

Abstract
d-Serine is a physiological co-agonist that activates N-methyl d-aspartate receptors (NMDARs) and is essential for neurotransmission, synaptic plasticity, and behavior. d-Serine may also trigger NMDAR-mediated neurotoxicity, and its dysregulation may play a role in neurodegeneration. d-Serine is synthesized by the enzyme serine racemase (SR), which directly converts l-serine to d-serine. However, many aspects concerning the regulation of d-serine production under physiological and pathological conditions remain to be elucidated. Here, we investigate possible mechanisms regulating the synthesis of d-serine by SR in paradigms relevant to neurotoxicity. We report that SR undergoes nucleocytoplasmic shuttling and that this process is dysregulated by several insults leading to neuronal death, typically by apoptotic stimuli. Cell death induction promotes nuclear accumulation of SR, in parallel with the nuclear translocation of GAPDH and Siah proteins at an early stage of the cell death process. Mutations in putative SR nuclear export signals (NESs) elicit SR nuclear accumulation and its depletion from the cytosol. Following apoptotic insult, SR associates with nuclear GAPDH along with other nuclear components, and this is accompanied by complete inactivation of the enzyme. As a result, extracellular d-serine concentration is reduced, even though extracellular glutamate concentration increases severalfold. Our observations imply that nuclear translocation of SR provides a fail-safe mechanism to prevent or limit secondary NMDAR-mediated toxicity in nearby synapses.
Keywords: apoptosis; cell death; excitotoxicity; glutamate receptor; N-methyl-d-aspartate receptor (NMDA receptor, NMDAR); nuclear translocation; D-serine; glycine; neurotoxicity; serine racemase
Introduction
d-Serine is an endogenous ligand for NMDA receptors mediating several physiological and pathological processes, including neurotransmission, memory formation, and neurotoxicity (1–3). d-Serine is synthesized from l-serine by the enzyme serine racemase (SR)3 (4–6). Most previous studies on SR and d-serine have focused on astrocytes (7–9). Astrocytic SR interacts with, and is activated by, Grip-1 and Pick-1 (10–12). Glial SR is also inhibited by phosphatidylinositol lipids, a process influenced by metabotropic glutamate receptor activation in astrocytes (13). Disc-1, a protein implicated in schizophrenia, binds and stabilizes SR in astrocytes but not in neurons (12). Recent studies, however, have indicated that SR is predominantly expressed by neurons (14–20) and that neurons account for the vast majority of cells containing d-serine in the cerebral cortex and hippocampus (15, 17). Neuronal SR activity is reduced by interaction with the synaptic protein Stargazin (21). In neurons, d-serine synthesis is enhanced by FBXO22, which prevents the association of SR to intracellular membranes (22), and by Golga3, which reduces the degradation of SR by the ubiquitin system (23).
SR knock-out (KO) mice display phenotypic abnormalities compatible with NMDAR hypofunction (24–26). Furthermore, they are resistant to NMDAR-mediated neurotoxicity and β-amyloid-induced cell death in vivo (27), and are less susceptible to stroke damage upon middle cerebral artery occlusion (20, 28).
In addition to promoting neuronal death, NMDAR overactivation elicits translocation of SR from the cytosol to the dendritic membrane leading to inhibition of d-serine synthesis (29). However, possible SR regulation within the wider context of cell death has not been previously investigated. We now raise the possibility that SR localization and activity may be regulated by broader mechanisms of cell death, such as apoptotic stimuli, in a manner that does not depend on direct NMDA receptor activation.
We report that a variety of apoptotic cell death insults induce SR translocation to the nucleus and that this process is associated with its inactivation, which may constitute a fail-safe mechanism for preventing neurotoxicity in vicinal cells. In addition to its role as a marker of neuronal death, SR translocation links cellular injury with d-serine production, with potential implications for the pathology of neurodegenerative diseases.
Experimental Procedures
Materials
l- and d-serine were purchased from Bachem. l-[3H]Serine and d-[3H]serine were purchased from American Radiolabeled Chemicals. Acetonitrile, NMDA, staurosporine, etoposide, thapsigargin, tetrahydrofuran, o-phthaldialdehyde, leptomycin B, β-mercaptoethanol (BME), and DAPI were purchased from Sigma. MK-801 was obtained from Tocris. Micrococcal nuclease, restriction enzymes, and protein A magnetic beads were purchased from New England Biolabs. A subcellular protein fractionation kit was purchased from Thermo Scientific. Basal medium Eagle's, minimum essential medium, fetal bovine serum, penicillin/streptomycin, penicillin/streptomycin/amphotericin, trypsin, and soybean trypsin inhibitor were obtained from Biological Industries (Kibbutz Beit Haemek, Israel). Normal goat serum was purchased from Jackson ImmunoResearch. B27 supplement was prepared in-house as described previously (30). Neuro-2A cells were obtained from ATCC (Manassas, VA).
Cell Cultures
Animals were killed by decapitation after isoflurane anesthesia with the approval of the committee for the supervision of animal experiments (Technion-Israel Institute of Technology). Serum-free neuronal cultures from the cerebral cortex were prepared from day E16 to E18 Sprague-Dawley rat embryos or C57Bl/6 mouse embryos, as described previously (18). These cultures were previously shown to contain less than 2% glial fibrillary acidic protein-positive glial cells (18, 31). The cells were plated in poly-d-lysine-coated (0.1 mg/ml) 6-well plates (Nunc) at a density of 2.0–2.2 × 106 cells/well. For immunocytochemistry, neurons were seeded on glass coverslips coated with poly-d-lysine (0.5 mg/ml) in 12-well plates at a concentration of 0.75–1.2 × 106 cells/well. The culture consisted of neurobasal (NB) medium supplemented with B27, 0.2 mm glutamine, and penicillin + streptomycin (NB/B27). On DIV1, media were changed to fresh NB/B27. Afterward, half of the culture media was replaced every 3 days with fresh NB/B27. Biochemical experiments were carried out on DIV10–12. Immunocytochemistry experiments were carried out on DIV8–10. Neuro-2A was cultured in DMEM supplemented with 10% FBS and 4 mm glutamine. Primary astroglial cultures were prepared from postnatal day 0 Sprague-Dawley rat pups as described previously (18). Astrocytes were maintained in BME supplemented with 10% FBS and 1 mm glutamine.
Western Blots
Mouse anti-β-actin (clone C4 1:1,000), mouse anti-GAPDH (clones 6C5 and 0411, 1:200), and rabbit anti-histone 2B (FL-126, 1:1000) were obtained from Santa Cruz Biotechnology. Rabbit anti-GAPDH (G9545, 1:10,000), mouse anti-β-tubulin (clone SAP 4G5, 1:10,000), and rabbit anti-GADD153/CHOP10 (G6916, 1:1000) were obtained from Sigma. Mouse anti-neuronal nucleus protein (NeuN)(clone A60, 1:500) was obtained from Chemicon. Mouse anti-lamin A/C (1:1000), rabbit anti-lamin A/C (1:1000), rabbit monoclonal anti-cleaved caspase 3 (clone 5A1E, 1:1000), and rabbit monoclonal anti-cleaved PARP1 (clone D64E10, 1:1000) were obtained from Cell Signaling. When compared with a global anti-caspase 3 antibody, anti-cleaved caspase 3 specifically recognizes the cleaved fragment and not the uncleaved protein (32–34). Likewise, when compared with a global anti-PARP-1 antibody, anti-cleaved PARP-1 specifically recognizes the cleaved fragment and not the uncleaved protein (35–37). Mouse anti-histone3 (1:1000) was obtained from Abcam. Rabbit anti-seven in absentia homologue (Siah) (1:500) was a gift from Dr. Simone Engelender (Technion-Israel Institute of Technology) and has been described in earlier studies (38, 39). Rabbit affinity-purified anti-SR antibody (0.2–0.3 μg/ml) (18) or guinea pig anti-SR have also been described elsewhere (40). Anti-rabbit, anti-mouse, and anti-guinea pig peroxide-conjugated IgG (1:20,000) was purchased from Jackson ImmunoResearch. EasyBlot peroxidase-conjugated anti-rabbit IgG (GTX221666-01, 1:1000) was obtained from GeneTex.
Immunocytochemistry
Cultured cells were fixed for 20 min with fresh 4% paraformaldehyde followed by three washes with PBS. Cells were blocked and permeabilized at room temperature for 90 min with PBS supplemented with 8% normal goat serum, 80 mm NaCl, 2 mg/ml IgG-free bovine serum albumin (BSA), and 0.1% Triton X-100. Nuclear localization was revealed by the use of serum anti-SR at 1:400 as described previously (18) and a monoclonal antibody against lamin A/C (1:200, Cell Signaling). Following primary antibody incubation for 16–20 h at 4 °C, co-localization was revealed with anti-rabbit Cy3 and anti-mouse Cy2 or anti-rabbit Cy2 and anti-mouse Cy3 secondary antibodies (Jackson ImmunoResearch) in PBS supplemented with 4% normal goat serum and 0.1% Triton X-100. After washing, 0.1 μg/ml DAPI was added for nuclear labeling, followed by additional washes. Controls were carried out by pre-absorption of the antibody with 10 μg of purified His-SR protein. Laser scanning confocal microscopy was carried out using an LSM 510 Meta laser scanning confocal system (Zeiss). Optical sections of 2–3 μm were performed at 1024 × 1024-pixel resolution using 1× zoom or 3× zoom. Acquisition was performed using 510 LSM software.
Time-lapse Microscopy
Neuronal cultures were prepared as described above except that they were seeded on an FD35-100 fluoro dish. On DIV5, the cells were transduced with lentivirus harboring GFP-SR as described below under “Lentivirus Production and Infection.” Neurons were imaged in a time-lapse system composed of a Zeiss Axio Observer inverted motorized fluorescent microscope, with an X-cite metal-halide light source and a high resolution black and white fluorescent camera (Zeiss Hrm). Cells were placed in the environmental chamber (at 37 °C humidity and 5% CO2) and imaged via an Axio Observer Z1, camera image 14-bit depth acquisition, and 16-bit grayscale pixel type, using a 20× objective (Cell Observer, Zeiss Inverted for Live Imaging). Acquisition was analyzed using Zeiss Axio Vision 4.8 software. Images were recorded from 10 different fields every 10 min. One micromolar staurosporine (STS) was added after 30 min (frame 3), and SR localization was monitored via imaging at a constant 10-min interval. After 141 frames (∼24 h), 10 μg/ml Hoechst 33342 (Thermo Scientific) was added to confirm nuclear localization.
