

Abstract
Superhydrophobic materials, with surfaces possessing permanent or metastable non-wetted states, are of interest for a number of biomedical and industrial applications. Here we describe how electrospinning or electrospraying a polymer mixture containing a biodegradable, biocompatible aliphatic polyester (e.g., polycaprolactone and poly(lactide-co-glycolide)), as the major component, doped with a hydrophobic copolymer composed of the polyester and a stearate-modified poly(glycerol carbonate) affords a superhydrophobic biomaterial. The fabrication techniques of electrospinning or electrospraying provide the enhanced surface roughness and porosity on and within the fibers or the particles, respectively. The use of a low surface energy copolymer dopant that blends with the polyester and can be stably electrospun or electrosprayed affords these superhydrophobic materials. Important parameters such as fiber size, copolymer dopant composition and/or concentration, and their effects on wettability are discussed. This combination of polymer chemistry and process engineering affords a versatile approach to develop application-specific materials using scalable techniques, which are likely generalizable to a wider class of polymers for a variety of applications.
Keywords: Bioengineering, Issue 102, Electrospinning, electrospraying, polycaprolactone, poly(lactide-co-glycolide), microfiber, nanofiber, microparticles, superhydrophobic, biomaterials, drug delivery, biodegradable, surface coatings.
Introduction
Superhydrophobic surfaces are generally categorized as exhibiting apparent water contact angles greater than 150° with low contact angle hysteresis. These surfaces are fabricated by introducing high surface roughness on low surface energy materials to establish a resulting air-liquid-solid interface that resists wetting1-6. Depending on the fabrication method, thin or multilayered superhydrophobic surfaces, multilayered superhydrophobic substrate coatings, or even bulk superhydrophobic structures can be prepared. This permanent or semi-permanent water repellency is a useful property that is employed to prepare self-cleaning surfaces7, microfluidic devices8, anti-fouling cell/protein surfaces9,10, drag-reducing surfaces11, and drug delivery devices12-15. Recently, stimuli-responsive superhydrophobic materials are described where the non-wetted to wetted state is triggered by chemical, physical, or environmental cues (e.g., light, pH, temperature, ultrasound, and applied electrical potential/current)14,16-20, and these materials are finding use for additional applications21-25.
The first synthetic superhydrophobic surfaces were prepared by treating material surfaces with methyldihalogenosilanes26, and were of limited value for biomedical applications, as the materials used were not suitable for in vivo use. Herein we describe the preparation of surface and bulk superhydrophobic materials from biocompatible polymers. Our approach entails electrospinning or electrospraying a polymer mixture containing a biodegradable, biocompatible aliphatic polyester as the major component, doped with a hydrophobic copolymer composed of the polyester and a stearate-modified poly(glycerol carbonate)27-30. The fabrication techniques provide the enhanced surface roughness and porosity on and within the fibers or the particles, respectively, while the use of a copolymer dopant provides a low surface energy polymer that blends with the polyester and can be stably electrospun or electrosprayed27,31,32.
Aliphatic biodegradable polyesters such as poly(lactic acid) (PLA), poly(glycolic acid) (PGA), poly(lactic acid-co-glycolic acid) (PLGA), and polycaprolactone (PCL) are polymers used in clinically-approved devices and prominent in biomedical materials research because of their non-toxicity, biodegradability, and ease of synthesis33. PGA and PLGA debuted in the clinic as bioresorbable sutures in the 1960’s and early 1970’s, respectively34-37. Since then, these poly(hydroxy acids) have been processed into a variety of other application-specific form factors, such as micro-38,39 and nanoparticles40,41, wafers/discs42, meshes27,43, foams44, and films45.
Aliphatic polyesters, as well as other polymers of biomedical interest, can be electrospun to produce nano- or microfiber mesh structures possessing a high surface area and porosity as well as tensile strength. Table 1 lists the synthetic polymers electrospun for various biomedical applications and their corresponding references. Electrospinning and electrospraying are rapid and commercially-scalable techniques. These two similar techniques rely on applying high voltage (electrostatic repulsion) to overcome surface tension of a polymer solution/melt in a syringe pump setup as it is directed toward a grounded target46,47. When this technique is used in conjunction with low surface energy polymers (hydrophobic polymers such as poly(caprolactone-co-glycerol monostearate)), the resulting materials exhibit superhydrophobicity.
To illustrate this general synthetic and materials processing approach to constructing superhydrophobic materials from biomedical polymers, we describe the synthesis of superhydrophobic polycaprolactone- and poly(lactide-co-glycolide)-based materials as representative examples. The respective copolymer dopants poly(caprolactone-co-glycerol monostearate) and poly(lactide-co-glycerol monostearate) are first synthesized, then blended with polycaprolactone and poly(lactide-co-glycolide), respectively, and finally electrospun or electrosprayed. The resulting materials are characterized by SEM imaging and contact angle goniometry, and tested for in vitro and in vivo biocompatibility. Finally, bulk wetting through three-dimensional superhydrophobic meshes is examined using contrast-enhanced microcomputed tomography.
Protocol
1. Synthesizing Functionalizable poly(1,3-glycerol carbonate-co-caprolactone)29 and poly(1,3-glycerol carbonate-co-lactide)27,28.
- Monomer synthesis.
- Dissolve cis-2-Phenyl-1,3-dioxan-5-ol (50 g, 0.28 mol, 1 eq.) in 500 ml dry tetrahydrofuran (THF) and stir on ice under nitrogen. Add potassium hydroxide (33.5 g, 0.84 mol, 3 eq.), finely crushed with a mortar and pestle. Place flask in ice bath.
- Add 49.6 ml benzyl bromide (71.32 g, 0.42 mol, 1.5 eq.) drop-wise with stirring on ice. Allow the reaction to warm to room temperature with stirring for 24 hr, under nitrogen.
- Add 150 ml distilled water to dissolve potassium hydroxide and remove the THF by rotary evaporation.
- Extract the remaining material with 200 ml dichloromethane (DCM) in a 1-L separatory funnel. Repeat the extraction twice.
- Dry the organic phase on sodium sulfate.
- Crystallize the product by adding 600 ml absolute ethanol to the solution, mixing well, and storing overnight at -20 °C. The product can be stored at -20 °C for several days before performing subsequent steps.
- Isolate product by vacuum-filtration through a Büchner funnel and dry on high-vacuum. The product can be stored for several days before performing subsequent steps. A typical yield for this step is ~80%.
- In a 1-L round-bottom flask, suspend the product obtained in step 1.1.7. in methanol (300 ml). Add 150 ml of 2 N hydrochloric acid. Reflux at 80 °C for 2 hr.
- Evaporate solvent and place under high-vacuum for 24 hr. The yield for this step is typically >98%.
- Dissolve product of 1.1.9 in THF (650 ml) and transfer to a 2-L round-bottom flask. Place flask on ice bath and stir under nitrogen. Add 22.4 ml ethyl chloroformate (25.6 g, 0.29 mol, 2 eq.) to flask under nitrogen.
- Add 32.8 ml triethylamine (0.29 mol, 2 eq.) to an addition funnel. Mix with an equal volume of THF. Place addition funnel on round-bottom flask and keep under nitrogen.
- With vigorous stirring, carefully dispense triethyamine/THF mixture drop-wise to the round-bottom flask on ice. CAUTION: this is an exothermic reaction. To prevent rapid temperature increase, add the triethylamine/THF solution no faster than 1 drop per second. After adding the full volume, stir the reaction for 4 hr, warming up to room temperature, or for 24 hr.
- Filter out the triethylamine hydrochloride salt using a Büchner funnel. Evaporate the solvent on a rotary evaporator.
- Add dichloromethane (200 ml) to the flask and heat gently until the residue is dissolved. Add 120 ml of diethyl ether while swirling. Store at -20 °C overnight to crystallize the product.
- Filter monomer crystals and re-crystallize before polymerizing. The monomer product can be stored sealed at room temperature for 2 weeks or at -20 °C indefinitely. Confirm product by 1H NMR, mass spectrometry, and elemental analysis. A typical yield for this final step in the monomer synthesis is between 40-60%.
- Copolymerization of D,L-lactide/ε-caprolactone with 5-benzyloxy-1,3-dioxan-2-one.
