

Summary
Objectives
To identify novel approaches to improve innate immunity in the lung following trauma complicated by hemorrhagic shock (T/HS) for prevention of nosocomial pneumonia.
Methods
We developed a rat model of T/HS followed by Pseudomonas aeruginosa (PA) pneumonia to assess the effect of alveolar epithelial cell (AEC) apoptosis, and its prevention by IL-6, on lung surfactant protein (SP)-D protein levels, lung bacterial burden, and survival from PA pneumonia, as well as to determine whether AEC apoptosis is a consequence of the unfolded protein response (UPR). Lung UPR transcriptome analysis was performed on rats subjected to sham, T/HS, and T/HS plus IL-6 protocols. Group comparisons were performed via Kaplan–Meier or ANOVA.
Results
T/HS decreased lung SP-D by 1.8-fold (p < 0.05), increased PA bacterial burden 9-fold (p < 0.05), and increased PA pneumonia mortality by 80% (p < 0.001). IL-6, when provided at resuscitation, normalized SP-D levels (p < 0.05), decreased PA bacterial burden by 4.8-fold (p < 0.05), and prevented all mortality from PA pneumonia (p < 0.001). The UPR transcriptome was significantly impacted by T/HS; IL-6 treatment normalized the T/HS-induced UPR transcriptome changes (p < 0.05).
Conclusions
Impaired innate lung defense occurs following T/HS and is mediated, in part, by reduction in SP-D protein levels, which, along with AEC apoptosis, may be mediated by the UPR, and prevented by use of IL-6 as a resuscitation adjuvant.
Keywords: Unfolded protein, response, Hemorrhagic shock, Pneumonia, Alveolar epithelial cell
Introduction
Nosocomial pneumonia is the most common cause of death in patients suffering trauma complicated by hemorrhagic shock (T/HS) who survive their initial injuries.1 While significant strides have been made in identifying the clinical findings and laboratory parameters associated with onset of pneumonia following traumatic injuries, the molecular basis for predisposition to pneumonia in T/HS is not fully understood. The concept of immune paralysis or immunodepression in patients following T/HS has growing support.2,3 However, details of how immunodepression develops and its subsequent impact on the host have not been fully elucidated, particularly with regards to innate immunity, nor have specific measures emerged to prevent it.
A key component of innate immune defense in the lung is surfactant protein (SP)-D. SP-D is a member of the collectin family of proteins, which have a carboxy-terminal domain with calcium-dependent lectin activity. This lectin domain mediates the lectin:pathogen interaction, leading to pathogen aggregation, opsonization and enhanced pathogen phagocytosis, as well as a direct bactericidal effect.4 SP-D has been shown to be critical in the innate host defense of the lung protecting against various inhaled pathogens and allergens.5,6 Indeed, SP-D null mice have demonstrated increased susceptibility to multiple pathogens,7 and SP-D has been shown to bind and aggregate Pseudomonas aeruginosa, one of the most commonly encountered pathogens in ventilator-associated pneumonia (VAP).8–10
SP-D, as with other surfactant proteins, is largely produced by type II alveolar epithelial cells (AECII).11 AECII are found within the alveolar space, forming the extensive alveolar epithelial lining of the lung in conjunction with type I alveolar epithelial cells (AECI). AECII constantly produce surfactant proteins, such as SP-D, that are extruded into the extracellular space in an exocytic fashion to help maintain the surfactant layer, a key component of innate lung defense.
Using a rat model of T/HS,12–15 we previously demonstrated that up to 15% of AECII undergo apoptosis in the acute post-resuscitative phase, and that AECII injury/apoptosis can be prevented when IL-6 is used as a resuscitative adjuvant through a Stat3-mediated mechanism.14 In this report, we investigated the hypothesis that AECII injury/apoptosis contributes to pneumonia susceptibility in T/HS and that this contribution is mediated, in part, through reductions in SP-D levels. We found that T/HS decreased lung SP-D levels by almost half, which was associated with a 9-fold increase in lung bacterial burden and a 80% increase in mortality from PA pneumonia. IL-6, when provided at resuscitation to T/HS rats, normalized lung SP-D levels, decreased bacterial burden, and prevented all mortality from PA pneumonia. Analysis of the UPR transcriptome supports the hypothesis that the UPR contributes to AECII apoptosis following T/HS and its prevention by IL-6. These findings provide new opportunities for preventing nosocomial pneumonia in shock/trauma patients including use of IL-6 as a resuscitation adjuvant or administration of clinically available proteostasis modulators.
Methods
Rat T/HS protocol
These studies were approved by the Baylor College of Medicine Institutional Review Board for animal experimentation (Protocol AN-1980) and conform to National Institutes of Health guidelines for the care and use of laboratory animals. Adult male Sprague–Dawley rats were obtained from Harlan (Indianapolis, IN). Rats were subjected to the sham or T/HS protocols, as described12–14,16,17 with modifications. Under inhaled isoflurane anesthesia, both superficial femoral arteries (SFA) were cannulated. The right SFA site was used for continuous blood pressure monitoring and the left SFA site was used for blood withdrawal and fluid administration. Animals subjected to T/HS, underwent an initial bleed of 2.25 ml/100 g body weight over 10 min to achieve a target mean arterial blood pressure (MAP) of 35 mmHg, maintained for a period of ∼3 h (mean duration = 191 ± 2.5 min) by episodically withdrawing or returning shed blood. Sham rats were anesthetized and cannulated for a total of ∼4 h (a time period encompassing both the hypotensive and resuscitation phases), but were not subjected to hemorrhage or resuscitation. At the end of the hypotensive period, rats in the T/HS-PBS groups were resuscitated with our standard protocol (infusion of the remaining shed blood and two times the total shed blood volume with Ringer's lactate over 30 min starting with a 0.1 ml bolus of PBS) or they received IL-6 (10 μg/kg in 0.1 ml PBS via intra-arterial catheter) at the start of standard resuscitation (T/HS-IL6). We previously performed dose-response-studies, in which four doses of IL-6 (1, 3, 10 and 30 μg/kg) were tested, and demonstrated that 10 μg/kg was the optimum concentration to prevent organ injury (Tweardy et al., 2002, unpublished). To assess the effect of T/HS without or with IL-6 on the UPR transcriptome, 4 rats from each group were sacrificed 1 h after the start of resuscitation while under anesthesia and their lungs were harvested for RNA isolation and microarray analysis. To assess the effect of sham or T/HS without or with IL-6 on susceptibility to pneumonia, femoral wounds were closed surgically and anesthesia was reversed. Animals were given analgesia, returned to their cages, observed overnight and allowed to ambulate and feed ad libitum before subjecting them to the PA pneumonia protocol (see below). The survival rate in rats subjected to the T/HS protocol without or with IL-6 is ∼100% at 24 h and beyond.