Purified Nuclei Preparation
Experiments were carried out on DIV12, and test compounds drugs were added to the culture medium unless otherwise specified. Cells were scraped in ice-cold buffer containing 20 mm Tris-HCl (pH 7.4) and 100 mm NaCl supplemented with protease inhibitor mixture (Mini-complete, Roche Applied Science) and then lysed by three freeze and thaw cycles consisting of immersion in liquid nitrogen followed by a 37 °C bath. An aliquot of the homogenate was put aside, and the remaining lysate was centrifuged at 1500 × g for 10 min (4 °C) to give a supernatant (S1) and a crude nuclear pellet (P1). S1 was put aside and P1 was further purified as described previously (41, 42)with the following modifications. Washed P1 fraction was suspended in nuclear lysis sucrose buffer consisting of 0.25% Triton X-100, 1 mm potassium phosphate (KPi) (pH 6.5), 1 mm MgCl2, and 1.32 m sucrose supplemented with protease inhibitors, followed by homogenization with 6–7 strokes in a 2-ml glass homogenizer. To obtain purified nuclei (PN), the suspension was layered over a 1.7 m sucrose cushion and centrifuged at 53,000 × g for 75 min at 4 °C. The PN were washed by resuspension in nuclear lysis sucrose buffer and centrifugation. To obtain cytosolic and membrane fractions, S1 was centrifuged at 200,000 × g for 30 min at 4 °C.
Purified nuclei from SR-WT and SR-KO mouse brains were isolated as described previously (42). Briefly, freshly dissected cortical tissue was homogenized in 0.32 m sucrose, 1 mm MgCl2, 1 mm KPi (pH 6.5), and 0.25% Triton X-100. The crude nuclear fraction was pelleted by centrifugation at 1250 × g for 10 min at 4 °C and washed twice in the same buffer. Subsequently, the crude nuclear fraction was suspended in 2 m sucrose buffer and layered over a 2.4 m sucrose cushion followed by centrifugation at 53,000 × g for 75 min at 4 °C. The purified nuclei were washed twice by resuspension in homogenization buffer and centrifuged.
Subnuclear Fractionation
Nuclear subfractionation was carried out as described previously (43) with the following modifications. Cells were lysed by five passages through a 27-gauge needle in hypotonic buffer consisting of 10 mm Tris-HCl (pH 7.4), 10 mm KCl, and 1 mm MgCl2 supplemented with protease inhibitor mixture. The lysate was centrifuged at 800 × g for 10 min to obtain crude nuclei (P1) and post-nuclear supernatant (S1). Subsequently, PN were isolated from P1 as described above. The PN fraction was lysed with buffer containing 50 mm Tris-HCl (pH 7.4), 1% Nonidet P-40, 1% glycerol, 300 mm NaCl, and protease inhibitor mixture for 30 min on ice with occasional mixing. The nuclear lysate was centrifuged at 800 × g for 10 min at 4 °C to obtain a chromatin-enriched pellet (P3). To release the chromatin-bound proteins, the P3 fraction was treated for 30 min at 37 °C with 1 unit of micrococcal nuclease in buffer containing 50 mm Tris-HCl (pH 7.4), 50 mm NaCl, and 5 mm CaCl2. The reaction was terminated by the addition of 1 mm EGTA. Soluble (S3) and insoluble (P4) components, representing chromatin and insoluble fraction consisting of nuclear lamina and matrix-associated proteins, respectively, were then separated by centrifugation at 800 × g for 10 min at 4 °C.
Lentiviral and DNA Constructs
For neuronal preferential expression, we replaced the EF1α promoter of the lentiviral pWPXLd vector (obtained from Dr. Didier Trono, École Polytechnique Fédérale de Lausanne (EPF)) with the human synapsin promoter generating the pWPXLd-hSyn vector. Briefly, we inserted an SbfI restriction site at the 3′ end of the EF1α promoter of the pWPXLd vector (at position 3282) by using complementary primers and a QuikChange II XL site-directed mutagenesis kit (Stratagene). Subsequently, the EF1α promoter was removed by cleavage at the SalI/SbfI sites and replaced by human synapsin promoter amplified from AAV-6P-SEWB vector (obtained from Dr. Sebastian Kugler, University of Goettingen). To express SR, we amplified GFP-SR by PCR from mouse SR-cloned into pEGFP-C1 vector (obtained from Dr. Michael Schell, Uniformed Services University). GFP-SR was subcloned into the pWPXLd-hSyn vector at BamHI and SpeI sites, replacing the GFP of the vector. SR mutants lacking NES consensus sequences were produced by PCR using a QuikChange II XL site-directed mutagenesis kit. All DNA constructs were verified by double-strand sequencing.
Lentivirus Production and Infection
GFP-SR and GFP-SR-6mut harboring mutations in putative nuclear export signals (L280Q, I282Q, V288Q, L290Q, L294Q, and V301Q) were subcloned into a pWPXLd-hSyn lentiviral vector. The virus was produced by PEI-mediated transfection of the lentiviral vector (13 μg), packaging system plasmid pCMV-dR8.74 (8.7 μg), and VSV-G envelope plasmid pMDG (4.6 μg) in HEK293T cells grown in 10-cm culture dishes. Virus was purified as described previously (44) with some modifications. Briefly, virus-containing medium was collected 48 and 72 h post-transfection, pooled, centrifuged at 1500 × g for 5 min to remove cell debris, and filtered through a 0.22-μm filter. Then viral stocks were concentrated by centrifugation at 50,000 × g for 2 h at 4 °C. The pellet was resuspended in 80–100 μl of NB/B27 medium. Virus stocks were stored at −70 °C until use. Virus multiplicity of infection was determined by infecting HEK293 cells and monitoring immunofluorescence for GFP.
Primary neuronal cultures were infected at DIV4 at a 20–40 multiplicity of infection. Expression of GFP-SR was monitored by immunocytochemistry and Western blot analysis with anti-SR antibody. We used cultures that exhibited about 40% infection efficiency on DIV12 to DIV14. Primary astroglial cultures were infected at DIV14 with 10 multiplicities of infection.
NMDAR-elicited Neurotoxicity
Neuronal primary cultures (DIV11–15) were cultured in 96-well plates using house-made neurobasal medium (31) prepared without the addition of glycine and l-serine. The medium was supplemented with 2% B27 and with 0.1 mm l-serine that was previously rendered free of contaminant d-serine by incubation with d-serine dehydratase (45). For the experiments, cells were transitioned to HCSS medium lacking magnesium and containing 20 mm HEPES-NaOH (pH 7.4), 1.8 mm CaCl2, 15 mm glucose, 5.4 mm KCl, and 120 mm NaCl, with or without 30 μm NMDA for 20 min. Then the medium was returned, and lactate dehydrogenase (LDH) release was monitored in sextuplicate using a TOX7 assay kit (Sigma) and an Infinite F200 plate reader (Tecan). The value of each well was normalized by the total LDH calculated after determining the activity remaining in the cell lysates.
Co-immunoprecipitation Experiments
A crude nuclear fraction of neuronal culture cells was obtained, and nuclei were disrupted by sonication in 10 mm Tris-HCl (pH 7.4), 10 mm KCl, 1.5 mm MgCl2, 2 mm DTT, and a Mini-complete protease inhibitor mixture. Next, the suspension was diluted in 40 mm Tris-HCl (pH 7.4), 150 mm NaCl, 0.5% Triton X-100, and 2 mm DTT (final concentrations). Two micrograms of purified rabbit anti-SR antibody were added by overnight rotation at 4 °C, and SR was immunoprecipitated with protein A magnetic beads. The beads were washed six times with the immunoprecipitation buffer, and SR was probed with guinea pig anti-SR, as described previously (40). Co-immunoprecipitation with lamin was revealed with mouse anti-lamin A/C. GAPDH co-immunoprecipitation was revealed with rabbit anti-GAPDH (Sigma) followed by Easy Blot anti-rabbit IgG (GeneTex). For GAPDH immunoprecipitation, the nuclear fraction was processed as above, except that 10 μg of purified anti-GAPDH (Sigma) or a rabbit IgG control was covalently coupled to protein A magnetic beads, as described previously (46).
Determination of d-Serine and SR Activity
To induce SR nuclear translocation and/or accumulation, neuronal cultures were treated with 1 μm STS or 20 nm LMB for 8 h in Neurobasal/B27 medium. Then the medium was replaced with a new one containing 5 mm l-serine lacking contaminant d-serine. After 12 h, the medium was collected, and extracellular d-serine and glutamate were monitored by HPLC analysis, as described previously (47).
Isolated cytosolic and nuclear fractions were assayed in vitro for d-serine synthesis in reaction medium containing 40 mm Tris-HCl (pH 8.0), 10 mm l-serine, 10 μm pyridoxal 5′-phosphate, 2 mm MgCl2, 0.5 mm ATPγS, 0.5% Triton X-100, 1 mm DTT, and 2 mm d-alanine (to prevent the degradation of d-serine by contaminant d-amino acid oxidase). The reaction was carried out under continuous agitation at 37 °C and stopped after 2–4 h by boiling for 5 min. 5% TCA was added to precipitate proteins, and d-serine synthesis was analyzed by HPLC as described previously (47). To determine the effect of soluble nucleoplasmic proteins on SR activity, HEK293 cells were transfected with mouse SR in pRK5-KS plasmid, and after 48 h, the cells were lysed by three cycles of freezing and thawing in lysis buffer containing 10 mm Tris-HCl (pH 7.4), 10 mm KCl, 1.5 mm MgCl2, 2 mm DTT, and a protease inhibitor mixture (mini complete EDTA-free, Roche Applied Science). Cytosolic fraction was isolated by 30 min of centrifugation at 200,000 × g. Nucleoplasm-enriched proteins were obtained by sonicating purified nuclei preparations of primary neuronal cultures in lysis buffer. Then Triton X-100 was added to 0.5% final concentration, followed by removal of insoluble nuclear envelope proteins by 20 min of centrifugation at 100,000 × g. The cytosolic fraction diluted at 0.01 mg of protein/ml was incubated for 2–6 h with nucleoplasmic protein extracts at 0.2–0.4 mg/ml. d-Serine production was monitored as described above.