- Heat silicone oil bath to 140 °C.
- Measure 2.1 g of 5-benzyloxy-1,3-dioxan-2-one (prepared in 1.1) and add it to a dry 100-ml round-bottom flask. If copolymerizing D,L-lactide, measure out 5.7 g and add to the flask now. Add a magnetic stir bar and seal the flask with a rubber stopper.
- Also measure 240 mg (an excess) of tin(II) ethylhexanoate in a small pear-shaped flask. This polymerization will result in a 20 mol% glycerol carbonate monomer composition. Adjust the masses of monomers to achieve different monomer compositions.
- Flush both flasks with nitrogen on a Schlenk manifold for 5 min and add 4.24 ml ε-caprolactone under nitrogen. Evacuate flasks’ atmosphere by applying high-vacuum (300 mTorr) for 15 min to remove trace water.
- Recharge the flasks’ atmosphere with nitrogen; repeat this cycle twice more.
- Mix 500 µl dry toluene with the tin catalyst under nitrogen.
- Place the monomer flask in the 140 °C oil bath and add catalyst once all solids have melted. The total volume of catalyst mixture delivered should be ~100 µl. Keep at 140 °C for no more than 24 hr, then cool the molten polymer to room temperature. Perform the subsequent steps immediately or at least 24 hr later.
- Dissolve the polymer in dichloromethane (50 ml) and precipitate into cold methanol (200 ml). Decant supernatant and dry under high-vacuum. The subsequent steps can be performed immediately or at any point. Store polymers in the freezer until further use. The typical polymerization yield/conversion is between 80-95%.
- Perform 1H NMR analysis to determine the co-monomer molar ratios. Dissolve polymer in deuterated chloroform (CDCl3) and integrate the benzylic proton shift of the carbonate monomer at 4.58-4.68 ppm; compare this peak area with that of the methylene peak at 2.3 ppm (PCL) and methyne peak at 5.2 ppm (PLGA).
- Polymer modification: deprotection and grafting.
- Dissolve polymer (~7 g) in 120 ml tetrahydrofuran (THF) in a high-pressure hydrogenation vessel. Weigh and add palladium-carbon catalyst (~2 g).
- Add hydrogen to the vessel using a hydrogenation apparatus. Hydrogenate at 50 psi for 4 hr. CAUTION: hydrogen gas is extremely flammable. Seek assistance from persons familiar with this procedure and always inspect the supply lines for possible leaks before performing this experiment.
- Filter out palladium-carbon catalyst using a packed bed of diatomaceous earth. Concentrate the polymer to ~50 ml under rotary evaporation and precipitate into cold methanol. CAUTION: Dry palladium particulates can spontaneously ignite. Keep a wet towel nearby in the event of a flare-up for smothering the flames. Add water to the palladium/carbon filter cake to keep it clumped and to prevent its ignition. Seek assistance from persons familiar with this procedure.
- Decant the supernatant and dry under high-vacuum. Confirm total conversion to free hydroxyl by noting the peak disappearance at 4.65 ppm (1H NMR in CDCl3). These polymers can be used immediately or saved for later use. Yields for this step are >90%.
- Dissolve the polymer and stearic acid (1.5 eq.) in 500 ml dry dichloromethane (DCM). Add N,N’-dicyclohexylcarbodiimide (DCC, 2.0 eq.) and 3 flakes of 4-dimethylaminopyridine. Stir under nitrogen at room temperature for 24 hr.
- Remove insoluble N,N’-dicyclohexylcarbourea through a series of repeated filtrations and concentrations. At the end, concentrate the solution to 50 ml.
- Precipitate polymer into cold methanol (~175 ml) and decant the supernatant. Dry the polymer under high-vacuum overnight. Subsequent use of these polymers can be performed at any time, but keep polymers in the freezer for long-term storage. The yield for this final modification step is generally between 85-90%.
2. Characterizing the Synthesized Copolymers
Weigh out ~10 mg polymer (record the actual mass) and add to aluminum sample pan, then hermetically seal it. Load sample pan and an unloaded (reference) pan into the differential scanning calorimeter.
Program a temperature ramp and cooling (“heat/cool/heat”) cycle: 1) heat from 20 °C to 225 °C at 10 °C/min, 2) cool to -75 °C at 5 °C/min, 3) heat to 225 °C at 10 °C/min.
Determine melting point (Tm), crystallization (Tc) and glass transition temperatures (Tg), and heat of fusion (ΔHf) from the thermal traces (if applicable).
Dissolve each synthesized copolymer in THF (1 mg/ml) and filter through a 0.02-µm PTFE filter. Inject the solution into a gel permeation chromatography system and compare retention time versus a range of polystyrene standards.
3. Preparing Polymer Solutions for Electrospinning/electrospraying27,31
- Dissolve polymer(s) at 10-40 wt% in suitable solvent, such as chloroform/methanol (5:1) for PCL or tetrahydrofuran/N,N-dimethylformamide (7:3) for PLGA, overnight. The mass of polymer required for this step will depend on the dimensions of the desired mesh. Note: For example, to produce a 10 cm x 10 cm mesh of approximately 300-micron thickness, 1 gram will typically be required. It is worth noting that material losses may occur in subsequent steps of this protocol, such as during solution transfer to the syringe (especially for viscous solutions), and from dead volumes present in the optional connector tubing and the needle housing itself, which will reduce the yield of the electrospinning process. These reductions in yield may result in up to 20% loss of material, and it is recommended to scale up 1.5-fold to anticipate these losses, and also those losses associated with optimizing the electrospinning parameters when attempting this procedure for the first time.
- Control fiber size by varying the total polymer concentration, with larger fibers expected from more concentrated solutions. For a modest enhancement of hydrophobicity, use 10% (by total polymer mass) superhydrophobic dopant. For extremely hydrophobic/superhydrophobic materials, use 30-50% dopant and/or reduce the total polymer concentration (i.e., reduce fiber size). Subsequent work with these solutions may be performed the next day or within one week thereafter.
- For electrospraying, prepare solutions at lower concentrations (i.e., 2-10%) in a suitable solvent such as chloroform. Like electrospinning, modulate particle size by varying the polymer concentration.
Vortex polymer solution to thoroughly mix. Allow large air bubbles to subside (5 min).
Load solution into a glass syringe. Depending on solution viscosity, it may be easiest to remove the plunger and pour the solution directly into the syringe. A piece of inert, flexible tubing may aid maneuverability within the electrospinning setup. Invert the syringe to displace air through the hose/needle assembly.
4. Electrospinning/electrospraying Polymer Solutions
Load syringe onto syringe pump, set total volume (e.g., 4.5 ml) and the rate (e.g., 5 ml/hr) at which to dispense this solution.
Cover the collector plate with aluminum foil to ease subsequent removal and transport. Secure the foil with masking tape along the outer edges.
- Attach the high voltage DC (HVDC) supply wire to needle tip. The distance of this needle tip to the collector is an important variable to consider because it 1) affects the electric field at a given voltage, and 2) impacts the evaporation of solvent and consequent drying of fibers during their collection.
- As a first attempt, use a tip-to-collector distance of 15 cm. CAUTION: High voltages and flammable solvents are involved in electrospinning/electrospraying. Provide adequate ventilation to outside exhaust, and never touch the syringe/needle or open the enclosure until absolutely certain the HVDC supply is off.
If electrospinning/electrospraying a large area of coverage, turn on rotating and translating collector drum. Otherwise, proceed to the next step.
Start the syringe pump.
Turn on and adjust the high voltage source to achieve an acceptable Taylor Cone. If the solution at the needle tip is sagging, increase the voltage. If multiple jets are forming, reduce the voltage. In addition to these adjustments, it may be necessary to adjust the tip-to-collector distance if the fibers/particles appear wet or if adjusting the voltage does not adequately solve a dragging droplet at the needle tip. Note: For detailed troubleshooting, see the comprehensive electrospinning optimization process by Leach and co-workers47. Electrospraying will generally involve higher voltages and lower solution concentrations than electrospinning.
Turn off the high voltage source and then the syringe pump and motorized drum (if applicable). Allow the electrospinning enclosure to continue ventilating for 30 min.
Remove meshes/coatings from collector. Allow trace solvents to evaporate in a hood overnight. Materials can be stored at room temperature for at least two weeks (PLGA) or two months (PCL). Steps 4.5-4.8 may be performed in any order.