Bacterial strain and inoculum preparation and quantification
P. aeruginosa (PA) strain ATCC-27853 (PA; a kind gift from Dr. John Alverdy, University of Chicago, IL) was used in all experiments. The target inoculum size of 3 × 107 CFU was determined as optimal in dose-survival experiments (0.03, 0.1, 0.3, and 1 × 109 CFU) in normal healthy Sprague–Dawley rats based on earlier studies in a rat intra-tracheal inoculum PA pneumonia model.18,19 The mortalities observed with each inoculum were 17%, 20%, 75%, and 100%, respectively. The 17% mortality observed with 3 × 107 CFU was assessed as optimum based on our earlier results demonstrating increased susceptibility to intraperitoneal Staphylococcus aureus infection in mice following T/HS.20 The target inoculum size (3 × 107 CFU) was obtained by a broth culture prepared by isolating a single colony from an agar plate grown at 37 °C for 15–17 h in trypticase-soy agar (TSA; Becton, Dickinson and Company. Sparks, MD, USA) and inoculating it into trypticase-soy broth (TSB; Becton, Dickinson and Company. Sparks, MD, USA). The broth was incubated at 37 °C and the optical density (OD) measured to achieve the OD corresponding to the target inoculum size, which was then confirmed by serial dilution and culture on TSA plates.
T/HS-pneumonia protocols
Twenty-four hours after being subjected to the sham or T/HS protocol without or with IL-6, rats were given a sublethal dose of PA (mean inoculum size 3.1 ± 0.2 × 107 CFU) through the transtracheal route. Briefly, a 1 cm incision in the anterior aspect of the neck was done under 2% isoflurane anesthesia, the fascia and muscle layers were dissected and the trachea exposed. The bacterial inoculum in a volume of 0.2 ml of PBS was transtracheally instilled through a 22-gauge needle inserted into the trachea, followed by 0.5 of air for uniform inoculum distribution. The incision was surgically closed. Rats were administered analgesia, allowed to recover in their cages, and observed every 6 h for 48 h to quantify survival (survival protocol; Fig. 1) or sacrificed 4 h after intratracheal inoculation and lungs harvested for lung bacterial burden quantification (bacterial burden protocol, Fig. 2). After sacrifice, lungs from rats subjected to the bacterial burden protocol were collected, weighed, and homogenized in 2 ml PBS. Serial log dilutions of organ homogenate (1:10, 1:100, and 1:1000) were made and plated on TSA plates in duplicate. Plates were incubated at 37 °C overnight after which bacterial CFU were counted. Results are presented as CFU/gm tissue weight.
Figure 1.
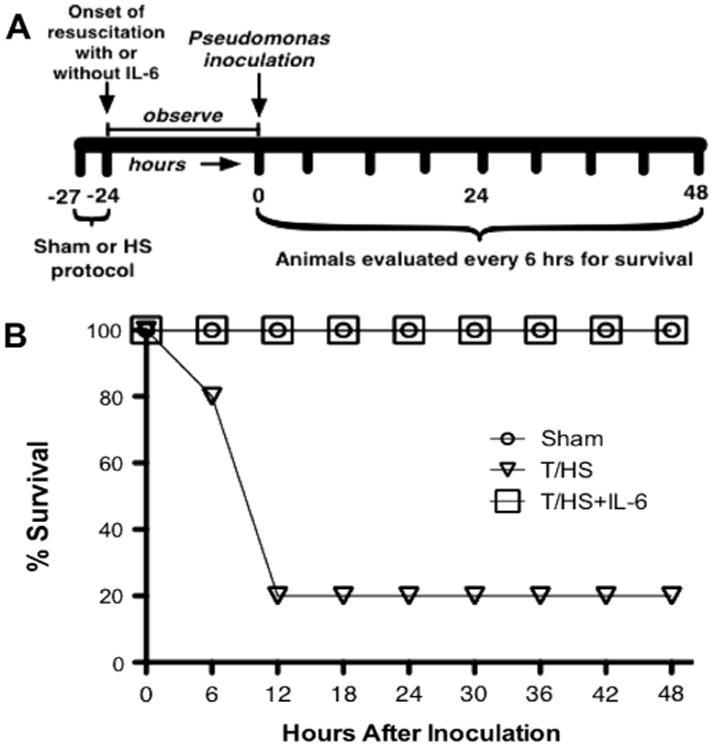
Effect of T-HS on mortality due to PA pneumonia. Panel A depicts the sequence of interventions in the rat pneumonia survival protocol. Rats (n = 10 per group) were subjected to either the sham [triangle], T/HS [square], or T/HS + IL-6 resuscitation [circle] protocol over 3 h followed 24 h later by transtracheal inoculation of PA, then observed 48 h for survival. Survival (panel B) of T-HS rats was reduced 80% compared to that of sham rats (p < 0.001, Kaplan–Meier analysis); reduction in survival by T/HS was reversed by administration of IL-6 (p < 0.001, Kaplan–Meier analysis).
Figure 2.
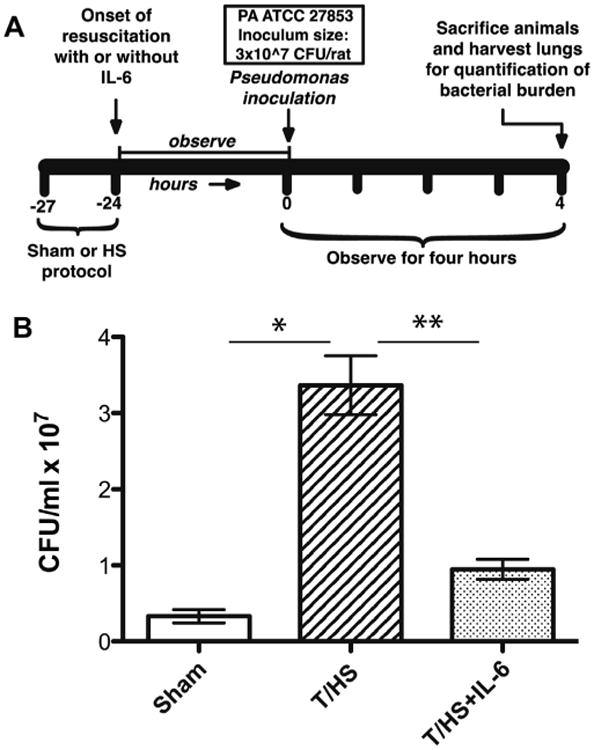
Effect of T-HS on lung bacterial burden in the PA pneumonia. Panel A depicts the sequence of interventions in the rat pneumonia bacterial burden protocol. Rats (n = 6 per group) were subjected to either the sham or T/HS protocol over 3 h near the end of which they either received or did not receive IL-6. Twenty-four hr later, rats received a transtracheal inoculation of PA followed by sacrifice 4 h later. Bacterial CFU were counted in lung homogenates. Data are presented as bacteria CFU/gram of lung tissue (mean ± SEM). Bars with paired single or double asterisks above (*, **) differ significantly (p < 0.05, ANOVA).
Lung protein extraction and protein quantitation
Frozen lungs of rats subjected to sham or T/HS protocol without or with IL-6 and harvested at 1 h after end of resuscitation were cut by cryotome, resuspended in high salt buffer, and sonicated in ice 3 times, 10 s each, as previously described.12–15 Samples were then centrifuged 15 min at 5000 RPM and the supernatant collected and evaluated by Bradford assay for total protein quantification.