Serine Uptake and Release
d-Serine and l-serine uptake were carried out as described previously (45, 48). Primary neuronal cultures were cultured in neurobasal, 2% B27 medium onto poly-d-lysine-coated 96-well plates (0.5 mg/ml). After 12–14 days in culture, the cells were incubated for 7 h with culture medium containing either vehicle (1% DMSO) or 1 μm STS. Then the culture medium was removed, and the cells were washed four times with HEPES-buffered saline solution (HBSS), containing 137 mm NaCl, 5.4 mm KCl, 0.34 mm K2HPO4, 0.44 mm KH2PO4, 0.41 mm MgSO4, 0.49 mm MgCl2, 1.07 mm CaCl2, 5.6 mm d-glucose, and 10 mm HEPES-NaOH (pH 7.4) at 23–25 °C. The uptake was started by addition of 80 μl of HBSS containing either 5 μm d-[3H]serine or l-[3H]serine (2 × 106 dpm/nmol). The cells were incubated both at 4 and 8 min at 23–25 °C, and the uptake was terminated by quickly washing the cells four times with ice-cold HBSS. The radioactivity was monitored after lysing the cells with 120 μl of water per well for 10 min. Blanks were carried out by incubating the cells with ice-cold uptake medium, and they typically accounted for less than 10% of the total radioactivity. The values were within the linear range of [3H]serine transport and were corrected for protein content in each well.
d-[3H]Serine release was carried out as described previously (45, 48). Primary neuronal cultures were incubated for 7 h in the presence or absence of STS as described above. Then the culture medium was removed by four washes with HBSS as described above, and the cells were preloaded for 30 min with 5 μm d-[3H]serine (4 × 106 DPM/nmol) in HBSS at 23–25 °C. Subsequently, the cultures were washed four times with ice-cold HBSS, and the release of d-[3H]serine was started by the addition of 200 μl of HBSS at 23–25 °C, containing either none or 1 mm l-serine. After 2 min, the release medium was transferred to another plate, and the radioactivity was counted on a scintillation counter. The linearity of d-[3H]serine release was assessed by comparing aliquots drawn at 1 and 2 min. Released d-[3H]serine (d-serineout) was monitored in the release medium, and the remaining intracellular d-[3H]serine (d-serinein) was determined after lysing the cells with 120 μl of water per well. In each experiment, the fractional d-serine release was calculated from at least sextuplicates by using the formula: (d-serineout/(d-serineout + d-serinein).
Statistical Analysis
Statistics were performed by means of repeated-measures analysis of variance and the Tukey multiple comparison test or by the two-tailed, paired Student's t test using GraphPad Prism software version 6.03 (GraphPad Inc.).
Results
SR Targeting Is Altered by Pathological Stimuli
We evaluated the effects of cell death-inducing agents that include endoplasmic reticulum (ER) stressors and apoptotic drugs on SR localization and d-serine synthesis. We found that a major part of SR accumulates in the nuclear fraction of primary cultured neurons (Fig. 1A, upper panel) in response to the ER stressor BME (49). The elevation in levels of nuclear SR is an early event that initiates after 1 h and progresses simultaneously with a reduction in cytosolic levels. After a 12-h treatment, the cytosolic levels of SR were barely visible (Fig. 1A, middle panel), and this was associated with a 3–4-fold increase in nuclear SR (Fig. 1A, graphs). No changes in total SR levels were observed in the cell homogenate (Fig. 1A, lower panel). Furthermore, loading controls such as neuronal nucleus protein (NeuN), β-actin, and β-tubulin were apparently unchanged in the different fractions (Fig. 1A).
FIGURE 1.

Translocation of SR to the nucleus upon cell death insults. A, primary cortical neuronal cultures (DIV10) treated with 4 mm BME display increased levels of SR in the purified nuclear fraction, associated with a pronounced reduction in cytosolic SR (Cyt) monitored by Western blot analysis. BME did not change SR levels in total homogenate (Hom). SR translocation was associated with induction of GADD153 expression, and a concomitant increase in Siah at the PN. Cell death induction was determined by cleavage of caspase 3 (cCasp3) and PARP1 (cPARP1) monitored in the homogenate fraction. Loading controls NeuN (PN), β-actin (Cyt), and β-tubulin (Hom) were unchanged. Graphs depict the densitometric quantification of SR normalized for loading controls in the cytosol and purified nucleus or the levels of cCasp3 and cleaved poly(ADP-ribose) polymerase 1 in homogenates, monitored after 12 h in the absence or presence of BME. The data represent the average ± S.E. of five independent experiments with different culture preparations. B, primary neuronal cultures express SR (red) in the cytosol and neuronal processes as shown by confocal laser microscopy (upper panels). A 10-h treatment with BME elicited translocation of SR to the nucleus where it co-localized with DAPI (blue), which labels cell nuclei (lower panels). Scale bar, 20 μm. The panels represent three experiments with different culture preparations. C, treatment of neurons with staurosporine (STS) or etoposide (ETO) for 8 h elicited similar effects as BME, without inducing the ER stress marker GADD153. Conversely, a 10-h treatment with thapsigargin (thaps) induced GADD153 expression but without eliciting SR translocation or cleavage of caspase 3 or PARP1. Graphs depict the densitometric quantification of SR normalized for loading controls in purified nucleus with and without the different treatments. The data represent the average ± S.E. of 11 (1 μm STS), 5 (1.2 μm thapsigargin), and 4 (100 μm etoposide) independent experiments with different culture preparations. D, neurons treated with H2O2 for 2 h, after which the medium was replaced, and cellular fractions were analyzed after 8 h. Graph depicts the densitometric quantification of SR normalized for loading controls in purified nucleus with and without H2O2. The data represent the average ± S.E. of three independent experiments with different culture preparations. E, NMDAR activation induced SR accumulation in the nucleus; neurons were treated with 100 μm NMDA for 30 min, and the medium was replaced and cellular fractions analyzed after 8 h. Graphs depict the densitometric quantification of SR normalized for loading controls in cytosol and purified nucleus, in the absence or presence of NMDA. The data represent the average ± S.E. of five independent experiments with different culture preparations. CTL, control. ***, **, *, different from control at p < 0.001, 0.01, and 0.05, respectively. n.s., not significantly significant.
ER stress such as that promoted by BME also leads to cell death via an apoptotic mechanism (50). To evaluate the cell death process, we monitored the cleavage of caspase 3, a process required for the activation and execution of apoptosis-associated proteolysis, DNA fragmentation, and changes in cell morphology (51–54). We also quantified the cleavage of poly(ADP-ribose) polymerase (PARP-1), which is a target of caspases during apoptosis (52, 54) and is involved in cell death mechanisms (55). Accordingly, we detected a time-dependent increase in caspase 3 cleavage in total cell homogenate along with SR accumulation in the nucleus (Fig. 1A, lower panel and graph), indicating that BME treatment also induced cell death. Likewise, PARP-1 cleavage was equally evident following BME treatment (Fig. 1A, lower panel and graph). To verify concomitant ER stress, we monitored the levels of growth arrest- and DNA damage-inducible gene 153 (GADD153), also known as C/EBP homologous protein (CHOP), a transcription factor that is normally expressed at very low levels but is robustly induced under ER stress (56, 57). GADD153/CHOP is required for ER stress-mediated apoptosis, regulating the levels of several genes related to cell survival, and it is therefore a useful marker of ER stress (57–59). As expected, GADD153/CHOP levels increased upon BME treatment; this was observed after 6 h (Fig. 1A, upper panel).
Translocation of Siah together with GAPDH to the nucleus plays a prominent role in neuronal apoptosis (60). We detected a time-dependent increase in nuclear Siah levels upon BME treatment (Fig. 1A, upper panel), thus serving as a cell death indicator along with cleaved caspase 3 and PARP-1.
We confirmed nuclear SR accumulation by immunochemistry experiments. BME treatment indeed elicited robust translocation of SR to the nucleus of primary cortical neurons, as revealed by substantial co-localization with DAPI (Fig. 1B, lower panels). In untreated neurons, SR was distributed mainly in the cytosol and dendritic processes (Fig. 1B, upper panels). Thus, nuclear accumulation of SR is not an artifact of subcellular fractionation experiments in vitro and constitutes the major intracellular targeting of SR upon toxic stimuli.
Cell Death Stimuli Promote SR Accumulation in the Nucleus
We next set out to determine the type of stimuli capable of driving SR into the nucleus. Similar to that observed with BME, incubation of primary neuronal cultures with STS, a classical apoptotic drug (61, 62), triggered pronounced nuclear accumulation of SR (Fig. 1C, upper panel, lanes 2 and 3, and graph) without affecting the total levels of SR in the cells (Fig. 1C, lower panel). Similar nuclear translocation was observed for GAPDH, known to translocate to the nucleus along with Siah during apoptosis (60), and this was associated with prominent cleavage of caspase 3 and PARP-1, whereas loading controls were apparently unaltered (Fig. 1C). In contrast to BME, however, addition of STS does not induce ER stress, as revealed by lack of GADD153 induction (Fig. 1C, upper panels). Treatment with etoposide, a topoisomerase II inhibitor and apoptotic drug (63, 64), induced SR and GAPDH nuclear accumulation associated with caspase 3 and PARP-1 cleavage, without inducing ER stress (Fig. 1C, lanes 6 and 7, and graph). In contrast, the ER stress inducer thapsigargin (56, 65) did not cause caspase 3 or PARP-1 cleavage while increasing the levels of the ER stress marker GADD153 (Fig. 1C, lanes 4 and 5). However, thapsigargin treatment did not increase nuclear SR (Fig. 1C, graph). Thus, it is likely that the nuclear SR accumulation caused by the ER stressor BME (Fig. 1A) is due to cell death induction, rather than to the ER stress itself.