5. Characterizing Fiber and Particle Size by Light and Scanning Electron Microscopy
- Light microscopy
- If producing an electrospun mesh, cut and mount thin portions of it on a glass slide.
- Observe fiber diameter, node characteristics (blobs or discrete), and fiber shape (i.e., beaded, flat, straight/wavy). Ideal electrospun mesh fibers are uniform, straight or wavy, and bead-free.
- Scanning electron microscopy (SEM)
- Cut and mount meshes or coated surfaces on aluminum SEM stubs using conductive copper tape. Electrospun fibers and electrosprayed coatings can also be observed by SEM by directly depositing fibers/particles onto the tape in advance.
- Coat the meshes/coatings with a thin (~4 nm) layer of Au/Pd through sputter coating.
- Load stubs into SEM chamber and observe at 1-2 keV. A 250X magnification provides a general topographical assessment of the material, while higher magnifications reveal additional fiber and particle features such as hierarchal patterns for extremely superhydrophobic fibers and interconnectivity for particle coatings.
6. Determining Non-wetting Properties
- Advancing and receding water contact angle measurements using the volume variation method
- Cut thin (0.5 cm x 5 cm) strips of mesh or coated material (if possible) and place on the stage of a contact angle goniometer.
- Capture the water drop profile while dispensing it (from a 24 AWG syringe needle) on the material surface.
- To do this, start with an approximate 5-µl drop, and make contact with the material surface. Continue to slowly add volume (20-25 µl) and capture the droplet image, which represents the advancing water contact angle. The needle tip should be small compared to the droplet, and the capillary length should be greater than the droplet to minimize distortion of droplet shape.
- Withdraw this same drop while simultaneously capturing its drop profile. Repeat on discrete surface locations of several samples to report an average value—typically, 10 measurements of both advancing and receding contact angles are sufficient to characterize these materials.
- Determine critical surface tension of materials by modifying probing liquids.
- Prepare solutions varying in ethanol, propylene glycol, or ethylene glycol content, as these mixtures have known surface tensions99-101.
- Alternatively, use solvents with varying surface tensions—for example, water (72 mN/m), glycerol (64 mN/m), dimethyl sulfoxide (44 mN/m), benzyl alcohol (39 mN/m), 1,4-dioxane (33 mN/m), 1-octanol (28 mN/m), and acetone (25 mN/m). It is important to use solvents that will not dissolve the polymers, as these will confound results. Additionally, it is important to note that, in addition to surface tension, these liquids have different viscosities, which may impact contact angle measurements and is a limitation of this technique.
- Measure the contact angle of these solutions probed on the material surface. Plot contact angle as a function of surface tension.
7. Detecting Bulk Wetting of Meshes31
- Observe water infiltration into 3D meshes using micro-computed tomography (µCT).
- Prepare an 80 mg/ml solution of Ioxaglate (an iodinated contrast agent) in water.
- Submerge meshes in these solutions and incubate at 37 °C; periodically measure contrast agent (water) infiltration by µCT (18 µm3 voxel resolution) using a 70 kVP tube voltage, 114 µA current, and a 300 msec integration time.
- Using image processing software, measure pixel intensity throughout the thickness of the mesh, where bright pixels represent water infiltration. Select a pixel threshold value (~1500) for which higher intensity represents water infiltration.
8. Testing the Mechanical Properties of Meshes
Cut meshes to 1 cm x 7 cm and place between the grips of a tensile testing apparatus. Measure the exact width, length, and thickness.
Perform a ramp test of extension on three samples. Plot a stress-strain curve using these data to determine the elastic modulus, ultimate tensile strength, and elongation-at-break.
Representative Results
Through a series of chemical transformations, the functional carbonate monomer 5-benzyloxy-1,3-dioxan-2-one is synthesized as a white crystalline solid (Figure 1A). 1H NMR confirms the structure (Figure 1B) and mass spectrometry and elemental analysis confirm the composition. This solid is then copolymerized with either D,L-lactide or ε-caprolactone using a tin-catalyzed ring opening reaction at 140 °C. After purification by precipitation, the polymer composition is determined using 1H NMR analysis by integrating the benzylic proton chemical shift at 4.58-4.68 ppm and the characteristic methylene peak of caprolactone or methyne peak of lactide (2.3 or 5.2 ppm, respectively). Selective removal of the benzyl protecting group is achieved by Pd/C-catalyzed hydrogenolysis. Complete deprotection is confirmed by noting the disappearance of the benzyl peak in the 1H NMR spectra. Subsequent grafting of stearic acid onto the free hydroxyl group renders the final copolymers hydrophobic. These copolymers are white solids at room temperature (Figure 1C), and they are capable of being processed into films, electrospun meshes, and electrosprayed coatings (Figure 1D).
The copolymer composition (i.e., lactide/caprolactone to glycerol carbonate) is tuned by varying the corresponding monomer feed ratios. Varying the composition provides a means to synthesize copolymers with a range of thermal and/or mechanical properties. For example, thermal analysis using differential scanning calorimetry (DSC) reveals that PLA-PGC18 polymers containing 10, 20, 30, or 40 mol% PGC18 monomer gradually become more crystalline with increased PGC mol%. The thermal properties of PCL-PGC18 and PLA-PGC18 copolymers are summarized in Table 2.
The poly(glycerol-monostearate)-based copolymers have lower surface energy than their corresponding PCL or PLGA counterparts, as determined using contact angle measurements on smooth casted films (Figure 2A). While PCL possesses an advancing water contact angle of 84°, the advancing contact angle for PCL-PGC18 (80:20) is ~120°. Likewise, PLGA possesses an advancing contact angle of 71°, while PLA-PGC18 (90:10) and PLA-PGC18 (60:40) exhibit advancing contact angles of 99° and 105°, respectively. Blending PCL or PLGA with their corresponding copolymer dopants results in advancing contact angle values between those obtained for pure polymers and copolymers, and affords a facile means to tune hydrophobicity (Figure 2B). In this case, both copolymer dopant concentration (i.e., 10% or 30% wt/wt) and copolymer composition (i.e., PLA-PGC18 (90:10) or PLA-PGC18 (60:40) species) affect hydrophobicity, with greater PGC18 content yielding higher contact angles.
Doping the synthesized copolymers into a solution of PCL or PLGA and subsequently electrospinning the blends achieves fibrous meshes with tunable hydrophobicity. Figure 3A illustrates how doping in 30% PCL-PGC18 or PLA-PGC18 transitions meshes from hydrophobic to superhydrophobic. Superhydrophobicity is defined as an apparent water contact angle ≥ 150° with a low contact angle hysteresis—defined as the difference between advancing and receding water contact angle measurements. The increased surface roughness of electrospun meshes also increases the apparent water contact angle of these materials in comparison to smooth films. Wettability is tuned by varying the concentration of copolymer dopant. For example, electrospun pure PCL meshes with ~7 µm diameter fibers possess an apparent contact angle of 123°, while meshes doped with 10, 30, and 50% (wt/wt) PCL-PGC18 exhibit apparent contact angles of 143°, 150°, and 160° at comparable fiber diameters, respectively (Figure 3B). Wettability is also controlled by the choice of copolymer dopant species. In this case, 6.5-7.5-µm fiber PLGA meshes doped with 30% PLA-PGC18 (90:10) or 30% PLA-PGC18 (60:40) exhibit apparent contact angles of 133° or 154°, respectively (Figure 3C). Altering (i.e., reducing) the fiber size also enhances hydrophobicity independent of dopant selection and/or concentration. This dependence of apparent contact angle on fiber diameter is shown for both PCL and PLGA in Figure 3D. Similar to electrospinning, electrosprayed PCL and doped-PCL coatings also display contact angles that increase with doping percentage, and even higher contact angles than those obtained by electrospinning are achieved with this technique (Figure 3E). By probing the mesh surface with different liquids (which possess different surface tensions) and reporting the contact angle, a critical surface tension value at which the mesh rapidly wets is determined. Figure 3F is a modified Zisman curve illustrating the critical surface tension studies for PLGA meshes doped with 30% PLA-PGC18 (60:40) and PCL meshes doped with 30% PCL-PGC18.