Frozen lungs of rats subjected to sham or T/HS protocol without or with IL-6 and harvested at 1 h after end of resuscitation were cut by cryotome, resuspended in high salt buffer, and sonicated in ice 3 times, 10 s each, as previously described.12–15 Samples were then centrifuged 15 min at 5000 RPM and the supernatant collected and evaluated by Bradford assay for total protein quantification.
Myeloperoxidase (MPO) staining
Paraformaldehyde-fixed and paraffin-embedded lung sections were rehydrated from Xylene to PBS through a series of decreasing concentrations of ethanol and placed in a DAKO autostainer. MPO rabbit polyclonal antibody (Lab Vision, Corp.) was used at the provided concentration. The horseradish peroxidase (HRP) system for rabbit antibodies was used as per the manufacturer's instructions. Slides were counterstained with hematoxyllin. MPO-positive cells were assessed microscopically in 20 random 1000× high power fields (hpf) by an experienced histologist. Data is presented as the number of MPO-positive cells/hpf.
Immunoblotting
Levels of surfactant protein (SP)-D in high-salt protein extracts of frozen lungs were assessed by immunoblotting with mouse monoclonal antibody to SP-D (Santa Cruz Biotechnology, Santa Cruz CA). Protein samples—total lung protein (50 μg), recombinant rat SP-D (2.5 μg) and protein standards (SeeBlue® Plus2 Pre-stained Standards, Invitrogen; 7 μl) were separated by Tris-Glycine SDS-PAGE and transferred to a PVDF membrane. Recombinant rat SP-D was purified from CHO K1 cells and was variably glycosylated21; it was the kind gift of Dr. Erika Crouch, Washington University, St. Louis, MO. The membrane was incubated overnight with mouse monoclonal antibody and subsequently incubated with goat anti-mouse antibody conjugated with horseradish peroxidase (HRP; Zymed, San Francisco, CA) for 1 h. ECL agent (Amersham Biosciences, UK) was used for detection. Densitometry was performed using ImageJ 1.4g software (National Institutes of Health, Bethesda, MD).
Microarray analysis
Gene expression profiling was performed with the Affymetrix Rat Array RAE 230A genechip following Affymetrix protocols used within the Baylor College of Medicine Microarray Core Facility. Genechips were hybridized with RNA isolated from the lungs of rats within each of three groups—sham, T/HS-PBS and T/HS-IL6—one chip for each of four lung RNA samples per group. Total RNA was isolated from 4 to 5 micron cryotome sections of each lung using TRIzol® Reagent (Invitrogen, Carlsbad, California) single step RNA isolation protocol followed by purification with RNeasy® Mini Kit (QIAGEN, Hilden, Germany) as instructed by the manufacturer. We used GenespringGX (Agilent Technologies Inc, Santa Clara CA) software package for quality assessment, statistical analysis and annotation. Low-level analyses included background correction, quartile normalization and expression estimation using RMA-based analysis within Genespring. One-way analysis of variance (ANOVA) with contrasts was used for group comparisons on all genes and on the list of UPR entities. p-Values were adjusted for multiple comparisons using the Benjamini-Hockberg method. The adjusted p-values represent false discovery rates (FDR) and are estimates of the proportion of “significant” genes that are false or spurious “discoveries”. We used a False Discovery Rate (FDR) of 5% as cut-off. RAE 230A genechips each contained 15,923 probe sets representing 13,521 annotated genes or expressed sequence tags. A UPR gene entity list was created using both Ingenuity Pathway Analysis (IPA® Redwood City, CA) and the Gene Ontology Database®, with keywords “endoplasmic reticulum stress, unfolded protein response”.
Statistical analysis
Data are presented as mean ± standard error of the mean (SEM). Multiple group comparisons of means were done by one-way analysis of variance (ANOVA) and the Student–Newman–Keuls test. Survival analysis was done by Kaplan–Meier test.
Results
T/HS increased mortality and bacterial burden in a model of P. aeruginosa pneumonia
To determine the effect of T/HS on susceptibility to PA pneumonia, we developed a pre-clinical model of T/HS combined with PA pneumonia in rats (Fig. 1A). Rats were subjected to the sham or T/HS protocol followed 24-h later by transtracheal inoculation of PA. After inoculation, rats were observed for survival for 72 h. Remarkably, survival in T/HS rats was decreased by 80% compared to sham rat group (p < 0.001, Kaplan–Meier analysis; Fig. 1B).
To determine if the cause of increased mortality observed in T/HS rats is due to increased bacterial burden in the lungs of these animals, we measured bacterial numbers in the homogenates of lungs of the rats subjected to sham or T/HS protocols 4 h after transtracheal inoculation of PA (Fig. 2). The PA CFU/gram of lung tissue was 9-fold higher in the rats subjected to T/HS protocol compared to sham rats (p < 0.05, ANOVA) strongly suggesting that the increased mortality of T/HS mice following PA inoculation was due to increased bacterial burden.
To assess if the increased bacterial burden in T/HS rats is due to a decrease in infiltrating neutrophils resulting in impaired clearance of bacteria, we performed myeloperoxidase staining of lung sections 4 h after inoculation with PA. The number of MPO-positive cells in the lungs of PA-infected T/HS rats (35 ± 5 cells per 1000× field; mean ± SEM) was identical to the number of MPO-positive cells in the lungs of PA-infected sham rats (34 ± 4 cells per 1000× field; mean ± SEM). In addition to there being no difference between groups in the number of MPO-positive cell within the lung, the distribution of MPO-positive cells within the alveoli and lung interstitium was similar (data not shown). Thus, the increased bacterial burden in T/HS rats was not due to differences in the quantity or distribution of PMN recruitment into the lung.
We previously demonstrated that AECII were the predominant cell type that underwent apoptosis following T/HS.14 Since AECII produce SP-D, which increases PMN-mediated phagocytosis of bacteria including PA, we assessed SP-D levels in the lungs of T/HS and sham rats (Fig. 3). SP-D levels in the lungs of rats subjected to T/HS were reduced by 50% compared to lungs of sham rats at 24 h (p < 0.05, ANOVA). Thus, our data suggest that the increase in bacterial burden in the lungs of T/HS rats is due to reduced levels of SP-D in the lungs.
Figure 3.
