian target of rapamycin regulates Nox4-mediated podocyte depletion in diabetic renal injury. Diabetes 62, 2935–2947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 34. Park K. S., Wiederkehr A., Kirkpatrick C., Mattenberger Y., Martinou J. C., Marchetti P., Demaurex N., and Wollheim C. B. (2008) Selective actions of mitochondrial fission/fusion genes on metabolism-secretion coupling in insulin-releasing cells. J. Biol. Chem. 283, 33347–33356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tabatabaian F., Dougherty K., Di Fulvio M., and Gomez-Cambronero J. (2010) Mammalian target of rapamycin (mTOR) and S6 kinase down-regulate phospholipase D2 basal expression and function. J. Biol. Chem. 285, 18991–19001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Briaud I., Dickson L. M., Lingohr M. K., McCuaig J. F., Lawrence J. C., and Rhodes C. J. (2005) Insulin receptor substrate-2 proteasomal degradation mediated by a mammalian target of rapamycin (mTOR)-induced negative feedback down-regulates protein kinase B-mediated signaling pathway in β-cells. J. Biol. Chem. 280, 2282–2293 [DOI] [PubMed] [Google Scholar]
- 37. Ma L., Chen Z., Erdjument-Bromage H., Tempst P., and Pandolfi P. P. (2005) Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 121, 179–193 [DOI] [PubMed] [Google Scholar]
- 38. Ma L., Teruya-Feldstein J., Bonner P., Bernardi R., Franz D. N., Witte D., Cordon-Cardo C., and Pandolfi P. P. (2007) Identification of S664 TSC2 phosphorylation as a marker for extracellular signal-regulated kinase mediated mTOR activation in tuberous sclerosis and human cancer. Cancer Res. 67, 7106–7112 [DOI] [PubMed] [Google Scholar]
- 39. Das F., Ghosh-Choudhury N., Bera A., Dey N., Abboud H. E., Kasinath B. S., and Choudhury G. G. (2013) Transforming growth factor β integrates Smad 3 to mechanistic target of rapamycin complexes to arrest deptor abundance for glomerular mesangial cell hypertrophy. J. Biol. Chem. 288, 7756–7768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lamouille S., Connolly E., Smyth J. W., Akhurst R. J., and Derynck R. (2012) TGF-β-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J. Cell Sci. 125, 1259–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lamouille S., and Derynck R. (2007) Cell size and invasion in TGF-β-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J. Cell Biol. 178, 437–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rozen-Zvi B., Hayashida T., Hubchak S. C., Hanna C., Platanias L. C., and Schnaper H. W. (2013) TGF-β/Smad3 activates mammalian target of rapamycin complex-1 to promote collagen production by increasing HIF-1α expression. Am. J. Physiol. Renal Physiol. 305, F485–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim S. I., Kwak J. H., Na H. J., Kim J. K., Ding Y., and Choi M. E. (2009) Transforming growth factor-β (TGF-β1) activates TAK1 via TAB1-mediated autophosphorylation, independent of TGF-β receptor kinase activity in mesangial cells. J. Biol. Chem. 284, 22285–22296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Moustakas A., and Heldin C. H. (2005) Non-Smad TGF-β signals. J. Cell Sci. 118, 3573–3584 [DOI] [PubMed] [Google Scholar]
- 45. Eid A. A., Ford B. M., Block K., Kasinath B. S., Gorin Y., Ghosh-Choudhury G., Barnes J. L., and Abboud H. E. (2010) AMP-activated protein kinase (AMPK) negatively regulates Nox4-dependent activation of p53 and epithelial cell apoptosis in diabetes. J. Biol. Chem. 285, 37503–37512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Eid A. A., Gorin Y., Fagg B. M., Maalouf R., Barnes J. L., Block K., and Abboud H. E. (2009) Mechanisms of podocyte injury in diabetes: role of cytochrome P450 and NADPH oxidases. Diabetes 58, 1201–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang Z., Wei X., Zhang Y., Ma X., Li B., Zhang S., Du P., Zhang X., and Yi F. (2009) NADPH oxidase-derived ROS contributes to upregulation of TRPC6 expression in puromycin aminonucleoside-induced podocyte injury. Cell. Physiol. Biochem. 24, 619–626 [DOI] [PubMed] [Google Scholar]