SEM imaging reveals that the meshes are the result of entangled microfibers. This technique is also useful for determining fiber or particle size, homogeneity, and interconnectivity. Figure 4A shows PCL + 30% PCL-PGC18 meshes with fiber diameters of 1-2 µm and 4-5 µm, while Figure 4B shows PLGA + 10% PLA-PGC18 meshes varying in fiber size from ~3 µm to ~7 µm. Electrosprayed coatings of PCL and PCL + 50% PCL-PGC18 are presented in Figure 4C, while electrosprayed coatings of PCL + 30% PCL-PGC18 of varying particle size are presented in Figure 4D.
Superhydrophobic PCL- and PLGA-based meshes are non-cytotoxic to NIH/3T3 fibroblasts (Figure 5A) and are well-tolerated in C57BL/6 mice, with modest fibrous encapsulation. Compared to non-porous films (not shown), meshes display a greater degree of cellular infiltration (i.e., macrophages) after 4 weeks’ implantation (Figure 5B-E)27. While the cytocompatibility/biocompatibility of superhydrophobic meshes is similar to non-superhydrophobic meshes, the in vitro performance of superhydrophobic meshes can be superior in drug delivery applications. Due to their slow wetting, superhydrophobic meshes are capable of sustaining drug release for significantly longer durations than non-superhydrophobic meshes, since drug release cannot occur without water contact. The in vitro drug release efficacy studies demonstrating this principle are described elsewhere12,13.
The wetting of the electrospun meshes can be followed non-destructively over time using microcomputed tomography and the commercially-available iodinated contrast agent Ioxaglate. The mesh is placed in an aqueous solution containing the contrast agent and imaged over time. As shown in Figure 6A the pure PCL mesh rapidly wets as water infiltrates the bulk material in the first day. In contrast, the meshes doped with 30% PCL-PGC18 remain non-wetted for >75 days, with air remaining within the bulk structure (Figure 6B). These results illustrate the importance of superhydrophobic bulk materials for non-wetting applications.
Lastly, the mechanical properties of electrospun meshes are determined from tensile testing. Table 3 shows representative mechanical data for PCL, PLGA, and their respective doped meshes (fiber size = 7 µm for all meshes) obtained from their stress-strain curves. As the percentage of doping increases, the elastic moduli (E) and ultimate tensile strengths of meshes tend to decrease.
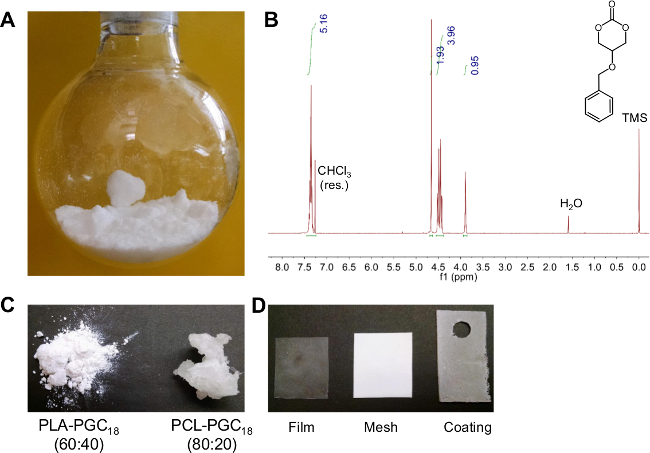 Figure 1. Monomer/polymer synthesis, characterization, and subsequent processing into films, electrospun meshes, and electrosprayed coatings. (A) Purified monomer is a white crystalline solid at room temperature; (B) corresponding 1H NMR spectra for monomer; (C) photograph of purified polymers PLA-PGC18 (left) and PCL-PGC18 (right); (D) photograph of PCL doped with 30% (wt/wt) PCL-PGC18 and processed into a (from left to right): film, electrospun mesh, and electrosprayed coating.
Figure 1. Monomer/polymer synthesis, characterization, and subsequent processing into films, electrospun meshes, and electrosprayed coatings. (A) Purified monomer is a white crystalline solid at room temperature; (B) corresponding 1H NMR spectra for monomer; (C) photograph of purified polymers PLA-PGC18 (left) and PCL-PGC18 (right); (D) photograph of PCL doped with 30% (wt/wt) PCL-PGC18 and processed into a (from left to right): film, electrospun mesh, and electrosprayed coating.
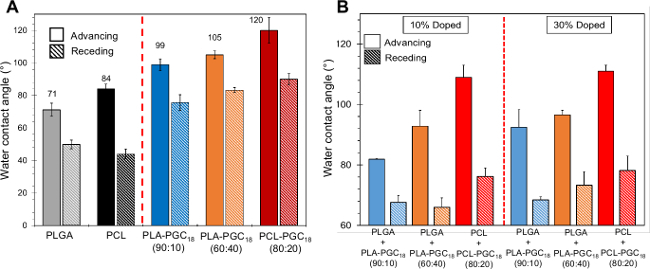 Figure 2. Advancing and receding water contact angles on polymer/copolymer films. (A) Advancing and receding water contact angle measurements for undoped PCL and PLGA smooth films compared to those for pure PCL-PGC18 and pure PLA-PGC18 smooth films; (B) advancing and receding contact angle measurements for doped PCL and PLGA films. Please click here to view a larger version of this figure.
Figure 2. Advancing and receding water contact angles on polymer/copolymer films. (A) Advancing and receding water contact angle measurements for undoped PCL and PLGA smooth films compared to those for pure PCL-PGC18 and pure PLA-PGC18 smooth films; (B) advancing and receding contact angle measurements for doped PCL and PLGA films. Please click here to view a larger version of this figure.
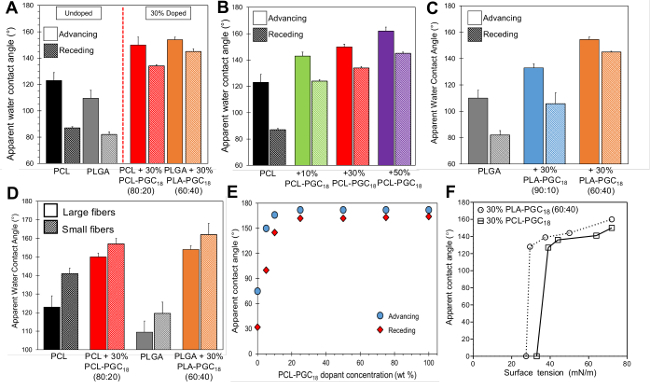 Figure 3. The processes of electrospinning and electrospraying generate rough surfaces that further enhance the hydrophobicity of PCL and PLGA. (A) Contact angle for electrospun PCL and PCL meshes doped with 30% PCL-PGC18 (80:20) meshes (fiber diameter ≈ 2.5 µm); PLGA meshes and PLGA meshes doped with 30% PLA-PGC18 (60:40) meshes (fiber diameter ≈ 6.5 µm), with both systems showing a transition from hydrophobic to superhydrophobic; (B) contact angles for PCL meshes as a function of increasing dopant copolymer concentration; (C) contact angles for PLGA meshes of ~6.5 µm diameter as a function of copolymer composition; (D) wettability as a function of fiber diameter for PCL (600 nm and 2.5 µm) and PLGA-based meshes (2.5 and 6.5 µm); (E) contact angles for electrosprayed PCL-based coatings as a function of copolymer doping concentration; (F) modified Zisman curves showing critical surface tension studies for PLGA meshes doped with 30% PLA-PGC18 (60:40) (circles with dashed connecting line) and PCL meshes doped with 30% PCL-PGC18 (squares with solid connecting line). Please click here to view a larger version of this figure.
Figure 3. The processes of electrospinning and electrospraying generate rough surfaces that further enhance the hydrophobicity of PCL and PLGA. (A) Contact angle for electrospun PCL and PCL meshes doped with 30% PCL-PGC18 (80:20) meshes (fiber diameter ≈ 2.5 µm); PLGA meshes and PLGA meshes doped with 30% PLA-PGC18 (60:40) meshes (fiber diameter ≈ 6.5 µm), with both systems showing a transition from hydrophobic to superhydrophobic; (B) contact angles for PCL meshes as a function of increasing dopant copolymer concentration; (C) contact angles for PLGA meshes of ~6.5 µm diameter as a function of copolymer composition; (D) wettability as a function of fiber diameter for PCL (600 nm and 2.5 µm) and PLGA-based meshes (2.5 and 6.5 µm); (E) contact angles for electrosprayed PCL-based coatings as a function of copolymer doping concentration; (F) modified Zisman curves showing critical surface tension studies for PLGA meshes doped with 30% PLA-PGC18 (60:40) (circles with dashed connecting line) and PCL meshes doped with 30% PCL-PGC18 (squares with solid connecting line). Please click here to view a larger version of this figure.