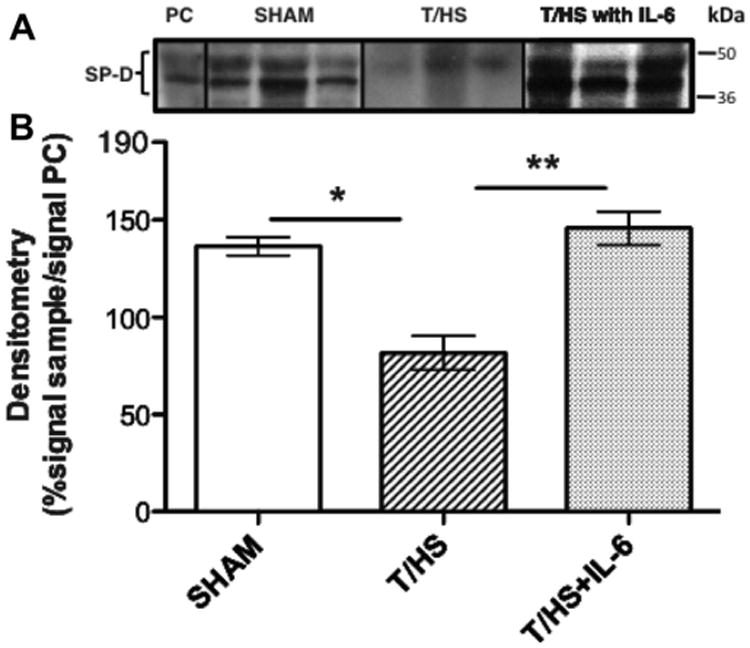
Effect of T-HS on lung surfactant protein-D (SP-D) levels. Rat recombinant SP-D (2.5 μg; positive control, PC) or whole lung protein extracts (50 μg) from rats (n = 3 per group) subjected to sham or T/HS protocol were separated by SDS-PAGE and immunoblotted with monoclonal antibodies to SP-D (panel A). The band signal intensity was quantitated by densitometry and reported as a ratio of signal intensity in sample to signal intensity in positive control (PC) times 100. Bars with paired single or double asterisks above (*, **) differ significantly (p < 0.05, ANOVA).
Administration of IL-6 as a resuscitation adjuvant in T/H rats prevented P. aeruginosa pneumonia mortality, reduced bacterial lung burden, and normalized SP-D levels in the lung
We previously demonstrated that IL-6 administration as a resuscitation adjuvant induced activation of Stat3, particularly Stat3α, within lung parenchymal cells resulting in protection against alveolar epithelial cell apoptosis following T/HS.14 To assess if this prevention of apoptosis by IL-6 also affected nosocomial pneumonia mortality, rats were subjected to our T/HS protocol with IL-6 administered as a resuscitation adjuvant, followed 24 h later by transtracheal inoculation of PA (Fig. 1B). Impressively, animals that received IL-6 were completely protected against mortality (p < 0.001, Kaplan–Meier analysis). When we assessed how IL-6 affected bacterial burden (Fig. 2B), we found that lung bacterial burden was decreased by 4.8-fold in T/HS-IL6 rats compared to lung bacterial burden in T/HS-PBS rats (p < 0.05, ANOVA) to levels statistically indistinguishable from sham rats inoculated with PA.
To determine if lung bacterial burden was reduced in IL-6-treated T/HS rats as a result of restoration of SP-D levels, we assessed the effect of IL-6 treatment on SP-D levels in the lung. Densitometry analysis of immunoblots of whole lung homogenates from IL-6-treated T/HS rats (Fig. 3) revealed that IL-6 treatment prevented the reduction in pulmonary SP-D seen in T/HS animals (p < 0.05, ANOVA) with SP-D protein levels in the lungs of IL-6-treated T/HS rats being equivalent to sham animals.
The UPR is significantly altered in the lung following T-HS and demonstrated normalization when IL-6 is used as a resuscitation adjuvant
The molecular mechanisms underlying T/HS-induced AECII apoptosis are not understood. We previously investigated the potential contribution of the classical intrinsic and extrinsic pathways to lung apoptosis in T/HS and demonstrated that T/HS altered the expression of many of intrinsic and extrinsic apoptosis pathway-related genes and that IL-6 treatment normalized expression of the majority of those genes altered by T/HS through a Stat3-dependent mechanism.14 However, which genes were critical for apoptosis of AECII and its prevention, if any, were not specifically delineated in these studies.
The unfolded protein response (UPR) is a critical homeostatic mechanism for highly secretory cells such as AECII. Recently, the UPR has been described as a major cause of apoptosis when secretory cells are exposed to overwhelming or prolonged endoplasmic reticulum (ER) stress as would occur in T/HS. Since a central feature of the UPR is activation of several key transcription factors such as DDIT3 (CHOP), ATF4, ATF6, and XBP1, we investigated the impact of T/HS on the ER stress response at the transcriptome level by mining oligonucleotide microarray (Affymetrix) data previously obtained and archived by us from the lungs of 3 groups of rats: Sham, T/HS-PBS, and T/HS-IL6, sacrificed 1 h after the end of resuscitation.14 A broad 185-gene UPR-associated entity list was generated following an extensive literature review and with the help of Ingenuity Pathway Analysis (IPA®). Of the 185-gene UPR set, 113 distinct gene entities were annotated and expressed across the chips after spot duplicates were removed. Importantly, the three experimental groups self organized based on this UPR-associated gene entity list expression (Fig. 4).
Figure 4.

Self-organizing heat map of UPR transcriptomes. Self-organizing heat map of UPR-associated gene entity list mRNA levels demonstrates clustering of experimental groups (n = 4 per group) and relative relatedness based on their expression profiles. Levels of relative expression range from bright green (−2-fold) to bright red (+2-fold). Genes marked with an asterisk (*) represent the first twelve pro-apoptotic genes listed in Table 1.
In order to determine the effect of the UPR on the observed T/HS-induced lung apoptosis and its prevention with IL-6, we performed intergroup comparisons of the transcriptome profiles within these groups. Within the list of 113 genes expressed by all groups, 65 (57%) were significantly impacted by T/HS when compared to Sham animals (ANOVA, p < 0.05). When we assessed for IL-6 responsive transcripts that proved significantly impacted by T/HS, we found that 53 (47%) entities were significantly altered in both T/HS vs. Sham and T/HS-IL6 vs. T/HS comparator groups. Thirty-two of these UPR-associated genes demonstrated ≥1.5-fold change in T/HS vs. Sham, 14 of which are considered pro-apoptotic (Table 1). Taking the known apoptotic function of these genes into context, we demonstrated that 86% (12 of 14) of the UPR-associated genes with known pro-apoptotic function were up-regulated following T/HS and subsequently normalized with IL-6. Among the most up-regulated genes within this intergroup comparison were pro-apoptotic UPR members, eukaryotic translation initiation factor 2-alpha kinase 2 (EI-F2AK2; increased 4.7 fold by T/HS), DNA-damage-inducible transcript 3 (DDIT3; increased 2.5 fold by T/HS; also known as C/EBP-homologous protein, CHOP). Canonical UPR members, X-box binding protein 1 (XBP1) and activating transcription factor 4 (ATF4) also were both increased by 1.5 fold by T/HS when compared to sham (Table 1). In animals in which pulmonary cell apoptosis was prevented by receiving IL-6 at resuscitation, we find that these potentially pro-apoptotic UPR member transcripts (EIF2AK2, DDIT3, XBP1, and ATF4) are reduced to levels statistically indistinguishable from sham levels, strongly suggesting a contribution to prevention of pulmonary cell apoptosis (Table 1).