Oxidative stress induced by H2O2 also leads to cell death and nuclear accumulation of SR (Fig. 1D and graph). In addition, we found that NMDAR activation induces nuclear accumulation of SR, followed by a reduction in cytosolic stores (Fig. 1E). Altogether, these data indicate that SR accumulates in the nucleus under a wide variety of cell death stimuli.
SR Undergoes Nucleocytoplasmic Shuttling
Does SR undergo nucleocytoplasmic shuttling under basal conditions? In silico analyses failed to indicate possible nuclear localization signals within SR (66, 67). To evaluate nucleocytoplasmic shuttling of SR, we treated primary neuronal cultures with LMB, an established inhibitor of the chromosome region maintenance 1 (CRM1)-dependent nuclear export system (68, 69), which is the major pathway for the export of proteins out of the nucleus (68). We found that 20 nm LMB augmented the steady-state levels of nuclear SR severalfold, without changing the loading control (Fig. 2A). Immunocytochemical experiments in primary neuronal cultures treated with LMB confirmed the increase in nuclear SR observed in the biochemical experiments (Fig. 2B). These data support the notion that SR undergoes functional export from the nucleus under physiological circumstances. Furthermore, using SR-KO mice as negative controls, we purified brain cell nuclei and found the presence of nuclear SR in the purified nuclear fraction (Fig. 2C).
FIGURE 2.

SR nuclear export inhibition and identification of nuclear export signals at SR. A, primary neuronal cultures at DIV10–12 were treated with LMB (20 nm) for 1 or 4 h or with STS for 8 h. Nuclear SR and loading control were monitored by Western blot analysis in purified nuclear fraction (PN). B, primary neuronal cultures were treated with LMB (20 nm) for 4 h and analyzed by immunocytochemistry and confocal laser microscopy. Untreated cells (control (CTL), upper panels) and LMB-treated cells (lower panels) are shown. Scale bar = 50 μm. C, purified nuclei from cortical brain tissue of wild type (WT) mice contain SR. Nuclei purified from SR-KO mice did not exhibit immunoreactivity to SR. NeuN served as loading control. D, five classes of putative nuclear export signals (NESs) were identified in the C terminus of SR. The hydrophobic amino acids that are critical for the NESs are highlighted in red. The underlined amino acids were mutated to Gln (glutamine) and corresponded to Leu-280 and Ile-282 (2mut), Leu-280, Ile-282, Val-288, and Leu-290 (4mut) or Leu-280, Ile-282, Val-288, Leu-290, Leu-294, and Val-301 (6mut), which disrupted 3, 5, and 9 of the putative NESs, respectively. E, mutations in putative NESs promoted nuclear accumulation of the protein. SR mutants harboring the mutations described in D were transfected into Neuro-2a cells, and levels of SR in the nuclear fraction and cytosol were monitored by Western blot analysis 48 h after transfection. Mutations in SR NESs promoted nuclear SR accumulation (upper panel), associated with a reduction in cytosolic SR (middle panel). No significant changes in total SR levels in the homogenate were observed (lower panel). PN, purified nucleus; Cyt, cytosol; H2B, histone 2B; Hom, total cell homogenate. The panels represent at least three experiments with different preparations.
Classic nuclear export requires NES sequences that are enriched in hydrophobic residues that interact directly with the CRM1/exportin1 system, the main pathway for the export of proteins out of the nucleus (68). We found that SR harbors at least nine putative NESs in its C terminus, belonging to five classes of NES consensus sequences (70). To disrupt the potential NESs, we mutated two to six conserved hydrophobic residues to glutamine (Fig. 2D, underlined residues). Transfection of mouse neuroblastoma Neuro-2A cells with SR variants led to an increase in the steady-state levels of nuclear SR and a concomitant depletion of cytosolic stores, without the addition of any cell death stimuli (Fig. 2E, compare lane 1 with lanes 2–4). SR nuclear accumulation increases in proportion to the number of hydrophobic putative residues that were mutated to glutamine, with no changes in total cell expression (Fig. 2E). The data indicate that the mutations disrupted functional NESs, which are critical for SR nucleocytoplasmic shuttling.
We sought to monitor SR translocation in live cells using primary neuronal cultures infected with recombinant lentivirus harboring GFP-SR. Neurons expressing GFP-SR behaved in the same fashion as endogenous SR (Fig. 3A, upper panels). Upon STS addition, nuclear accumulation of GFP-SR was apparent, accompanied by major cytosolic depletion. Endogenous SR (Fig. 3A, SRend.) also accumulated in the nucleus following stimuli. Time-lapse microscopy indicated robust accumulation of GFP-SR in the nucleus ∼1 h after the addition of STS, whereas nearly all the enzyme accumulated in the nucleus after 12 h (Fig. 3B and supplemental Movie). To confirm the data obtained with Neuro-2A, we infected neurons with lentiviruses harboring GFP-SR and GFP-SR-NES 6mut. We found that GFP-SR is mostly cytosolic (Fig. 3C, upper panels), whereas the GFP-SR 6mut builds up in the neuronal nuclei (Fig. 3C).
FIGURE 3.
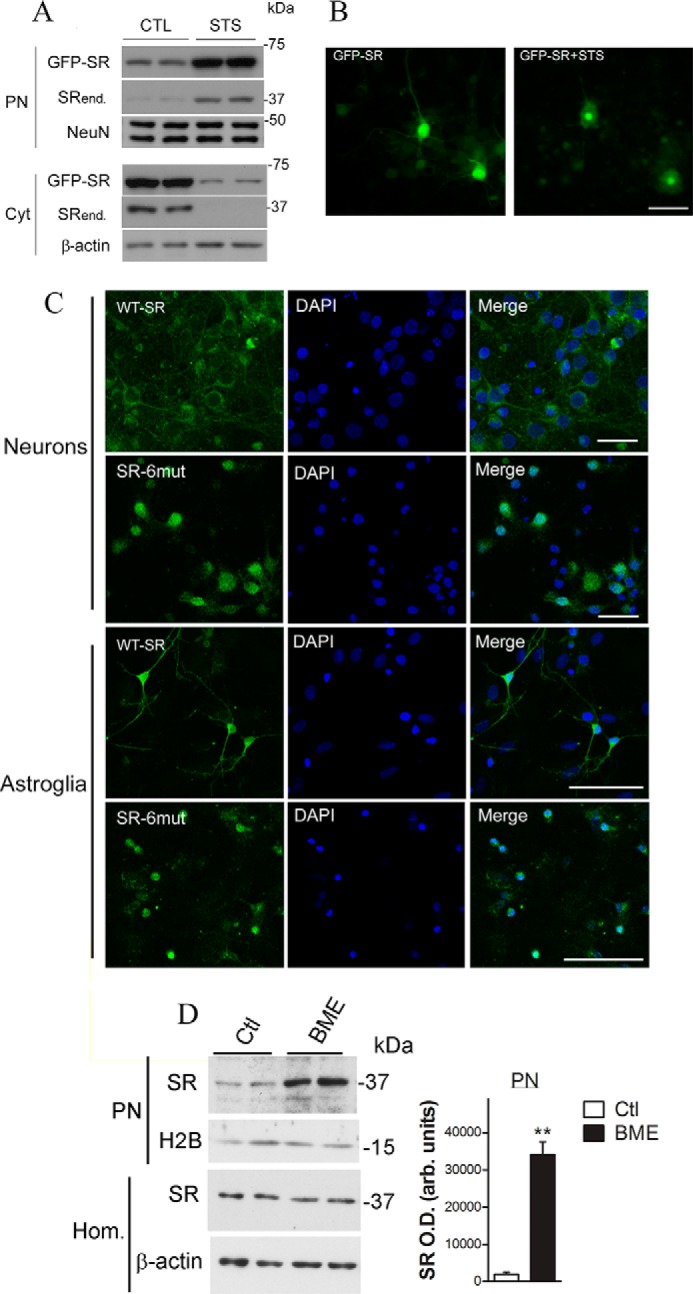
Lentivirus-infected primary neuronal cultures exhibited SR nuclear accumulation. A, primary neuronal cultures, DIV5, were infected with lentivirus harboring GFP-SR. On DIV10, neurons were treated with STS (1 μm) for 8 h. Nuclear (PN) and cytosolic (Cyt) fractions were monitored by Western blot analysis and probed with SR and loading controls. Translocation of GFP-SR to the nucleus took place simultaneously with a reduction in cytosolic levels. Endogenous SR (SRend.) also exhibited nuclear accumulation. B, live neurons (DIV10) were imaged every 10 min. After 30 min, STS (1 μm) was added to the medium, and the cells were further imaged for up to 24 h. GFP-SR was evenly distributed within the neuron (left panel), and complete translocation of GFP-SR to the nucleus was imaged 12 h after addition of STS (right panel). Scale bar, 50 μm. C, primary cortical neuronal cultures (DIV5) or primary astrocytes (DIV14) were incubated for 6 days with lentiviruses harboring GFP-SR WT (upper panels) or GFP-SR 6mut (lower panels). Both neurons and astrocytes expressing SR containing six NES mutations (SR-6mut) exhibited nuclear accumulation, as revealed by co-localization with DAPI and confocal laser microscopy. Scale bar, 35 μm. The panels represent at least three experiments with different preparations. D, BME treatment (12 h) promoted nuclear accumulation of SR and GAPDH in primary astrocyte cultures. Data represent average ± S.E. of four experiments with different astrocyte cultures. **, different from control at p < 0.001. Cyt, cytosolic fraction; H2B, histone H2B; Hom., homogenate; PN, purified nucleus.
Because cultured astroglia also express SR (18), we investigated whether astrocytes also exhibit nuclear SR accumulation. Lentiviral infection of the nuclear export-deficient SR mutant (SR-6mut) demonstrated nuclear accumulation of SR in the astrocytes as well, indicating nucleocytoplasmic shuttling of SR (Fig. 3C). Upon apoptotic stimuli, we detected robust accumulation of endogenous SR in the purified nuclear fraction of the primary astrocyte cultures, without apparent changes in loading control (Fig. 3D). Therefore, both neuronal SR and glial SR undergo nucleocytoplasmic shuttling that is dysregulated upon apoptotic stimulus.