 Figure 4. SEM imaging of electrospun meshes and electrosprayed coatings reveals fiber/particle size and morphology. (A) Small-diameter PCL + 30% PCL-PGC18 fibers (1-2 µm) and corresponding large-diameter microfiber (4-5 µm) mesh (left and right, respectively), scale bar = 10 µm; (B) small-diameter PLGA + 10% PLA-PGC18 (90:10) (2.5-3.5 µm) microfiber and large diameter (6.5-7.5 µm) microfiber meshes (left and right, respectively; scale bar = 10 µm); (C) electrosprayed particles consisting of pure PCL (left), PCL + 50% PCL-PGC18 (right), scale bar = 20 µm; (D) electrosprayed PCL + 30% PCL-PGC18 particles of small (left) and large (right) radii (scale bar = 2 µm). Please click here to view a larger version of this figure.
Figure 4. SEM imaging of electrospun meshes and electrosprayed coatings reveals fiber/particle size and morphology. (A) Small-diameter PCL + 30% PCL-PGC18 fibers (1-2 µm) and corresponding large-diameter microfiber (4-5 µm) mesh (left and right, respectively), scale bar = 10 µm; (B) small-diameter PLGA + 10% PLA-PGC18 (90:10) (2.5-3.5 µm) microfiber and large diameter (6.5-7.5 µm) microfiber meshes (left and right, respectively; scale bar = 10 µm); (C) electrosprayed particles consisting of pure PCL (left), PCL + 50% PCL-PGC18 (right), scale bar = 20 µm; (D) electrosprayed PCL + 30% PCL-PGC18 particles of small (left) and large (right) radii (scale bar = 2 µm). Please click here to view a larger version of this figure.
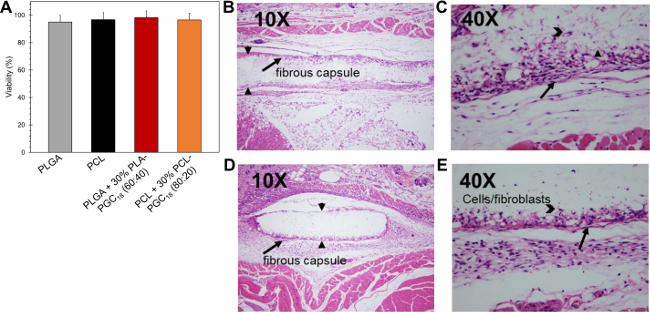 Figure 5. In vitro and in vivo cell viability/biocompatibility of electrospun superhydrophobic meshes. (A) In vitro cell assay of NIH/3T3 fibroblast viability upon 24-hr incubation with PCL, PLGA, and doped meshes; (B and C) histological (H&E) specimens of in vivo foreign body response to superhydrophobic PLGA + 30 wt% PLA-PGC18 (60:40) electrospun meshes after 4 weeks’ subcutaneous implantation in C57BL/6 mice at 10X (B) and 40X (C) magnification; (D and E) response to implanted pure PLGA electrospun meshes at 10X (D) and 40X (E) magnification. Please click here to view a larger version of this figure.
Figure 5. In vitro and in vivo cell viability/biocompatibility of electrospun superhydrophobic meshes. (A) In vitro cell assay of NIH/3T3 fibroblast viability upon 24-hr incubation with PCL, PLGA, and doped meshes; (B and C) histological (H&E) specimens of in vivo foreign body response to superhydrophobic PLGA + 30 wt% PLA-PGC18 (60:40) electrospun meshes after 4 weeks’ subcutaneous implantation in C57BL/6 mice at 10X (B) and 40X (C) magnification; (D and E) response to implanted pure PLGA electrospun meshes at 10X (D) and 40X (E) magnification. Please click here to view a larger version of this figure.
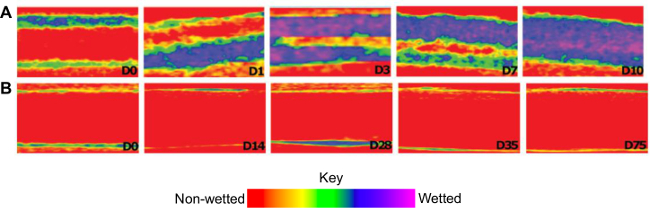 Figure 6. Contrast-enhanced microcomputed tomography (µCT) characterization of the bulk wetting of superhydrophobic meshes. The iodinated CT contrast agent Ioxaglate (80 mgI/ml) in water serves as a non-invasive marker of water infiltrating (A) non-superhydrophobic PCL meshes and (B) superhydrophobic PCL + 30% PCL-PGC18 meshes. Color map indicates non-wetted mesh as red and transitioning from yellow to green to blue/purple as wetting progresses. Please click here to view a larger version of this figure.
Figure 6. Contrast-enhanced microcomputed tomography (µCT) characterization of the bulk wetting of superhydrophobic meshes. The iodinated CT contrast agent Ioxaglate (80 mgI/ml) in water serves as a non-invasive marker of water infiltrating (A) non-superhydrophobic PCL meshes and (B) superhydrophobic PCL + 30% PCL-PGC18 meshes. Color map indicates non-wetted mesh as red and transitioning from yellow to green to blue/purple as wetting progresses. Please click here to view a larger version of this figure.
| Electrospun Synthetic Polymers: | Reference(s): |
| Poly(lactide-co-glycolide) | 27,36,43,48-52 |
| Polyglycolide | 52,53 |
| Poly(lactide-co-caprolactone) | 54-57 |
| Polycaprolactone | 13,58-66 |
| Polylactide | 52,67 |
| Poly(vinyl alcohol) | 68-71 |
| Poly(ethylene glycol)/block copolymers | 72,73 |
| Poly(ester urethane)s | 74-78 |
| Poly(trimethylene carbonate) | 79 |
| Poly(dimethyl siloxane) | 80,81 |
| Poly(ethylene-co-vinyl acetate) | 82 |
| Polyvinylpyrrolidone | 83 |
| Polyamide(s) | 84-86 |
| Polyhydroxybutryate | 87,88 |
| Polyphosphazene(s) | 89,90 |
| Poly(propylene carbonate) | 91-93 |
| Polyethyleneimine | 94,95 |
| Poly(γ-glutamic acid) | 96 |
| Silicate | 97,98 |
Table 1: Examples of synthetic biomedical polymers that have been electrospun for biomedical applications, with accompanying references.
| Copolymer | Conversion (%) | Lactidea | Glycerola | Mn (g/mol)b | Mw/Mn | Tg (°C)c | Tm (°C) | Tc (°C) | ∆Hf (J/g) |
| PLA-PGC18 (90:10) | 92 | 89 | 11 | 12,512 | 1.5 | 28 | - | - | - |
| PLA-PGC18 (80:20) | 96 | 78 | 23 | 10,979 | 1.5 | 17 | 33 | 11 | 3 |
| PLA-PGC18 (70:30) | 90 | 66 | 34 | 17,305 | 1.5 | * | 40 | 17 | 23 |
| PLA-PGC18 (60:40) | 86 | 54 | 47 | 13,226 | 1.6 | * | 43 | 27 | 32 |
| PCL-PGC18 (80:20) | 99 | (caprolactone) 81 | 19 | 21,100 | 1.7 | -53 | 31 | 19 | 55 |
Table 2: Characterization of synthesized copolymers. aMole %; bAs determined by size-exclusion chromatography (THF, 1.0 mL/min); Mn = number average molecular weight, Mw/Mn = dispersity. cTg = glass transition temperature; Tm= melting temperature; Tc = crystallization temperature; ΔHf = heat of fusion. d No Tg was observed for these semicrystalline polymers over the temperature range from -75 °C to 225 °C.
| Mesh Composition | Elastic Modulus (E) (MPa) | Ultimate Tensile Strength (MPa) |
| PCLa | 15.3 | 1.5 |
| + 10% PCL-PGC18 | 10.8 | 1.5 |
| + 30% PCL-PGC18 | 3.5 | 0.8 |
| PLGAb | 84.9 | 2.6 |
| + 10% PLA-PGC18 (60:40) | 40.3 | 0.8 |
| + 30% PLA-PGC18 (60:40) | 10.1 | 0.3 |
Table 3: Representative tensile properties of electrospun meshes. aFiber size for PCL and PCL-based meshes ≈ 7 µm. bFiber size for PLGA and PLGA-based meshes ≈ 7 µm.