Table 1.
Fold change comparisons (T/HS vs. Sham, T/HS + IL6 vs. T/HS, and T/HS + IL6 vs. Sham) for mRNA transcript levels with an absolute fold change of 1. 5-fold or greater T/HS vs. Sham comparison (p < 0.05, ANOVA).
| Gene symbol | Apoptotic role | Fold change | ||
|---|---|---|---|---|
|
| ||||
| T/HS vs. Sham | T/HS + IL6 vs. T/HS | T/HS + IL6 vs. Sham | ||
| Eif2ak2 | pro | 4.7 | −4.2 | 1.1 |
| Tnf | pro | 2.8 | −1.7 | 1.6* |
| Ppp1r15a | pro | 2.8 | −1.3 | 2.1* |
| Ddit3 | pro | 2.5 | −3.6 | −1.4 |
| Casp12 | pro | 2.4 | −2.3 | 1.0 |
| Bid | pro | 2.1 | −1.9 | 1.1 |
| Casp3 | pro | 1.6 | −2.1 | −1.3 |
| Casp7 | pro | 1.6 | −1.6 | 1.0 |
| Atf4 | pro | 1.5 | −1.4 | 1.1 |
| Eif2s1 | pro | 1.5 | −1.2 | 1.3 |
| Tp53 | pro | 1.5 | −1.4 | 1.1 |
| Xbp1 | pro | 1.5 | −1.2 | 1.3 |
| Ccnd1 | pro | −1.8 | 1.2 | −1.5* |
| Pik3ip1 | pro | −2.3 | 1.7 | −1.4 |
| Apobec1 | anti | 3.2 | −3.9 | −1.2 |
| Psmb10 | anti | 2.6 | −2.7 | 1.0 |
| Psmb9 | anti | 2.5 | −3.3 | −1.3 |
| Psme2 | anti | 2.3 | −2.3 | 1.0 |
| Psmb2 | anti | 1.9 | −1.4 | 1.4 |
| Psmb8 | anti | 1.9 | −2.7 | −1.4 |
| Psma3 | anti | 1.8 | −1.3 | 1.4 |
| Psma1 | anti | 1.8 | −1.6 | 1.1 |
| Aars | anti | 1.8 | −1.7 | 1.1 |
| Psma5 | anti | 1.7 | −1.5 | 1.1 |
| Psma7 | anti | 1.7 | −1.5 | 1.1 |
| Psme1 | anti | 1.6 | −1.9 | −1.2 |
| Psmb3 | anti | 1.6 | −1.4 | 1.1 |
| Psma6 | anti | 1.5 | −1.1 | 1.4 |
| Vcp | anti | 1.5 | −1.3 | 1.2 |
| Hspb7 | anti | 1.5 | 2.0 | 3.0* |
| Hspalb | anti | −1.5 | 2.2 | 1.5* |
| Tor1b | unknown | 2.2 | −2.3 | 1.0 |
p < 0.05;
all other T/HS + IL6 vs. Sham comparisons p > 0.05.
Discussion
To investigate the impact of T/HS on the innate host defense of the lung, we developed a rodent model of secondary P. aeruginosa pneumonia following T/HS. We demonstrated in this model that T/HS increased lung bacterial burden 9-fold and resulted in an 80% increase in mortality. Neutrophil recruitment to the lung was not altered in infected T/HS lungs compared to infected sham lung to explain impaired bacterial clearance; rather, lung SP-D protein levels were decreased by nearly 50%. Use of IL-6 as a resuscitation adjuvant prevented the decrease in lung SP-D, reduced lung PA bacterial burden nearly 5-fold and completely prevented PA-mediated mortality. The UPR transcriptome of the lung was found to be significantly impacted by T/HS and to be normalized when IL-6 is given as a resuscitative adjuvant. These findings indicate that there is a maladaptive reduction in innate lung defense in T/HS mediated by a reduction in SP-D, which accompanies AECII apoptosis and contributes to increased susceptibility to pneumonia. AECII apoptosis may be mediated by the UPR, which, along with increased susceptibility to pneumonia, may be prevented by use of IL-6 as a resuscitative adjuvant.
AECII are referred to by some authors22,23 as the “Defender of the Alveolus”. They contribute to the mechanical viability of the alveolus by producing the pulmonary surfactant layer and are responsible for restoring injured alveolar epithelium. In addition, they have a unique role in the innate immunity of the lung. AECII have been shown to contribute to host defense by secreting anti-inflammatory and anti-microbial proteins into the alveolar space.22,24 Most notable of these anti-microbial proteins are the collectins, SP-A and SP-D. SP-D, in particular, is known to enhance phagocytosis of Pseudomonas.25,26 Given our previous findings that AECII was the pulmonary cell type undergoing the majority of the apoptosis caused by T/HS,14 we hypothesized that loss of these cells may contribute to impaired innate host defense of the lung following T/HS. We began to address the role of the AECII in innate host defense by determining levels of SP-D in the lung of animals in our model of T/HS. SP-D is a good marker of AECII function as these cells are the predominant source of SP-D production. SP-D was reduced by nearly 50% in the whole lung of animals undergoing T/HS when compared to sham, which provides a unifying explanation for our observed findings that a relative deficiency of SP-D in the lung allows for increased PA growth leading to increased bacterial burden and increased pneumonia mortality.
Supporting the hypothesis that decreased SP-D predisposes to pneumonia is a recent study demonstrating that children with absent SP-D more frequently have pneumonia27 and a recent study of acute lung injury caused by intestinal ischemia/reperfusion injury, which demonstrated a significant reduction of SP-D in the lung by immunohistochemistry staining.28 Similarly, other investigators have shown that early in the evolution of acute lung injury, alveolar epithelial cell death leads to decreased production and increased clearance of SP-D.29,30
We have previously demonstrated the ability of IL-6-stimulated Stat3 to prevent AECII apoptosis following T/HS.14 The exact mechanism of this protection is not fully understood despite prior investigation into the intrinsic and extrinsic cell death pathways.14 Given the critical role of the UPR in highly secretory cell types such as AECII and its ability to drive these types of cells into apoptosis, we investigated the impact of T/HS with and without IL-6 on the UPR. Our investigation into the UPR suggests that this pathway contributes to the concert of stimuli that leads to cell death within the lung following T/HS. Previous work has demonstrated that the UPR is activated by trauma with hemorrhage demonstrating increased expression of ATF6, PERK, IREα, and CHOP and is associated with increased apoptosis within the liver.31 Utilizing whole organ transcriptomic analysis, we examined the impact of T/HS on the UPR in the lung one hour following resuscitation, when apoptosis is maximal.14 We identified several canonical members of the UPR with pro-apoptotic functions that demonstrate changes in transcript levels in response to T/HS and IL-6 intervention, which suggest this pathway may contribute to pro-apoptotic signaling. The most significantly impacted UPR members across all experimental comparisons were Eif2ak2, ATF4, CHOP (DDIT3), and XBP-1. ATF4 and XBP-1 have both been shown to be transcriptional activators of CHOP, which has been shown to be a potent stimulator of apoptosis through its downstream targets in many models.32–35 Of note, previous work has shown that CHOP signaling mediates LPS-induced lung injury in a mouse model of sepsis, and when over-expressed in lung cell lines leads to increased apoptosis,32 suggesting a role for CHOP in apoptosis of lung epithelial cells in settings of stress including T/HS.