Nuclear SR Targets
To further characterize the SR nuclear translocation, primary neuronal cultures were first treated with stimuli shown to elicit nuclear accumulation of SR, and the purified nuclear fraction was isolated. The nuclear pellet was subsequently extracted with increasing salt and detergent concentrations (Fig. 4A). We found that SR is not extracted from the purified nuclear fraction even under high stringency conditions, indicating a strong association with nuclear components. Like SR, GAPDH is not extracted from the nuclear fraction under these conditions (Fig. 4A). As expected, lamin A/C remained in the nuclear fraction as well (Fig. 4A, bottom panel).
FIGURE 4.

Distribution of nuclear SR and complex with nuclear GAPDH and lamin A/C. A, SR is strongly bound to the nucleus. Primary neural cultures at DIV12 were treated with 1 μm STS. Cells were lysed after 8 h, and the nuclear fraction was purified as described under “Experimental Procedures.” Proteins bound to purified nuclei (PN) were extracted in buffers containing 20 mm Tris-HCl (pH 7.4) and increasing concentrations of NaCl (up to 0.5 m) and Triton X-100 (up to 1%) for 10 min and centrifuged to recover the extracted nuclear pellets (P1 to P4) and supernatants (S1 to S4). Samples were probed for SR, GAPDH, and lamin A/C. B, SR is bound to the nuclear insoluble protein fraction consisting of nuclear lamina and matrix-associated proteins. PN were lysed and fractionated into chromatin-associated proteins and insoluble protein fraction as described under “Experimental Procedures.” Both SR and GAPDH were found in the insoluble fraction with virtually no signal in the chromatin fraction as revealed by Western blot analysis. Histone3 and lamin A/C served as loading controls. C, SR overlaps with lamin A/C in primary neuronal cultures. On DIV10, the cultures were treated with STS as revealed by confocal laser microscopy. Scale bar = 30 μm. D, immunoprecipitation (IP) of SR from the disrupted nuclear fraction suspension of primary neuronal cultures also brought down lamin A/C and GAPDH. Lamin A/C and GAPDH were not detectable when IgG was used as an immunoprecipitation control. E, immunoprecipitation of GAPDH from the disrupted nuclear fraction suspension of primary neuronal cultures also brought down lamin A/C and SR. Lamin A/C and SR were not detectable when IgG was used as an immunoprecipitation control. The results represent at least three experiments with different preparations.
To ascertain the subnuclear compartmentalization of SR, purified nuclei were separated into a chromatin-associated and insoluble nuclear protein fraction (Fig. 4B). Chromatin-associated proteins were investigated following treatment with micrococcal nuclease to release the chromatin-bound proteins. We found that virtually all SR and GAPDH are present in the insoluble fraction, which is devoid of histone 3 but contains lamin A and C (Fig. 4B). Accordingly, we also observed overlap between nuclear SR and lamin A and C in STS neurons treated by immunocytochemistry and confocal laser microscopy (Fig. 4C).
To evaluate whether SR binds to a complex containing lamin A and C, we carried out co-immunoprecipitation studies from a nuclear fraction disrupted by sonication and detergent treatment. We found that SR co-immunoprecipitates with lamin A and C (Fig. 4D), indicating an association of SR with nuclear components.
Cytosolic GAPDH has been recently shown to bind to SR and modulate its activity (71). Because we found that GAPDH translocates to the nucleus in the same time frame as SR (Fig. 1), we also investigated whether GAPDH associates with SR in the nuclear fraction. Immunoprecipitation of endogenous SR from the nuclear fraction of primary neuronal cultures brought down GAPDH as well (Fig. 4D). Conversely, immunoprecipitation of GAPDH from the nuclear fraction also brought down SR, as well as lamin A/C (Fig. 4E). The data are compatible with the notion that SR may exist as a complex with GAPDH along with additional nuclear components.
Effect of Nuclear SR in Cell Death Mechanisms
To investigate whether nuclear SR plays a direct role in cell death processes, we cultured primary cortical neurons from SR-KO mice and infected them with recombinant lentiviruses containing GFP, GFP-SR, or GFP-SR-6mut. In these experiments, we supplemented the culture media with an excess of glycine (50 μm) to saturate the NMDAR co-agonist site. This allowed us to investigate whether SR translocation per se is toxic, eliminating the effect of its known role in producing d-serine, which is required for NMDAR-mediated neurotoxicity (72). We found no evidence that expression of GFP-SR or nuclear export-deficient SR mutant in SR-KO mice cultures changes the basal cell death as determined by cleavage of caspase 3, PARP-1, and lamin A (Fig. 5, A and B). Furthermore, there was no change in STS-mediated neuronal cell death, as judged by the lack of effect on cell death markers.
FIGURE 5.

Effect of SR translocation in cell death mechanisms. A, primary neuronal cultures obtained from SR-KO mice were infected at DIV5 with lentivirus harboring GFP, GFP-SR, or GFP-SR 6mut. On DIV10, neurons were treated with STS (1 μm) for 8 h in the presence of saturating levels of the NMDAR co-agonist glycine (50 μm). Total cell homogenates were probed for cleaved caspase 3, PARP-1, and lamin A cleavage by Western blot analysis. Lower panel depicts GFP-SR expression in total cell homogenate by anti-SR antibody. B, ratio of cleaved caspase3, PARP-1, and lamin A between STS-treated and -untreated cells normalized by β-tubulin levels. C, primary neuronal cultures obtained from SR-KO mice were infected at DIV5 with lentivirus harboring GFP or GFP-SR 6mut. On DIV10, neurons were treated with STS (1 μm) for 8 h in the presence of 30 μm MK-801 to evaluate cell death that was not mediated via NMDARs. The results are expressed as the ratio of LDH release by STS and untreated cultures. D, SR-KO cultures are less susceptible to NMDAR-mediated cell death. LDH release was monitored from cultures treated with no NMDA, 30 μm NMDA, or NMDA plus 20 μm MK-801. E, cell death was restored to WT levels when SR-KO primary neuronal cultures were supplemented with 10 μm d-serine. The results are mean ± S.E. of four (A–C and E) or nine (D) experiments with different culture preparations. *, ***, different from control at p < 0.05 and 0.001, respectively.
We also monitored the release of LDH, a known marker of loss of membrane integrity as a result of neuronal cell death (73). When NMDARs were completely blocked by the selective NMDAR antagonist MK-801, lentivirus-mediated expression of GFP-SR 6mut, which exhibits robust nuclear accumulation, did not increase the extent of LDH release promoted by STS treatment in primary neuronal cultures (Fig. 5C). Therefore, SR translocation (cytosolic or nuclear) does not per se have a direct effect on the induced cell death processes under conditions where the NMDARs are either completely saturated (Fig. 5, A and B) or blocked by MK-801 (Fig. 5C).
However, when NMDARs were exposed to endogenous levels of co-agonists, SR activity was clearly associated with neurotoxicity following NMDAR activation (Fig. 5D). We found that SR-KO displayed a much lower rate of cell death when compared with WT (Fig. 5D). The effects were blocked by the selective NMDAR antagonist MK-801 (Fig. 5D). NMDAR neurotoxicity in SR-KO cultures was restored to WT levels by supplementing the culture medium with 10 μm d-serine (Fig. 5E), whereas WT cultures were little or not affected by additional d-serine (Fig. 5E). Therefore, although SR does not directly participate in cell death mechanisms, its inactivation prevents the neuronal demise via NMDAR stimulation.
Nucleus-associated SR Is Inactive Toward d-Serine Synthesis
Because SR is required for NMDAR activation (Fig. 5, D and E), we explored the possibility that SR nuclear translocation affects SR activity. Conceivably, the strong association of SR to GAPDH and nuclear lamina components may affect the catalytic activity of the enzyme and alter d-serine dynamics under cell death execution.
We found that primary neuronal cultures treated with LMB or STS exhibit much lower extracellular d-serine levels when compared with untreated cells (Fig. 6A). Along with a drastic reduction in d-serine, STS treatment also induced a 10-fold increase in extracellular glutamate, presumably because of cell death induction, whereas LMB treatment appeared to display only minimal effects on glutamate levels (Fig. 6B). Because extracellular glutamate on its own triggers the release of neuronal d-serine (18), the reduction in extracellular d-serine by STS and LMB suggests that nuclear translocation may inactivate d-serine production.
FIGURE 6.

Nuclear SR is inactive toward d-serine synthesis. A, primary neuronal cultures incubated with LMB (20 nm) or STS (1 μm) exhibited a reduction in the extracellular levels of d-serine. B, levels of glutamate in the culture media of treated cells. C, STS treatment did not affect d- or l-serine transport. Primary neuronal cultures were incubated for 7 h with either vehicle or 1 μm STS, and d- or l-serine uptake was monitored in HBSS containing 5 μm either d-[3H]serine or l-[3H]serine. D, STS did not affect basal or hetero-exchange-mediated d-serine release. Primary neuronal cultures were incubated for 7 h with either vehicle or 1 μm STS and preloaded with d-[3H]serine as described under “Experimental Procedures.” Subsequently, the fractional d-[3H]serine release was monitored by adding either no l-serine (Basal) or 1 mm l-serine (+d-Ser) to induce transport-mediated d-[3H]serine release for 2 min. E, nuclear SR is inactive toward d-serine synthesis. Primary neuronal cultures were treated with either no STS or 1 μm STS for 8 h followed by isolation of cytosolic and nuclear fractions, which were then assayed in vitro for d-serine synthesis. d-Serine production was normalized by the SR content in each fraction monitored by Western blot analysis and densitometry. F, cytosolic SR was not affected by soluble nucleoplasmic (NP) proteins. Cytosolic extracts of HEK293 cells transfected with mouse SR were incubated either with or without soluble nucleoplasma proteins extracted from purified nuclei from primary neuronal cultures. **, ***, different from control at p < 0.01 and 0.001, respectively. The results are mean ± S.E. of six (A and B) or four (C–F) different experiments with different preparations.
Neurons express plasma membrane antiporters that use both d-serine and l-serine as substrates, including the Asc-1 (Slc7a10) and members of the alanine, serine, cysteine, and threonine family (74–76). Asc-1 is the sole high affinity transporter for d-serine in the brain, and its knock-out disrupts both the transport of d-serine and l-serine in purified synaptic terminals (75, 77, 78). As control, we found that prolonged STS treatment did not affect the uptake kinetics of either l-[3H]serine or d-[3H]serine into primary neuronal cultures by plasma membrane transporters (Fig. 6C).