Discussion
Our approach to constructing superhydrophobic materials from biomedical polymers combines synthetic polymer chemistry with the polymer processing techniques of electrospinning and electrospraying. These techniques provide either fibers or particles, respectively. Specifically, polycaprolactone and poly(lactide-co-glycolide) based superhydrophobic materials are prepared using this strategy. By varying the hydrophobic copolymer composition, percent copolymer in the final polymer blend, fiber/particle size, overall polymer weight percent, and fabrication conditions, the wettability of the resulting electrospun/electrosprayed materials is controlled. The materials fabricated in this work are from non-toxic and biocompatible polymers, and possess a meta-stable air barrier in the presence of water.
The critical steps in this protocol involve 1) synthesizing copolymers using ring-opening polymerization, 2) electrospinning or electrospraying these copolymers with a corresponding biomedical polymer such as PCL or PLGA; and 3) characterizing their morphology, non-wetting behavior/hydrophobicity, mechanical properties, and in vitro/in vivo biocompatibility. If difficulties with polymer synthesis, modification, and/or electrospinning are encountered, the following techniques will help identify and troubleshoot these issues.
It is important to ensure the purity of the monomers and that they do not contain trace water, such as that from the atmosphere. The presence of water may prevent or terminate polymerization, result in low molecular weight polymers, or yield polymers with extremely broad molecular weight distributions. Always evacuate the contents of polymerization vessels and re-fill with dry nitrogen or argon, and perform all additions (monomers and catalysts) under dry, inert atmosphere. If polymerization appears incomplete or unsuccessful, it may be necessary to dry the reagents by distillation, or re-crystallize the monomers to improve purity. If de-benzylation of the resulting copolymer appears unsuccessful (as observed by subsequent 1H NMR analysis), it may be necessary to add more catalyst or use a different catalyst reagent. We specifically note here that unsuccessful deprotection has been observed with certain Pd/C catalysts, and it is best to use the one listed in the Table of Materials.
Several technical difficulties may be encountered during the electrospinning and electrospraying process. If the solution at the needle tip is sagging, increase the voltage. If multiple jets are forming, reduce the voltage. In addition to these adjustments, it may be necessary to adjust the tip-to-collector distance if the fibers/particles appear wet (in this case, increase the collection distance), or if adjusting the voltage does not adequately solve a dragging droplet at the needle tip, reduce the collection distance. If fibers are not forming, it may be necessary to increase the viscosity of the solution by increasing the polymer concentration; the same is true if the fibers appear to have a bead-on-string morphology. If the difficulties remain, it may be necessary to switch to a different electrospinning solvent. For more troubleshooting, Leach and coworkers47 provide a comprehensive troubleshooting guide to electrospinning.
While electrospinning and electrospraying are useful techniques for fabricating biomedical materials, they do have limitations. First, these techniques rely on a grounded target to collect fibers or particles, so electrical conductivity is an important parameter to consider. It may be difficult to electrospin or electrospray materials that are particularly good electrical insulators, since the polymer jet may be more attracted to areas surrounding these substrates. One possible solution involves securing less-conductive materials to conductive copper tape. Additionally, while we have been successful in electrospinning meshes up to 1 mm thick, the fabrication of extremely thick meshes may be hindered due to the insulating nature of the polymer coating on the collector. At this point, meshes may increase in surface area without much increase in their overall thickness. Second, depending on the size of mesh desired, a substantial amount of material is required to achieve sufficient solution viscosity (which is required for electrospinning, as chain entanglements are necessary for fiber formation). Therefore, electrospinning may not be a suitable option for precious materials; electrospraying generally uses lower concentrations and thus is less demanding in terms of the required quantity of material. If the sample quantity is very limited, it may be possible to reduce material loss by omitting connector tubing (which otherwise adds to overall dead volume). Lastly, the determination of critical surface tension relies on the use of various probing liquids, which also possess different viscosities. As such, this method has a potential limitation in that viscosity is also a contributing factor to these results.
Superhydrophobic materials are an exciting class of biomaterials, which are finding increased use for a range of applications in drug delivery, tissue engineering, wound healing, and anti-fouling. Several techniques exist for enhancing surface roughness to materials for biomimetic and non-wetting applications, such as layer-by-layer assembly15, micropatterning/microtexturing102, electrospinning1,5,13, and electrospraying32. Of these approaches, electrospinning and electrospraying are particularly attractive methods due to their scalability and general compatibility with underlying substrates. In conclusion, this strategy combining polymer chemistry and process engineering is a versatile and general one that will enable other researchers to prepare, characterize, and study new biomaterials where wettability of the materials is a key design feature.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
Funding was provided in part by BU and the NIH R01CA149561. The authors wish to thank the electrospinning/electrospraying team including Stefan Yohe, Eric Falde, Joseph Hersey, and Julia Wang for their helpful discussions and contributions to the preparation and characterization of superhydrophobic biomaterials.
References
- Li XM, Reinhoudt D, Crego-Calama M. What do we need for a superhydrophobic surface? A review on the recent progress in the preparation of superhydrophobic surfaces. Chem. Soc. Rev. 2007;36:1350–1368. doi: 10.1039/b602486f. [DOI] [PubMed] [Google Scholar]
- Crick CR, Parkin IP. Preparation and characterisation of super-hydrophobic surfaces. Chem. - Eur. J. 2010;16:3568–3588. doi: 10.1002/chem.200903335. [DOI] [PubMed] [Google Scholar]
- Genzer J, Efimenko K. Recent developments in superhydrophobic surfaces and their relevance to marine fouling: a review. Biofouling. 2006;22:339–360. doi: 10.1080/08927010600980223. [DOI] [PubMed] [Google Scholar]
- Marmur A. Super-hydrophobicity fundamentals: implications to biofouling prevention. Biofouling. 2006;22:107–115. doi: 10.1080/08927010600562328. [DOI] [PubMed] [Google Scholar]
- Sas I, Gorga RE, Joines JA, Thoney KA. Literature review on superhydrophobic self-cleaning surfaces produced by electrospinning. J. Polym. Sci., Part B: Polym. Phys. 2012;50:824–845. [Google Scholar]
- Zhang X, Shi F, Niu J, Jiang Y, Wang Z. Superhydrophobic surfaces: from structural control to functional application. J. Mat. Chem. 2008;18:621–633. [Google Scholar]
- Xue C-H, Li Y-R, Zhang P, Ma J-Z, Jia S-T. Washable and wear-resistant superhydrophobic surfaces with self-cleaning property by chemical etching of fibers and hydrophobization. ACS Appl. Mater. Interfaces. 2014;6:10153–10161. doi: 10.1021/am501371b. [DOI] [PubMed] [Google Scholar]
- Ou J, Perot B, Rothstein JP. Laminar drag reduction in microchannels using ultrahydrophobic surfaces. Phys. Fluids. 2004;16:4635–4643. [Google Scholar]
- Ko T-J, et al. Adhesion behavior of mouse liver cancer cells on nanostructured superhydrophobic and superhydrophilic surfaces. Soft Matter. 2013.