In addition to 12 of the 14 pro-apoptotic gene transcripts that were increased with T/HS vs. Sham and normalized with IL-6 (Table 1), a pattern consistent with their contributing to lung apoptosis in T/HS and the protective effect of IL-6, there were several anti-apoptotic gene transcripts that were increased with T/HS vs. Sham and also were normalized with IL-6 (Table 1). These changes likely are a component of the lung's efforts to maintain homeostasis; T/HS-induced lung apoptosis and its prevention by IL-6 were accomplished in spite of their modulation. However, one UPR-related anti-apoptosis gene, Hspb7, a member of the small heat shock protein family (Table 1), was increased by T/HS vs. Sham and was further increased in the T/HS + IL-6 group while another UPR-related anti-apoptosis gene, Hspa1b, a member of the heat shock protein 70 family (Table 1), was decreased by T/HS vs. Sham and was increased in the T/HS + IL-6 group. Modulations in these two anti-apoptosis genes, perhaps like the 12 pro-apoptotic genes discussed above, also may have contributed to T/HS-induced AECII apoptosis and its prevention by IL-6.
Our results also indicate that IL-6 provides protection against PA pneumonia following T/HS. This protection is due, at least in part, to the ability of IL-6-activated Stat3 to protect AECII against T/HS-induced apoptosis as we previously demonstrated.14 In this paper, we demonstrate that sparing the AECII from apoptosis maintains SP-D levels in the lung, which likely contributes to the protection against PA bacterial burden and mortality observed in our model of post-T/HS PA pneumonia.
Nosocomial pneumonia including ventilator-associated pneumonia (VAP) is one of the leading causes of healthcare-associated infection following severe trauma1 and the most common cause of death in patients surviving the original traumatic injury. VAP following severe trauma is most often due to PA8,36,37 with infection with antibiotic-resistant PA38,39 and other Gram-negative bacteria40 increasing in incidence at an alarming rate. Our model allows for investigation not only of the mechanisms of pathogenesis of pneumonia following T/HS, but also allows us to begin to investigate novel interventions that may prevent PA pneumonia following T/HS. One category of intervention involves agents that might prevent AECII apoptosis and the resultant SP-D deficiency, while another category of intervention involves restoration of impaired innate epithelial cell immunity within the lung. Within the first category is the use of IL-6 as a resuscitation adjuvant. We have established that IL-6 as a resuscitation adjuvant is of clear benefit in preventing organ apoptosis and inflammation in rat and porcine models of T/HS,12–15,41 as well as in reducing the severity of illness in pre-clinical models of bloodstream infections.20 In addition to potentially preventing AECII apoptosis and pneumonia susceptibility in T/HS patients, this intervention also may prevent heart and liver dysfunction in T/HS by preventing apoptosis of cardiomyocytes12 and hepatocytes.13 However, the FDA has not approved IL-6 for this or any other indication. An alternative intervention that may be able to prevent AECII apoptosis and subsequent decrease in SP-D is use of proteostasis modulators such as geranylgeranylacetone (GGA; teprenone; Selbex®). GGA is an antiulcer drug that has been used in Japan for over thirty years and has a favorable side effect profile. GGA induces expression of HSP70 and HSP90; HSP90, in particular, has been shown to downregulate apoptosis secondary to UPR.42,43 In fact, when given to rats during intracerebral hemorrhage, GGA decreased neuronal cell apoptosis and improved neurological recovery by increasing Stat3 activity.44 In studies underway, we are examining the potential benefit of GGA in our rat model of T/HS to determine if it can be used to prevent apoptosis of AECII, reduction in SP-D, and susceptibility to PA pneumonia.
Two potential interventions that may restore lung innate immunity following T/HS-induced AECII apoptosis also are suggested by our findings—one is aerosol administration of SP-D; the second is inhaled Pam2-ODN, a combination of Toll-like receptor (TLR) agonists. Surfactant therapy has been used extensively and successfully in reducing mortality from respiratory distress syndrome of the newborn.45 However, a significant proportion of infants born at less than 28 weeks' gestation develop neonatal chronic lung disease. Current surfactant therapies lack SP-D, yet animal models support a role for SP-D in reducing inflammation and infection in the lung, which suggests that supplementation of current surfactant therapies with recombinant forms of SP-D may help offset the risk of development of chronic lung disease. Thus, surfactant preparations containing SP-D may be available in the future to test in T/HS patients for the ability to reduce nosocomial pneumonia. Pam2-ODN consists of Pam2CSK4, a diacylated lipopeptide ligand for TLR2/6, combined with oligonucleotide (ODN) M362, a ligand for TLR9.46 Pam2-ODN has been demonstrated to broadly protect mice against otherwise lethal pneumonias including those caused by P. aeruginosa and Streptococcus pneumonia.46 Thus, an alternative to SP-D is Pam2-ODN inhalation to therapeutically boost residual lung epithelial cell intrinsic defenses following T/HS and potentially to protect T/HS patients from VAP.
Acknowledgments
Supported, in part, by grants HL07619 (DT), HL66991 (DT), and AI055413 (ST, AM) from the National Institutes of Health, grant W81XWH-11-2-0018 (DT) from the US Army and H48839 (AM) from the American Lung Association. We would like to acknowledge Dr. John Alverdy (University of Chicago, Chicago, IL) for kindly providing the PA ATCC-27853 strain used in our experiments and Dr. Erica C. Crouch (Washington University, St. Louis, MO) for rat re-combinant SP-D used in our experiments.