A major feature of Asc-1 and other putative plasma membrane d-serine transporters is their ability to release d-serine from cells in exchange for extracellular l-serine, as demonstrated in primary cultures, slices, and in vivo microdialysis (45, 48, 79–81). We found that STS treatment did not affect the basal or l-serine-induced release of preloaded d-[3H]serine from primary neuronal cultures (Fig. 6D). The data indicate that STS treatment did not impair plasma membrane antiporter systems of l- and d-serine.
Conceivably, nuclear localization directly inactivates SR. To directly test the activity of nuclear SR, we purified nuclei and cytosol from primary neuronal cultures pretreated with STS to elicit nuclear translocation of SR. We found that SR was completely inactive in the purified nuclear fraction, although its activity was preserved in the cytosol of STS-treated cells (Fig. 6E). Addition of soluble nucleoplasmic proteins to cytosolic SR did not inhibit the enzyme (Fig. 6F), indicating that the inhibition is only achieved when SR associates with the nuclear envelope components during the apoptotic signal in intact cells. The data indicate that nuclear accumulation abolishes the catalytic activity of SR and that this inhibition is not a side product of the cell death insult but of SR cellular localization.
Discussion
We described a novel mechanism of SR regulation in neurons, linking a variety of insults leading to neuronal death with d-serine production. Our study revealed the existence of a nucleocytoplasmic shuttling of SR, and it demonstrated how nuclear SR levels build up via pathologically relevant stimuli, providing a new marker of cell death. SR present in both neurons and astrocytes undergoes nuclear accumulation during apoptosis, indicating that this process may globally affect the production of d-serine.
We found that apoptotic stimuli that are not primarily initiated by NMDAR activation increase glutamate release from neurons but drastically reduce extracellular levels of d-serine. Thus, inactivation of SR during neurotoxic insult may serve as a fail-safe mechanism to prevent secondary NMDAR overactivation at vicinal neurons or synapses.
d-Serine is critical for NMDA receptor-mediated neurotoxicity both in vitro and in vivo (18, 27, 72, 82). To date, there are few signals connecting NMDAR activation to SR activity or d-serine dynamics. NMDAR activation promotes SR S-nitrosylation and inhibition, a process that is completely reversed by treatment with reducing agents such as DTT (83). Under our experimental conditions, DTT was included in our assays and did not prevent SR inactivation at the nuclear fraction. Thus, the inhibition of SR observed in this study could not be attributed to S-nitrosylation.
We observed SR translocation by classical apoptotic paradigms, such as staurosporine and etoposide, suggesting that apoptosis initiation likely promotes SR translocation. Cleavage of caspase 3, PARP1, and lamin A, along with the nuclear translocation of GAPDH and Siah processes, typically occurring during apoptosis (60), were also detected. However, our data do not conflict with the occurrence of apoptosis with other modes of cell death, because other insults that induced SR translocation (e.g. oxidative stress by H2O2) also promote necrotic cell death.
Inhibition of active nuclear export or mutations in SR NESs promotes the accumulation of nuclear SR, demonstrating that SR normally shuttles between the nucleus and the cytosol. It is well known that oxidative stress takes place during neuronal cell death and inhibits protein nuclear export by multiple mechanisms targeting CRM1 and nucleoporins, which are key in nuclear export (84). Thus, it is likely that cell death stimuli cause a more general dysregulation of the nuclear export system, which in turn will compartmentalize SR in the nucleus, where it strongly binds to lamin A- and C-containing protein complex, the major components of the nuclear lamina.
Moreover, SR may also accumulate in the nucleus as a consequence of increased nuclear import. It is noteworthy that we found that SR accumulates in the nucleus along with GAPDH and Siah. Cytosolic GAPDH was recently shown to interact with cytosolic SR and inhibit its activity by about 20–30% (71). Because GAPDH is transported into the nucleus during apoptosis (60), it is possible that SR shuttles to the nucleus bound to GAPDH. In agreement with this proposed mechanism, we found that nuclear SR co-immunoprecipitates with nuclear GAPDH along with lamin A/C.
Nuclear SR was shown to be completely inactive, as we could not recover any SR activity from purified nuclei of neurons. Nuclear SR is also resistant to detergent and salt extraction, indicating that the strong association with the nuclear envelope components, including GAPDH, may underlie SR inactivation.
d-Serine released to the media of neurons treated with apoptotic drugs or nuclear export inhibitor was markedly reduced because of inactivation of the nucleus-associated SR. Interestingly, we found that extracellular d-serine was paradoxically reduced, although extracellular glutamate levels increased 10-fold (Fig. 6, A and B), even though glutamate is known to promote d-serine release (8, 18). In agreement with the role of d-serine in neurotoxicity, we found that cultures from SR-KO mice are much less sensitive to NMDAR-mediated cell death. In this framework, nuclear compartmentalization and inhibition of SR may prevent the amplification of cell death by averting secondary NMDAR neurotoxicity in nearby neurons. The data also indicate that the rate of d-serine synthesis is the major determinant of extracellular d-serine and that d-serine release in itself is not a limiting step.
In addition to nuclear translocation, other mechanisms may contribute to feedback inhibition of NMDARs. Although we found no changes in total SR expression upon cell death insults, SR expression in cerebellar granule cells decreased during apoptosis (85). NMDAR activation was also shown to promote SR translocation from the cytosol to membranes, which also inhibits SR (29). Activation of multiple mechanisms to prevent d-serine synthesis during cell death insults highlights the importance of the d-serine pathway in neurotoxicity. Manipulation of SR intracellular targeting within the neuron may be of further use to aid drug development for prevalent diseases of the CNS that involve neurodegeneration, providing a means for the cells to down-regulate the production of d-serine and thus protect vicinal neurons from NMDAR-mediated neurotoxicity.
Author Contributions
G. K. and I. R. designed, performed, and analyzed the main body of the experiments. E. D. conceived the experiments. H. S. provided technical expertise and assistance with primary cultures. D. R. carried out the neurotoxicity experiments of Fig. 5, D and E. H. M. provided important reagents/expertise and interpreted the data. H. W. carried out experiments of serine transport and coordinated the study. H. W. and I. R. analyzed the data and wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Adi C. Segal for technical assistance, Edith Suss-Toby for expert confocal microscopy assistance, and Prof. Ralph Kehlenbach for helpful discussions.
This work was supported by the Israel Science Foundation, Legacy Heritage Fund, ALS Research Fund in the memory of Dr. David S. David, and the Allen and Jewel Prince Center for Neurodegenerative Processes of the Brain (to H. W.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Movie.
- SR
- serine racemase
- NMDAR
- N-methyl d-aspartate receptor
- BME
- β-mercaptoethanol
- LMB
- leptomycin B
- NES
- nuclear export signal
- NMDAR
- N-methyl-d-aspartate receptor
- PN
- purified nucleus
- Siah
- seven in absentia homologue
- STS
- staurosporine
- ER
- endoplasmic reticulum
- LDH
- lactate dehydrogenase
- ATPγS
- adenosine 5′-O-(thiotriphosphate)
- DIV
- days in vitro
- HBSS
- HEPES-buffered saline solution.