- 48. Chiluiza D., Krishna S., Schumacher V. A., and Schlöndorff J. (2013) Gain-of-function mutations in transient receptor potential C6 (TRPC6) activate extracellular signal-regulated kinases 1/2 (ERK1/2). J. Biol. Chem. 288, 18407–18420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yu L., Lin Q., Liao H., Feng J., Dong X., and Ye J. (2010) TGF-β1 induces podocyte injury through Smad3-ERK-NF-κB pathway and Fyn-dependent TRPC6 phosphorylation. Cell. Physiol. Biochem. 26, 869–878 [DOI] [PubMed] [Google Scholar]
- 50. Lee M. K., Pardoux C., Hall M. C., Lee P. S., Warburton D., Qing J., Smith S. M., and Derynck R. (2007) TGF-β activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 26, 3957–3967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Simeone D. M., Zhang L., Graziano K., Nicke B., Pham T., Schaefer C., and Logsdon C. D. (2001) Smad4 mediates activation of mitogen-activated protein kinases by TGF-β in pancreatic acinar cells. Am. J. Physiol. Cell Physiol. 281, C311–319 [DOI] [PubMed] [Google Scholar]
- 52. Xavier S., Niranjan T., Krick S., Zhang T., Ju W., Shaw A. S., Schiffer M., and Böttinger E. P. (2009) TβRI independently activates Smad- and CD2AP-dependent pathways in podocytes. J. Am. Soc. Nephrol. 20, 2127–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Abe Y., Sakairi T., Beeson C., and Kopp J. B. (2013) TGF-beta1 stimulates mitochondrial oxidative phosphorylation and generation of reactive oxygen species in cultured mouse podocytes, mediated in part by the mTOR pathway. Am. J. Physiol. Renal Physiol. 305, F1477–1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tejada T., Catanuto P., Ijaz A., Santos J. V., Xia X., Sanchez P., Sanabria N., Lenz O., Elliot S. J., and Fornoni A. (2008) Failure to phosphorylate AKT in podocytes from mice with early diabetic nephropathy promotes cell death. Kidney Int. 73, 1385–1393 [DOI] [PubMed] [Google Scholar]
- 55. Sedeek M., Callera G., Montezano A., Gutsol A., Heitz F., Szyndralewiez C., Page P., Kennedy C. R., Burns K. D., Touyz R. M., and Hébert R. L. (2010) Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: implications in type 2 diabetic nephropathy. Am. J. Physiol. Renal Physiol. 299, F1348–1358 [DOI] [PubMed] [Google Scholar]
- 56. Bondi C. D., Manickam N., Lee D. Y., Block K., Gorin Y., Abboud H. E., and Barnes J. L. (2010) NAD(P)H oxidase mediates TGF-β1-induced activation of kidney myofibroblasts. J. Am. Soc. Nephrol. 21, 93–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Carrière A., Cargnello M., Julien L. A., Gao H., Bonneil E., Thibault P., and Roux P. P. (2008) Oncogenic MAPK signaling stimulates mTORC1 activity by promoting RSK-mediated raptor phosphorylation. Curr. Biol. 18, 1269–1277 [DOI] [PubMed] [Google Scholar]
- 58. Carriere A., Romeo Y., Acosta-Jaquez H. A., Moreau J., Bonneil E., Thibault P., Fingar D. C., and Roux P. P. (2011) ERK1/2 phosphorylate Raptor to promote Ras-dependent activation of mTOR complex 1 (mTORC1). J. Biol. Chem. 286, 567–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dong J., and Pan D. (2004) Tsc2 is not a critical target of Akt during normal Drosophila development. Genes Dev. 18, 2479–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hahn-Windgassen A., Nogueira V., Chen C. C., Skeen J. E., Sonenberg N., and Hay N. (2005) Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J. Biol. Chem. 280, 32081–32089 [DOI] [PubMed] [Google Scholar]
- 61. Vander Haar E., Lee S. I., Bandhakavi S., Griffin T. J., and Kim D. H. (2007) Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 9, 316–323 [DOI] [PubMed] [Google Scholar]
- 62. Thakur S., Viswanadhapalli S., Kopp J. B., Shi Q., Barnes J. L., Block K., Gorin Y., and Abboud H. E. (2015) Activation of AMP-activated protein kinase prevents TGF-β1-induced epithelial-mesenchymal transition and myofibroblast activation. Am. J. Pathol. 185, 2168–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang D., Boerner S. A., Winkler J. D., and LoRusso P. M. (2007) Clinical experience of MEK inhibitors in cancer therapy. Biochim. Biophys. Acta 1773, 1248–1255 [DOI] [PubMed] [Google Scholar]