References
- 1.Sauaia A, Moore FA, Moore EE, Moser KS, Brennan R, Read RA, et al. Epidemiology of trauma deaths: a reassessment. The Journal of Trauma. 1995 Feb;38(2):185–93. doi: 10.1097/00005373-199502000-00006. [DOI] [PubMed] [Google Scholar]
- 2.Angele MK, Faist E. Clinical review: immunodepression in the surgical patient and increased susceptibility to infection. Critical Care (London, England) 2002 Aug;6(4):298–305. doi: 10.1186/cc1514. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kimura F, Shimizu H, Yoshidome H, Ohtsuka M, Miyazaki M. Immunosuppression following surgical and traumatic injury. Surgery Today. 2010 Sep;40(9):793–808. doi: 10.1007/s00595-010-4323-z. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu H, Kuzmenko A, Wan S, Schaffer L, Weiss A, Fisher JH, et al. Surfactant proteins A and D inhibit the growth of gram-negative bacteria by increasing membrane permeability. The Journal of Clinical Investigation. 2003 Jun;111(10):1589–602. doi: 10.1172/JCI16889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crouch E, Wright JR. Surfactant proteins a and d and pulmonary host defense. Annual Review of Physiology. 2001;63:521–54. doi: 10.1146/annurev.physiol.63.1.521. Review. [DOI] [PubMed] [Google Scholar]
- 6.Wright JR. Immunoregulatory functions of surfactant proteins. Nature Reviews Immunology. 2005;5(1):58–68. doi: 10.1038/nri1528. Review. [DOI] [PubMed] [Google Scholar]
- 7.Shepherd VL. Distinct roles for lung collectins in pulmonary host defense. American Journal of Respiratory Cell and Molecular Biology. 2002 Apr 01;26(3):257–60. doi: 10.1165/ajrcmb.26.3.f227. Review. [DOI] [PubMed] [Google Scholar]
- 8.Babcock HM, Zack JE, Garrison T, Trovillion E, Kollef MH, Fraser VJ. Ventilator-associated pneumonia in a multi-hospital system: differences in microbiology by location. Infection Control and Hospital Epidemiology: the Official Journal of the Society of Hospital Epidemiologists of America. 2003 Nov 01;24(11):853–8. doi: 10.1086/502149. Comparative Study. [DOI] [PubMed] [Google Scholar]
- 9.Douda DN, Jackson R, Grasemann H, Palaniyar N. Innate immune collectin surfactant protein D simultaneously binds both neutrophil extracellular traps and carbohydrate ligands and promotes bacterial trapping. Journal of Immunology (Baltimore, Md: 1950) 2011 Aug 15;187(4):1856–65. doi: 10.4049/jimmunol.1004201. [DOI] [PubMed] [Google Scholar]
- 10.Griese M, Starosta V. Agglutination of Pseudomonas aeruginosa by surfactant protein D. Pediatric Pulmonology. 2005 Nov 01;40(5):378–84. doi: 10.1002/ppul.20295. [DOI] [PubMed] [Google Scholar]
- 11.Voorhout WF, Veenendaal T, Kuroki Y, Ogasawara Y, van Golde LM, Geuze HJ. Immunocytochemical localization of surfactant protein D (SP-D) in type II cells, Clara cells, and alveolar macrophages of rat lung. The Journal of Histochemistry and Cytochemistry: Official Journal of the Histochemistry Society. 1992 Oct 01;40(10):1589–97. doi: 10.1177/40.10.1527377. [DOI] [PubMed] [Google Scholar]
- 12.Alten JA, Moran A, Tsimelzon AI, Mastrangelo MA, Hilsenbeck SG, Poli V, et al. Prevention of hypovolemic circulatory collapse by IL-6 activated Stat3. PLoS One. 2008;3(2):e1605. doi: 10.1371/journal.pone.0001605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moran A, Akcan Arikan A, Mastrangelo MA, Wu Y, Yu B, Poli V, et al. Prevention of trauma and hemorrhagic shock-mediated liver apoptosis by activation of stat3alpha. International Journal of Clinical and Experimental Medicine. 2008;1(3):213–47. [PMC free article] [PubMed] [Google Scholar]
- 14.Moran A, Tsimelzon AI, Mastrangelo MA, Wu Y, Yu B, Hilsenbeck SG, et al. Prevention of trauma/hemorrhagic shock-induced lung apoptosis by IL-6-mediated activation of Stat3. Clinical and Translational Science. 2009 Feb;2(1):41–9. doi: 10.1111/j.1752-8062.2008.00076.x. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moran A, Thacker SA, Arikan AA, Mastrangelo MA, Wu Y, Yu B, et al. IL-6-mediated activation of Stat3alpha prevents trauma/hemorrhagic shock-induced liver inflammation. PLoS One. 2011;6(6):e21449. doi: 10.1371/journal.pone.0021449. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hierholzer C, Kalff JC, Omert L, Tsukada K, Loeffert JE, Watkins SC, et al. Interleukin-6 production in hemorrhagic shock is accompanied by neutrophil recruitment and lung injury. American Journal of Physiology. 1998;275(3 Pt 1):L611–21. doi: 10.1152/ajplung.1998.275.3.L611. [DOI] [PubMed] [Google Scholar]
- 17.Ono M, Yu B, Hardison EG, Mastrangelo MA, Tweardy DJ. Increased susceptibility to liver injury after hemorrhagic shock in rats chronically fed ethanol: role of nuclear factor-kappa B, interleukin-6, and granulocyte colony-stimulating factor. Shock. 2004 Jun;21(6):519–25. doi: 10.1097/01.shk.0000126905.75237.07. [DOI] [PubMed] [Google Scholar]
- 18.Vanderzwan J, McCaig L, Mehta S, Joseph M, Whitsett J, McCormack DG, et al. Characterizing alterations in the pulmonary surfactant system in a rat model of Pseudomonas aeruginosa pneumonia. The European Respiratory Journal: Official Journal of the European Society for Clinical Respiratory Physiology. 1998 Dec 01;12(6):1388–96. doi: 10.1183/09031936.98.12061388. [DOI] [PubMed] [Google Scholar]
- 19.Webert KE, Vanderzwan J, Duggan M, Scott JA, McCormack DG, Lewis JF, et al. Effects of inhaled nitric oxide in a rat model of Pseudomonas aeruginosa pneumonia. Critical Care Medicine. 2000 Jul;28(7):2397–405. doi: 10.1097/00003246-200007000-00035. Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- 20.Arikan AA, Yu B, Mastrangelo MA, Tweardy DJ. Interleukin-6 treatment reverses apoptosis and blunts susceptibility to intra-peritoneal bacterial challenge following hemorrhagic shock. Critical Care Medicine. 2006 Mar;34(3):771–7. doi: 10.1097/01.ccm.0000201901.30292.c2. Evaluation Studies Research Support, N.I.H., Extramural. [DOI] [PubMed] [Google Scholar]
- 21.Crouch E, Persson A, Chang D, Heuser J. Molecular structure of pulmonary surfactant protein D (SP-D) The Journal of Biological Chemistry. 1994 Jun 24;269(25):17311–9. Research Support, U.S. Gov't, P.H.S. [PubMed] [Google Scholar]
- 22.Mason RJ. Biology of alveolar type II cells. Respirology (Carlton, Vic) 2006 Feb;11 Suppl:S12–5. doi: 10.1111/j.1440-1843.2006.00800.x. Review. [DOI] [PubMed] [Google Scholar]
- 23.Mason RJ, Williams MC. Type II alveolar cell. Defender of the alveolus. The American Review of Respiratory Disease. 1977 Jul;115(6 Pt 2):81–91. doi: 10.1164/arrd.1977.115.S.81. [DOI] [PubMed] [Google Scholar]