References
- 1. Billard J. M. (2013) Serine racemase as a prime target for age-related memory deficits. Eur. J. Neurosci. 37, 1931–1938 [DOI] [PubMed] [Google Scholar]
- 2. Wolosker H., Dumin E., Balan L., and Foltyn V. N. (2008) d-Amino acids in the brain: d-serine in neurotransmission and neurodegeneration. FEBS J. 275, 3514–3526 [DOI] [PubMed] [Google Scholar]
- 3. Mothet J. P., Parent A. T., Wolosker H., Brady R. O. Jr., Linden D. J., Ferris C. D., Rogawski M. A., and Snyder S. H. (2000) d-Serine is an endogenous ligand for the glycine site of the N-methyl-d-aspartate receptor. Proc. Natl. Acad. Sci. U.S.A. 97, 4926–4931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wolosker H., Sheth K. N., Takahashi M., Mothet J. P., Brady R. O. Jr., Ferris C. D., and Snyder S. H. (1999) Purification of serine racemase: biosynthesis of the neuromodulator d-serine. Proc. Natl. Acad. Sci. U.S.A. 96, 721–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wolosker H., Blackshaw S., and Snyder S. H. (1999) Serine racemase: a glial enzyme synthesizing d-serine to regulate glutamate-N-methyl-d-aspartate neurotransmission. Proc. Natl. Acad. Sci. U.S.A. 96, 13409–13414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. De Miranda J., Santoro A., Engelender S., and Wolosker H. (2000) Human serine racemase: molecular cloning, genomic organization and functional analysis. Gene 256, 183–188 [DOI] [PubMed] [Google Scholar]
- 7. Panatier A., Theodosis D. T., Mothet J. P., Touquet B., Pollegioni L., Poulain D. A., and Oliet S. H. (2006) Glia-derived d-serine controls NMDA receptor activity and synaptic memory. Cell 125, 775–784 [DOI] [PubMed] [Google Scholar]
- 8. Mothet J. P., Pollegioni L., Ouanounou G., Martineau M., Fossier P., and Baux G. (2005) Glutamate receptor activation triggers a calcium-dependent and SNARE protein-dependent release of the gliotransmitter d-serine. Proc. Natl. Acad. Sci. U.S.A. 102, 5606–5611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Henneberger C., Papouin T., Oliet S. H., and Rusakov D. A. (2010) Long-term potentiation depends on release of d-serine from astrocytes. Nature 463, 232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fujii K., Maeda K., Hikida T., Mustafa A. K., Balkissoon R., Xia J., Yamada T., Ozeki Y., Kawahara R., Okawa M., Huganir R. L., Ujike H., Snyder S. H., and Sawa A. (2006) Serine racemase binds to PICK1: potential relevance to schizophrenia. Mol. Psychiatry 11, 150–157 [DOI] [PubMed] [Google Scholar]
- 11. Kim P. M., Aizawa H., Kim P. S., Huang A. S., Wickramasinghe S. R., Kashani A. H., Barrow R. K., Huganir R. L., Ghosh A., and Snyder S. H. (2005) Serine racemase: activation by glutamate neurotransmission via glutamate receptor interacting protein and mediation of neuronal migration. Proc. Natl. Acad. Sci. U.S.A. 102, 2105–2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ma T. M., Abazyan S., Abazyan B., Nomura J., Yang C., Seshadri S., Sawa A., Snyder S. H., and Pletnikov M. V. (2013) Pathogenic disruption of DISC1-serine racemase binding elicits schizophrenia-like behavior via d-serine depletion. Mol. Psychiatry 18, 557–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mustafa A. K., van Rossum D. B., Patterson R. L., Maag D., Ehmsen J. T., Gazi S. K., Chakraborty A., Barrow R. K., Amzel L. M., and Snyder S. H. (2009) Glutamatergic regulation of serine racemase via reversal of PIP2 inhibition. Proc. Natl. Acad. Sci. U.S.A. 106, 2921–2926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Balu D. T., and Coyle J. T. (2012) Neuronal d-serine regulates dendritic architecture in the somatosensory cortex. Neurosci. Lett. 517, 77–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Balu D. T., Takagi S., Puhl M. D., Benneyworth M. A., and Coyle J. T. (2014) d-Serine and serine racemase are localized to neurons in the adult mouse and human forebrain. Cell. Mol. Neurobiol. 34, 419–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Benneyworth M. A., Li Y., Basu A. C., Bolshakov V. Y., and Coyle J. T. (2012) Cell selective conditional null mutations of serine racemase demonstrate a predominate localization in cortical glutamatergic neurons. Cell. Mol. Neurobiol. 32, 613–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ehmsen J. T., Ma T. M., Sason H., Rosenberg D., Ogo T., Furuya S., Snyder S. H., and Wolosker H. (2013) d-Serine in glia and neurons derives from 3-phosphoglycerate dehydrogenase. J. Neurosci. 33, 12464–12469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kartvelishvily E., Shleper M., Balan L., Dumin E., and Wolosker H. (2006) Neuron-derived d-serine release provides a novel means to activate N-methyl-d-aspartate receptors. J. Biol. Chem. 281, 14151–14162 [DOI] [PubMed] [Google Scholar]
- 19. Miya K., Inoue R., Takata Y., Abe M., Natsume R., Sakimura K., Hongou K., Miyawaki T., and Mori H. (2008) Serine racemase is predominantly localized in neurons in mouse brain. J. Comp. Neurol. 510, 641–654 [DOI] [PubMed] [Google Scholar]
- 20. Abe T., Suzuki M., Sasabe J., Takahashi S., Unekawa M., Mashima K., Iizumi T., Hamase K., Konno R., Aiso S., and Suzuki N. (2014) Cellular origin and regulation of d- and l-serine in in vitro and in vivo models of cerebral ischemia. J. Cereb. Blood Flow Metab. 34, 1928–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ma T. M., Paul B. D., Fu C., Hu S., Zhu H., Blackshaw S., Wolosker H., and Snyder S. H. (2014) Serine racemase regulated by binding to Stargazin and PSD-95: potential N-methyl-d-aspartate-α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (NMDA-AMPA) glutamate neurotransmission cross-talk. J. Biol. Chem. 289, 29631–29641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dikopoltsev E., Foltyn V. N., Zehl M., Jensen O. N., Mori H., Radzishevsky I., and Wolosker H. (2014) FBXO22 is required for optimal synthesis of the NMDA receptor co-agonist d-serine. J. Biol. Chem. 289, 33904–33915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dumin E., Bendikov I., Foltyn V. N., Misumi Y., Ikehara Y., Kartvelishvily E., and Wolosker H. (2006) Modulation of d-serine levels via ubiquitin-dependent proteasomal degradation of serine racemase. J. Biol. Chem. 281, 20291–20302 [DOI] [PubMed] [Google Scholar]
- 24. Balu D. T., Li Y., Puhl M. D., Benneyworth M. A., Basu A. C., Takagi S., Bolshakov V. Y., and Coyle J. T. (2013) Multiple risk pathways for schizophrenia converge in serine racemase knockout mice, a mouse model of NMDA receptor hypofunction. Proc. Natl. Acad. Sci. U.S.A. 110, E2400–E2409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Basu A. C., Tsai G. E., Ma C. L., Ehmsen J. T., Mustafa A. K., Han L., Jiang Z. I., Benneyworth M. A., Froimowitz M. P., Lange N., Snyder S. H., Bergeron R., and Coyle J. T. (2009) Targeted disruption of serine racemase affects glutamatergic neurotransmission and behavior. Mol. Psychiatry 14, 719–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Labrie V., Fukumura R., Rastogi A., Fick L. J., Wang W., Boutros P. C., Kennedy J. L., Semeralul M. O., Lee F. H., Baker G. B., Belsham D. D., Barger S. W., Gondo Y., Wong A. H., and Roder J. C. (2009) Serine racemase is associated with schizophrenia susceptibility in humans and in a mouse model. Hum. Mol. Genet. 18, 3227–3243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Inoue R., Hashimoto K., Harai T., and Mori H. (2008) NMDA- and β-amyloid 1–42-induced neurotoxicity is attenuated in serine racemase knock-out mice. J. Neurosci. 28, 14486–14491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mustafa A. K., Ahmad A. S., Zeynalov E., Gazi S. K., Sikka G., Ehmsen J. T., Barrow R. K., Coyle J. T., Snyder S. H., and Doré S. (2010) Serine racemase deletion protects against cerebral ischemia and excitotoxicity. J. Neurosci. 30, 1413–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Balan L., Foltyn V. N., Zehl M., Dumin E., Dikopoltsev E., Knoh D., Ohno Y., Kihara A., Jensen O. N., Radzishevsky I. S., and Wolosker H. (2009) Feedback inactivation of d-serine synthesis by NMDA receptor-elicited translocation of serine racemase to the membrane. Proc. Natl. Acad. Sci. U.S.A. 106, 7589–7594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Price P. J., and Brewer G. J. (2001) in Protocols for Neural Cell Culture (Fedoroff S., and Richardson A., eds) pp. 255–264, Human Press, Totowa, NJ [Google Scholar]
- 31. Brewer G. J., Torricelli J. R., Evege E. K., and Price P. J. (1993) Optimized survival of hippocampal neurons in B27-supplemented neurobasal, a new serum-free medium combination. J. Neurosci. Res. 35, 567–576 [DOI] [PubMed] [Google Scholar]
- 32. Zhang Y., Leavitt B. R., van Raamsdonk J. M., Dragatsis I., Goldowitz D., MacDonald M. E., Hayden M. R., and Friedlander R. M. (2006) Huntingtin inhibits caspase-3 activation. EMBO J. 25, 5896–5906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li X., Lao Y., Zhang H., Wang X., Tan H., Lin Z., and Xu H. (2015) The natural compound Guttiferone F sensitizes prostate cancer to starvation induced apoptosis via calcium and JNK elevation. BMC Cancer 15, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schoenmann Z., Assa-Kunik E., Tiomny S., Minis A., Haklai-Topper L., Arama E., and Yaron A. (2010) Axonal degeneration is regulated by the apoptotic machinery or a NAD+-sensitive pathway in insects and mammals. J. Neurosci. 30, 6375–6386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tanaka K., Sasayama T., Irino Y., Takata K., Nagashima H., Satoh N., Kyotani K., Mizowaki T., Imahori T., Ejima Y., Masui K., Gini B., Yang H., Hosoda K., Sasaki R., Mischel P. S., and Kohmura E. (2015) Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. J. Clin. Invest. 125, 1591–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shackelford D. B., Abt E., Gerken L., Vasquez D. S., Seki A., Leblanc M., Wei L., Fishbein M. C., Czernin J., Mischel P. S., and Shaw R. J. (2013) LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 23, 143–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen S., Evans H. G., and Evans D. R. (2012) FLASH knockdown sensitizes cells to Fas-mediated apoptosis via down-regulation of the anti-apoptotic proteins, MCL-1 and Cflip short. PLoS ONE 7, e32971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rott R., Szargel R., Haskin J., Shani V., Shainskaya A., Manov I., Liani E., Avraham E., and Engelender S. (2008) Monoubiquitylation of α-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J. Biol. Chem. 283, 3316–3328 [DOI] [PubMed] [Google Scholar]