- Lourenco BN, et al. Wettability influences cell behavior on superhydrophobic surfaces with different topographies. Biointerphases. 2012;7 doi: 10.1007/s13758-012-0046-6. [DOI] [PubMed] [Google Scholar]
- Srinivasan S, et al. Drag reduction for viscous laminar flow on spray-coated non-wetting surfaces. Soft Matter. 2013;9:5691–5702. [Google Scholar]
- Yohe ST, Colson YL, Grinstaff MW. Superhydrophobic materials for tunable drug release: using displacement of air to control delivery rates. J. Am. Chem. Soc. 2012;134:2016–2019. doi: 10.1021/ja211148a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yohe ST, Herrera VLM, Colson YL, Grinstaff MW. 3D superhydrophobic electrospun meshes as reinforcement materials for sustained local drug delivery against colorectal cancer cells. J. Control. Release. 2012;162:92–101. doi: 10.1016/j.jconrel.2012.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yohe ST, Kopechek JA, Porter TM, Colson YL, Grinstaff MW. Triggered drug release from superhydrophobic meshes using high-intensity focused ultrasound. Adv. Healthcare Mater. 2013;2:1204–1208. doi: 10.1002/adhm.201200381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manna U, Kratochvil MJ, Lynn DM. Superhydrophobic polymer multilayers that promote the extended, long-term release of embedded water-soluble agents. Adv. Mater. 2013;25:6405–6409. doi: 10.1002/adma.201302561. [DOI] [PubMed] [Google Scholar]
- Ju G, Cheng M, Shi F. A pH-responsive smart surface for the continuous separation of oil/water/oil ternary mixtures. NPG Asia Mater. 2014;6:e111. [Google Scholar]
- Lim HS, Han JT, Kwak D, Jin M, Cho K. Photoreversibly switchable superhydrophobic surface with erasable and rewritable pattern. J. Am. Chem. Soc. 2006;128:14458–14459. doi: 10.1021/ja0655901. [DOI] [PubMed] [Google Scholar]
- Macias-Montero M, Borras A, Alvarez R, Gonzalez-Elipe AR. Following the wetting of one-dimensional photoactive surfaces. Langmuir. 2012;28:15047–15055. doi: 10.1021/la3028918. [DOI] [PubMed] [Google Scholar]
- Sun T, et al. Reversible switching between superhydrophilicity and superhydrophobicity. Angew. Chem. Int. Ed. 2004;43:357–360. doi: 10.1002/anie.200352565. [DOI] [PubMed] [Google Scholar]
- Verplanck N, Coffinier Y, Thomy V, Boukherroub R. Wettability switching techniques on superhydrophobic surfaces. Nanoscale Res. Lett. 2007;2:577–596. [Google Scholar]
- Deng D, et al. Hydrophobic meshes for oil spill recovery devices. ACS Appl. Mater. Interfaces. 2013;5:774–781. doi: 10.1021/am302338x. [DOI] [PubMed] [Google Scholar]
- Ebrahimi A, et al. Nanotextured superhydrophobic electrodes enable detection of attomolar-scale DNA concentration within a droplet by non-faradaic impedance spectroscopy. Lab Chip. 2013;13:4248–4256. doi: 10.1039/c3lc50517k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guix M, et al. Superhydrophobic alkanethiol-coated microsubmarines for effective removal of oil. ACS Nano. 2012;6:4445–4451. doi: 10.1021/nn301175b. [DOI] [PubMed] [Google Scholar]
- Korhonen JT, Kettunen M, Ras RHA, Ikkala O. Hydrophobic nanocellulose aerogels as floating, sustainable, reusable, and recyclable oil absorbents. ACS Appl. Mater. Interfaces. 2011;3:1813–1816. doi: 10.1021/am200475b. [DOI] [PubMed] [Google Scholar]
- Wu Y, Hang T, Komadina J, Ling H, Li M. High-adhesive superhydrophobic 3D nanostructured silver films applied as sensitive, long-lived, reproducible and recyclable SERS substrates. Nanoscale. 2014;6:9720–9726. doi: 10.1039/c4nr02198c. [DOI] [PubMed] [Google Scholar]
- Norton FJ, inventor. Waterproofing treatment of materials. 2386259 A. US Patent. 1945
- Kaplan JA, et al. Imparting superhydrophobicity to biodegradable poly(lactide-co-glycolide) electrospun meshes. Biomacromolecules. 2014;15:2548–2554. doi: 10.1021/bm500410h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray WC, Grinstaff MW. Polycarbonate and poly(carbonate−ester)s synthesized from biocompatible building blocks of glycerol and lactic acid. Macromolecules. 2003;36:3557–3562. [Google Scholar]
- Wolinsky JB, Ray WC, Colson YL, Grinstaff MW. Poly(carbonate ester)s based on units of 6-hydroxyhexanoic acid and glycerol. Macromolecules. 2007;40:7065–7068. [Google Scholar]
- Wolinsky JB, Yohe ST, Colson YL, Grinstaff MW. Functionalized hydrophobic poly(glycerol-co-ε-caprolactone) depots for controlled drug release. Biomacromolecules. 2012;13 doi: 10.1021/bm201443m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yohe ST, Freedman JD, Falde EJ, Colson YL, Grinstaff MW. A mechanistic study of wetting superhydrophobic porous 3D meshes. Adv. Funct. Mater. 2013;23:3628–3637. doi: 10.1002/adfm.201203111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yohe ST, Grinstaff MW. A facile approach to robust superhydrophobic 3D coatings via connective-particle formation using the electrospraying process. Chem. Commun. 2013;49:804–806. doi: 10.1039/c2cc38012a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian HY, Tang ZH, Zhuang XL, Chen XS, Jing XB. Biodegradable synthetic polymers: Preparation, functionalization and biomedical application. Prog. Polym. Sci. 2012;37:237–280. [Google Scholar]
- Emil SE, Albert PR, inventors. Surgical sutures. 3297033 A. US Patent. 1967
- Greenberg JA, Clark RM. Advances in suture material for obstetric and gynecologic surgery. Rev. Obstet. Gynecol. 2009;2:146–158. [PMC free article] [PubMed] [Google Scholar]
- Weldon CB, et al. Electrospun drug-eluting sutures for local anesthesia. J. Control. Release. 2012;161:903–909. doi: 10.1016/j.jconrel.2012.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright J, Hoffman A. Chapter 2. Long Acting Injections and Implants. In: Wright JC, Burgess DJ, editors. Advances in Delivery Science and Technology. Springer; 2012. pp. 11–24. [Google Scholar]
- Wischke C, Schwendeman SP. Principles of encapsulating hydrophobic drugs in PLA/PLGA microparticles. Int. J. Pharm. 2008;364:298–327. doi: 10.1016/j.ijpharm.2008.04.042. [DOI] [PubMed] [Google Scholar]
- Xie JW, Tan RS, Wang CH. Biodegradable microparticles and fiber fabrics for sustained delivery of cisplatin to treat C6 glioma in vitro. J. Biomed. Mater. Res., Part A. 2008;85A:897–908. doi: 10.1002/jbm.a.31499. [DOI] [PubMed] [Google Scholar]
- Danhier F, et al. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release. 2012;161:505–522. doi: 10.1016/j.jconrel.2012.01.043. [DOI] [PubMed] [Google Scholar]
- Korin N, et al. Shear-activated nanotherapeutics for drug targeting to obstructed blood vessels. Science. 2012;337:738–742. doi: 10.1126/science.1217815. [DOI] [PubMed] [Google Scholar]
- Lee JS, et al. Evaluation of in vitro and in vivo antitumor activity of BCNU-Ioaded PLGA wafer against 9L gliosarcoma. Eur. J. Pharm. Biopharm. 2005;59:169–175. doi: 10.1016/j.ejpb.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Liu H, Wang SD, Qi N. Controllable structure, properties, and degradation of the electrospun PLGA/PLA-blended nanofibrous scaffolds. J. Appl. Polym. Sci. 2012;125:E468–E476. [Google Scholar]
- Ong BYS, et al. Paclitaxel delivery from PLGA foams for controlled release in post-surgical chemotherapy against glioblastoma multiforme. Biomaterials. 2009;30:3189–3196. doi: 10.1016/j.biomaterials.2009.02.030. [DOI] [PubMed] [Google Scholar]
- Paun IA, Moldovan A, Luculescu CR, Staicu A, Dinescu MMAPLE. deposition of PLGA:PEG films for controlled drug delivery: Influence of PEG molecular weight. Appl. Surf. Sci. 2012;258:9302–9308. [Google Scholar]
- Reneker DH, Yarin AL, Zussman E, Xu H. Aref H, Van der Giessen E, editors. Electrospinning of nanofibers from polymer solutions and melts. Advances in Applied Mechanics. 2007. pp. 43–195.