- 24.Nayak A, Dodagatta-Marri E, Tsolaki AG, Kishore U. An insight into the diverse roles of surfactant proteins, SP-A and SP-D in innate and adaptive immunity. Frontiers in Immunology. 2012;3:131. doi: 10.3389/fimmu.2012.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lim BL, Wang JY, Holmskov U, Hoppe HJ, Reid KB. Expression of the carbohydrate recognition domain of lung surfactant protein D and demonstration of its binding to lipopolysaccharides of gram-negative bacteria. Biochemical and Biophysical Research Communications. 1994 Aug 15;202(3):1674–e80. doi: 10.1006/bbrc.1994.2127. [DOI] [PubMed] [Google Scholar]
- 26.Restrepo CI, Dong Q, Savov J, Mariencheck WI, Wright JR. Surfactant protein D stimulates phagocytosis of Pseudomonas aeruginosa by alveolar macrophages. American Journal of Respiratory Cell and Molecular Biology. 1999 Nov;21(5):576–85. doi: 10.1165/ajrcmb.21.5.3334. [DOI] [PubMed] [Google Scholar]
- 27.Griese M, Steinecker M, Schumacher S, Braun A, Lohse P, Heinrich S. Children with absent surfactant protein D in bronchoalveolar lavage have more frequently pneumonia. Pediatr Allergy Immunol. 2008 Nov;19(7):639–47. doi: 10.1111/j.1399-3038.2007.00695.x. [DOI] [PubMed] [Google Scholar]
- 28.Guzel A, Kanter M, Guzel A, Pergel A, Erboga M. Anti-inflammatory and antioxidant effects of infliximab on acute lung injury in a rat model of intestinal ischemia/reperfusion. Journal of Molecular Histology. 2012 Jul;43(3):361–9. doi: 10.1007/s10735-012-9396-0. [DOI] [PubMed] [Google Scholar]
- 29.Herbein JF, Wright JR. Enhanced clearance of surfactant protein D during LPS-induced acute inflammation in rat lung. American Journal of Physiology Lung Cellular and Molecular Physiology. 2001 Jul;281(1):L268–77. doi: 10.1152/ajplung.2001.281.1.L268. [DOI] [PubMed] [Google Scholar]
- 30.Cheng IW, Ware LB, Greene KE, Nuckton TJ, Eisner MD, Matthay MA. Prognostic value of surfactant proteins A and D in patients with acute lung injury. Critical Care Medicine. 2003 Feb;31(1):20–7. doi: 10.1097/00003246-200301000-00003. [DOI] [PubMed] [Google Scholar]
- 31.Jian B, Hsieh CH, Chen J, Choudhry M, Bland K, Chaudry I, et al. Activation of endoplasmic reticulum stress response following trauma-hemorrhage. Biochimica et Biophysica Acta. 2008 Nov 01;1782(11):621–6. doi: 10.1016/j.bbadis.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Endo M, Oyadomari S, Suga M, Mori M, Gotoh T. The ER stress pathway involving CHOP is activated in the lungs of LPS-treated mice. Journal of Biochemistry. 2005 Oct 01;138(4):501–7. doi: 10.1093/jb/mvi143. [DOI] [PubMed] [Google Scholar]
- 33.Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes & Development. 1998 May 01;12(7):982–95. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes & Development. 2004 Dec 15;18(24):3066–77. doi: 10.1101/gad.1250704. Comparative Study. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ji C, Mehrian-Shai R, Chan C, Hsu YH, Kaplowitz N. Role of CHOP in hepatic apoptosis in the murine model of intragastric ethanol feeding. Alcoholism, Clinical and Experimental Research. 2005 Aug 01;29(8):1496–503. doi: 10.1097/01.alc.0000174691.03751.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Richards MJ, Edwards JR, Culver DH, Gaynes RP. Nosocomial infections in pediatric intensive care units in the United States. National Nosocomial Infections Surveillance System. Pediatrics. 1999 May 01;103(4):e39. doi: 10.1542/peds.103.4.e39. [DOI] [PubMed] [Google Scholar]
- 37.Foglia E, Meier MD, Elward A. Ventilator-associated pneumonia in neonatal and pediatric intensive care unit patients. Clinical Microbiology Reviews. 2007 Jul 01;20(3):409–25. doi: 10.1128/CMR.00041-06. [Review]. [table of contents] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun HY, Fujitani S, Quintiliani R, Yu VL. Pneumonia due to Pseudomonas aeruginosa: part II: antimicrobial resistance, pharmacodynamic concepts, and antibiotic therapy. Chest. 2011 May;139(5):1172–85. doi: 10.1378/chest.10-0167. Review. [DOI] [PubMed] [Google Scholar]
- 39.Fujitani S, Sun HY, Yu VL, Weingarten JA. Pneumonia due to Pseudomonas aeruginosa: part I: epidemiology, clinical diagnosis, and source. Chest. 2011 Apr;139(4):909–19. doi: 10.1378/chest.10-0166. Review. [DOI] [PubMed] [Google Scholar]
- 40.Grgurich PE, Hudcova J, Lei Y, Sarwar A, Craven DE. Management and prevention of ventilator-associated pneumonia caused by multidrug-resistant pathogens. Expert Review of Respiratory Medicine. 2012 Nov;6(5):533–55. doi: 10.1586/ers.12.45. Review. [DOI] [PubMed] [Google Scholar]
- 41.Brundage SI, Zautke NA, Holcomb JB, Spain DA, Lam JC, Mastrangelo MA, et al. Interleukin-6 infusion blunts proinflammatory cytokine production without causing systematic toxicity in a swine model of uncontrolled hemorrhagic shock. The Journal of Trauma. 2004 Nov;57(5):970–7. doi: 10.1097/01.ta.0000141970.68269.ac. discussion 7–8. [DOI] [PubMed] [Google Scholar]
- 42.Marcu MG, Doyle M, Bertolotti A, Ron D, Hendershot L, Neckers L. Heat shock protein 90 modulates the unfolded protein response by stabilizing IRE1alpha. Molecular and Cellular Biology. 2002 Dec;22(24):8506–13. doi: 10.1128/MCB.22.24.8506-8513.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gallerne C, Prola A, Lemaire C. Hsp90 inhibition by PU-H71 induces apoptosis through endoplasmic reticulum stress and mitochondrial pathway in cancer cells and overcomes the resistance conferred by Bcl-2. Biochimica et Biophysica Acta. 2013 Jun;1833(6):1356–66. doi: 10.1016/j.bbamcr.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 44.Sinn DI, Chu K, Lee ST, Song EC, Jung KH, Kim EH, et al. Pharmacological induction of heat shock protein exerts neuroprotective effects in experimental intracerebral hemorrhage. Brain Research. 2007 Mar 2;1135(1):167–76. doi: 10.1016/j.brainres.2006.11.098. Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- 45.Clark HW. Untapped therapeutic potential of surfactant proteins: is there a case for recombinant SP-D supplementation in neonatal lung disease? Neonatology. 2010 Jun;97(4):380–7. doi: 10.1159/000297770. Review. [DOI] [PubMed] [Google Scholar]
- 46.Evans SE, Scott BL, Clement CG, Larson DT, Kontoyiannis D, Lewis RE, et al. Stimulated innate resistance of lung epithelium protects mice broadly against bacteria and fungi. American Journal of Respiratory Cell and Molecular Biology. 2010;42(1):40–50. doi: 10.1165/rcmb.2008-0260OC. [DOI] [PMC free article] [PubMed] [Google Scholar]