- 39. Szargel R., Rott R., Eyal A., Haskin J., Shani V., Balan L., Wolosker H., and Engelender S. (2009) Synphilin-1A inhibits seven in absentia homolog (SIAH) and modulates α-synuclein monoubiquitylation and inclusion formation. J. Biol. Chem. 284, 11706–11716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Inoue R., Yoshihisa Y., Tojo Y., Okamura C., Yoshida Y., Kishimoto J., Luan X., Watanabe M., Mizuguchi M., Nabeshima Y., Hamase K., Matsunaga K., Shimizu T., and Mori H. (2014) Localization of serine racemase and its role in the skin. J. Invest. Dermatol. 134, 1618–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hara M. R., Thomas B., Cascio M. B., Bae B. I., Hester L. D., Dawson V. L., Dawson T. M., Sawa A., and Snyder S. H. (2006) Neuroprotection by pharmacologic blockade of the GAPDH death cascade. Proc. Natl. Acad. Sci. U.S.A. 103, 3887–3889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mullen R. J., Buck C. R., and Smith A. M. (1992) NeuN, a neuronal specific nuclear protein in vertebrates. Development 116, 201–211 [DOI] [PubMed] [Google Scholar]
- 43. Wysocka J., Reilly P. T., and Herr W. (2001) Loss of HCF-1-chromatin association precedes temperature-induced growth arrest of tsBN67 cells. Mol. Cell. Biol. 21, 3820–3829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tiscornia G., Singer O., and Verma I. M. (2006) Production and purification of lentiviral vectors. Nat. Protoc. 1, 241–245 [DOI] [PubMed] [Google Scholar]
- 45. Rosenberg D., Artoul S., Segal A. C., Kolodney G., Radzishevsky I., Dikopoltsev E., Foltyn V. N., Inoue R., Mori H., Billard J. M., and Wolosker H. (2013) Neuronal d-serine and glycine release via the Asc-1 transporter regulates NMDA receptor-dependent synaptic activity. J. Neurosci. 33, 3533–3544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Harlow E., and Lane D. (eds) (1988) Antibodies: A Laboratory Manual, 1st Ed., pp. 343–345, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 47. Radzishevsky I., and Wolosker H. (2012) An enzymatic-HPLC assay to monitor endogenous d-serine release from neuronal cultures. Methods Mol. Biol. 794, 291–297 [DOI] [PubMed] [Google Scholar]
- 48. Rosenberg D., Kartvelishvily E., Shleper M., Klinker C. M., Bowser M. T., and Wolosker H. (2010) Neuronal release of d-serine: a physiological pathway controlling extracellular d-serine concentration. FASEB J. 24, 2951–2961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim I., Xu W., and Reed J. C. (2008) Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 7, 1013–1030 [DOI] [PubMed] [Google Scholar]
- 50. Tabas I., and Ron D. (2011) Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 13, 184–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nicholson D. W., and Thornberry N. A. (1997) Caspases: killer proteases. Trends Biochem. Sci. 22, 299–306 [DOI] [PubMed] [Google Scholar]
- 52. Nicholson D. W., Ali A., Thornberry N. A., Vaillancourt J. P., Ding C. K., Gallant M., Gareau Y., Griffin P. R., Labelle M., and Lazebnik Y. A. (1995) Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 376, 37–43 [DOI] [PubMed] [Google Scholar]
- 53. Jänicke R. U., Sprengart M. L., Wati M. R., and Porter A. G. (1998) Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J. Biol. Chem. 273, 9357–9360 [DOI] [PubMed] [Google Scholar]
- 54. Slee E. A., Adrain C., and Martin S. J. (2001) Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J. Biol. Chem. 276, 7320–7326 [DOI] [PubMed] [Google Scholar]
- 55. Yu S. W., Wang H., Poitras M. F., Coombs C., Bowers W. J., Federoff H. J., Poirier G. G., Dawson T. M., and Dawson V. L. (2002) Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 297, 259–263 [DOI] [PubMed] [Google Scholar]
- 56. Yoshida H., Okada T., Haze K., Yanagi H., Yura T., Negishi M., and Mori K. (2001) Endoplasmic reticulum stress-induced formation of transcription factor complex ERSF including NF-Y (CBF) and activating transcription factors 6α and 6β that activates the mammalian unfolded protein response. Mol. Cell. Biol. 21, 1239–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Oyadomari S., and Mori M. (2004) Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 11, 381–389 [DOI] [PubMed] [Google Scholar]
- 58. Zinszner H., Kuroda M., Wang X., Batchvarova N., Lightfoot R. T., Remotti H., Stevens J. L., and Ron D. (1998) CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 12, 982–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ohoka N., Yoshii S., Hattori T., Onozaki K., and Hayashi H. (2005) TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 24, 1243–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hara M. R., Agrawal N., Kim S. F., Cascio M. B., Fujimuro M., Ozeki Y., Takahashi M., Cheah J. H., Tankou S. K., Hester L. D., Ferris C. D., Hayward S. D., Snyder S. H., and Sawa A. (2005) S-Nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 7, 665–674 [DOI] [PubMed] [Google Scholar]
- 61. Sakahira H., Enari M., and Nagata S. (1998) Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature 391, 96–99 [DOI] [PubMed] [Google Scholar]
- 62. Vander Heiden M. G., Chandel N. S., Williamson E. K., Schumacker P. T., and Thompson C. B. (1997) Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell 91, 627–637 [DOI] [PubMed] [Google Scholar]
- 63. Yang J., Liu X., Bhalla K., Kim C. N., Ibrado A. M., Cai J., Peng T. I., Jones D. P., and Wang X. (1997) Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 275, 1129–1132 [DOI] [PubMed] [Google Scholar]
- 64. Mizumoto K., Rothman R. J., and Farber J. L. (1994) Programmed cell death (apoptosis) of mouse fibroblasts is induced by the topoisomerase II inhibitor etoposide. Mol. Pharmacol. 46, 890–895 [PubMed] [Google Scholar]
- 65. Kaufman R. J. (1999) Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 13, 1211–1233 [DOI] [PubMed] [Google Scholar]
- 66. Kosugi S., Hasebe M., Tomita M., and Yanagawa H. (2009) Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc. Natl. Acad. Sci. U.S.A. 106, 10171–10176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nguyen Ba A. N., Pogoutse A., Provart N., and Moses A. M. (2009) NLStradamus: a simple hidden markov model for nuclear localization signal prediction. BMC Bioinformatics 10, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hutten S., and Kehlenbach R. H. (2007) CRM1-mediated nuclear export: to the pore and beyond. Trends Cell Biol. 17, 193–201 [DOI] [PubMed] [Google Scholar]
- 69. Wolff B., Sanglier J. J., and Wang Y. (1997) Leptomycin B is an inhibitor of nuclear export: inhibition of nucleo-cytoplasmic translocation of the human immunodeficiency virus type 1 (HIV-1) Rev protein and Rev-dependent mRNA. Chem. Biol. 4, 139–147 [DOI] [PubMed] [Google Scholar]
- 70. Kosugi S., Hasebe M., Matsumura N., Takashima H., Miyamoto-Sato E., Tomita M., and Yanagawa H. (2009) Six classes of nuclear localization signals specific to different binding grooves of importin α. J. Biol. Chem. 284, 478–485 [DOI] [PubMed] [Google Scholar]
- 71. Suzuki M., Sasabe J., Miyoshi Y., Kuwasako K., Muto Y., Hamase K., Matsuoka M., Imanishi N., and Aiso S. (2015) Glycolytic flux controls d-serine synthesis through glyceraldehyde-3-phosphate dehydrogenase in astrocytes. Proc. Natl. Acad. Sci. U.S.A. 112, E2217–2224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Shleper M., Kartvelishvily E., and Wolosker H. (2005) d-Serine is the dominant endogenous coagonist for NMDA receptor neurotoxicity in organotypic hippocampal slices. J. Neurosci. 25, 9413–9417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hartley D. M., Kurth M. C., Bjerkness L., Weiss J. H., and Choi D. W. (1993) Glutamate receptor-induced 45Ca2+ accumulation in cortical cell culture correlates with subsequent neuronal degeneration. J. Neurosci. 13, 1993–2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Fukasawa Y., Segawa H., Kim J. Y., Chairoungdua A., Kim D. K., Matsuo H., Cha S. H., Endou H., and Kanai Y. (2000) Identification and characterization of a Na+-independent neutral amino acid transporter that associates with the 4F2 heavy chain and exhibits substrate selectivity for small neutral d- and l-amino acids. J. Biol. Chem. 275, 9690–9698 [DOI] [PubMed] [Google Scholar]
- 75. Rutter A. R., Fradley R. L., Garrett E. M., Chapman K. L., Lawrence J. M., Rosahl T. W., and Patel S. (2007) Evidence from gene knockout studies implicates Asc-1 as the primary transporter mediating d-serine reuptake in the mouse CNS. Eur. J. Neurosci. 25, 1757–1766 [DOI] [PubMed] [Google Scholar]
- 76. Yamamoto T., Nishizaki I., Nukada T., Kamegaya E., Furuya S., Hirabayashi Y., Ikeda K., Hata H., Kobayashi H., Sora I., and Yamamoto H. (2004) Functional identification of ASCT1 neutral amino acid transporter as the predominant system for the uptake of l-serine in rat neurons in primary culture. Neurosci. Res. 49, 101–111 [DOI] [PubMed] [Google Scholar]
- 77. Radzishevsky I., Sason H., and Wolosker H. (2013) d-Serine: physiology and pathology. Curr. Opin. Clin. Nutr. Metab. Care 16, 72–75 [DOI] [PubMed] [Google Scholar]
- 78. Safory H., Neame S., Shulman Y., Zubedat S., Radzishevsky I., Rosenberg D., Sason H., Engelender S., Avital A., Hülsmann S., Schiller J., and Wolosker H. (2015) The alanine-serine-cysteine-1 (Asc-1) transporter controls glycine levels in the brain and is required for glycinergic inhibitory transmission. EMBO Rep. 16, 590–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Maucler C., Pernot P., Vasylieva N., Pollegioni L., and Marinesco S. (2013) In vivo d-serine hetero-exchange through alanine-serine-cysteine (ASC) transporters detected by microelectrode biosensors. ACS Chem. Neurosci. 4, 772–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. O'Brien K. B., and Bowser M. T. (2006) Measuring d-serine efflux from mouse cortical brain slices using online microdialysis-capillary electrophoresis. Electrophoresis 27, 1949–1956 [DOI] [PubMed] [Google Scholar]
- 81. Ribeiro C. S., Reis M., Panizzutti R., de Miranda J., and Wolosker H. (2002) Glial transport of the neuromodulator d-serine. Brain Res. 929, 202–209 [DOI] [PubMed] [Google Scholar]
- 82. Katsuki H., Nonaka M., Shirakawa H., Kume T., and Akaike A. (2004) Endogenous d-serine is involved in induction of neuronal death by N-methyl-d-aspartate and simulated ischemia in rat cerebrocortical slices. J. Pharmacol. Exp. Ther. 311, 836–844 [DOI] [PubMed] [Google Scholar]
- 83. Mustafa A. K., Kumar M., Selvakumar B., Ho G. P., Ehmsen J. T., Barrow R. K., Amzel L. M., and Snyder S. H. (2007) Nitric oxide S-nitrosylates serine racemase, mediating feedback inhibition of d-serine formation. Proc. Natl. Acad. Sci. U.S.A. 104, 2950–2955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Crampton N., Kodiha M., Shrivastava S., Umar R., and Stochaj U. (2009) Oxidative stress inhibits nuclear protein export by multiple mechanisms that target FG nucleoporins and Crm1. Mol. Biol. Cell 20, 5106–5116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Esposito S., Pristerà A., Maresca G., Cavallaro S., Felsani A., Florenzano F., Manni L., Ciotti M. T., Pollegioni L., Borsello T., and Canu N. (2012) Contribution of serine racemase/d-serine pathway to neuronal apoptosis. Aging Cell 11, 588–598 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.