- Leach MK, Feng Z-Q, Tuck SJ, Corey JM. Electrospinning fundamentals: optimizing solution and apparatus parameters. J. Vis. Exp. 2011. [DOI] [PMC free article] [PubMed]
- Oh JH, Park KM, Lee JS, Moon HT, Park KD. Electrospun microfibrous PLGA meshes coated with in situ cross-linkable gelatin hydrogels for tissue regeneration. Curr. Appl. Phys. 2012;12:S144–S149. [Google Scholar]
- Kim TG, Park TG. Biomimicking extracellular matrix: cell adhesive RGD peptide modified electrospun poly(D,L-lactic-co-glycolic acid) nanofiber mesh. Tissue Eng. 2006;12:221–233. doi: 10.1089/ten.2006.12.221. [DOI] [PubMed] [Google Scholar]
- Stitzel J, et al. Controlled fabrication of a biological vascular substitute. Biomaterials. 2006;27:1088–1094. doi: 10.1016/j.biomaterials.2005.07.048. [DOI] [PubMed] [Google Scholar]
- Liang D, et al. In vitro non-viral gene delivery with nanofibrous scaffolds. Nucleic Acids Res. 2005;33:e170. doi: 10.1093/nar/gni171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Y, Min B-M, Lee SJ, Lee TS, Park WH. In vitro degradation behavior of electrospun polyglycolide, polylactide, and poly(lactide-co-glycolide) J. Appl. Polym. Sci. 2005;95:193–200. [Google Scholar]
- Boland ED, Wnek GE, Simpson DG, Pawlowski KJ, Bowlin GL. Tailoring tissue engineering scaffolds using electrostatic processing techniques: a study of poly(glycolic acid) electrospinning. J. Macromol. Sci., Part A: Pure Appl. Chem. 2001;38:1231–1243. [Google Scholar]
- Inoguchi H, Tanaka T, Maehara Y, Matsuda T. The effect of gradually graded shear stress on the morphological integrity of a huvec-seeded compliant small-diameter vascular graft. Biomaterials. 2007;28:486–495. doi: 10.1016/j.biomaterials.2006.09.020. [DOI] [PubMed] [Google Scholar]
- Xu CY, Inai R, Kotaki M, Ramakrishna S. Aligned biodegradable nanofibrous structure: a potential scaffold for blood vessel engineering. Biomaterials. 2004;25:877–886. doi: 10.1016/s0142-9612(03)00593-3. [DOI] [PubMed] [Google Scholar]
- Mun CH, et al. Three-dimensional electrospun poly(lactide-co-varepsilon-caprolactone) for small-diameter vascular grafts. Tissue Eng. Part A. 2012;18:1608–1616. doi: 10.1089/ten.TEA.2011.0695. [DOI] [PubMed] [Google Scholar]
- Inai R, Kotaki M, Ramakrishna S. Deformation behavior of electrospun poly(L-lactide-co-ε-caprolactone) nonwoven membranes under uniaxial tensile loading. J. Polym. Sci., Part B: Polym. Phys. 2005;43:3205–3212. [Google Scholar]
- Cao H, McHugh K, Chew SY, Anderson JM. The topographical effect of electrospun nanofibrous scaffolds on the in vivo and in vitro foreign body reaction. J. Biomed.Mater.Res.,PartA. 2010;93A:1151–1159. doi: 10.1002/jbm.a.32609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham QP, Sharma U, Mikos AG. Electrospun poly(epsilon-caprolactone) microfiber and multilayer nanofiber/microfiber scaffolds: characterization of scaffolds and measurement of cellular infiltration. Biomacromolecules. 2006;7:2796–2805. doi: 10.1021/bm060680j. [DOI] [PubMed] [Google Scholar]
- Jiang H, Zhao P, Zhu K. Fabrication and characterization of zein-based nanofibrous scaffolds by an electrospinning method. Macromol. Biosci. 2007;7:517–525. doi: 10.1002/mabi.200600277. [DOI] [PubMed] [Google Scholar]
- Zhang YZ, Venugopal J, Huang ZM, Lim CT, Ramakrishna S. Characterization of the surface biocompatibility of the electrospun PCL-collagen nanofibers using fibroblasts. Biomacromolecules. 2005;6:2583–2589. doi: 10.1021/bm050314k. [DOI] [PubMed] [Google Scholar]
- Jiang H, Hu Y, Zhao P, Li Y, Zhu K. Modulation of protein release from biodegradable core-shell structured fibers prepared by coaxial electrospinning. J. Biomed. Mater. Res., Part B: Appl. Biomat. 2006;79:50–57. doi: 10.1002/jbm.b.30510. [DOI] [PubMed] [Google Scholar]
- Jiang H, et al. A facile technique to prepare biodegradable coaxial electrospun nanofibers for controlled release of bioactive agents. J. Control. Release. 2005;108:237–243. doi: 10.1016/j.jconrel.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Zhang YZ, et al. Coaxial electrospinning of (fluorescein isothiocyanate-conjugated bovine serum albumin)-encapsulated poly(epsilon-caprolactone) nanofibers for sustained release. Biomacromolecules. 2006;7:1049–1057. doi: 10.1021/bm050743i. [DOI] [PubMed] [Google Scholar]
- Schnell E, et al. Guidance of glial cell migration and axonal growth on electrospun nanofibers of poly-epsilon-caprolactone and a collagen/poly-epsilon-caprolactone blend. Biomaterials. 2007;28:3012–3025. doi: 10.1016/j.biomaterials.2007.03.009. [DOI] [PubMed] [Google Scholar]
- Ma Z, He W, Yong T, Ramakrishna S. Grafting of gelatin on electrospun poly(caprolactone) nanofibers to improve endothelial cell spreading and proliferation and to control cell Orientation. Tissue Eng. 2005;11:1149–1158. doi: 10.1089/ten.2005.11.1149. [DOI] [PubMed] [Google Scholar]
- Peesan M, Rujiravanit R, Supaphol P. Electrospinning of hexanoyl chitosan/polylactide blends. J. Biomater. Sci., Polym. Ed. 2006;17:547–565. doi: 10.1163/156856206776986251. [DOI] [PubMed] [Google Scholar]
- Jia Y-T, et al. Fabrication and characterization of poly (vinyl alcohol)/chitosan blend nanofibers produced by electrospinning method. Carbohydr. Polym. 2007;67:403–409. [Google Scholar]
- Kenawy E-R, Abdel-Hay FI, El-Newehy MH, Wnek GE. Controlled release of ketoprofen from electrospun poly(vinyl alcohol) nanofibers. Mater. Sci. Eng., A. 2007;459:390–396. [Google Scholar]
- Zhang C, Yuan X, Wu L, Han Y, Sheng J. Study on morphology of electrospun poly(vinyl alcohol) mats. Eur. Polym. J. 2005;41:423–432. [Google Scholar]
- Hong KH. Preparation and properties of electrospun poly(vinyl alcohol)/silver fiber web as wound dressings. Polym. Eng. Sci. 2007;47:43–49. [Google Scholar]
- Bhattarai SR, et al. Novel biodegradable electrospun membrane: scaffold for tissue engineering. Biomaterials. 2004;25:2595–2602. doi: 10.1016/j.biomaterials.2003.09.043. [DOI] [PubMed] [Google Scholar]
- Grafahrend D, et al. Biofunctionalized poly(ethylene glycol)-block-poly(ε-caprolactone) nanofibers for tissue engineering. J. Mater. Sci.: Mater. Med. 2008;19:1479–1484. doi: 10.1007/s10856-007-3299-8. [DOI] [PubMed] [Google Scholar]
- Riboldi SA, Sampaolesi M, Neuenschwander P, Cossu G, Mantero S. Electrospun degradable polyesterurethane membranes: potential scaffolds for skeletal muscle tissue engineering. Biomaterials. 2005;26:4606–4615. doi: 10.1016/j.biomaterials.2004.11.035. [DOI] [PubMed] [Google Scholar]
- Gugerell A, et al. Electrospun poly(ester-urethane)- and poly(ester-urethane-urea) fleeces as promising tissue engineering scaffolds for adipose-derived stem cells. PLoS ONE. 2014;9:e90676. doi: 10.1371/journal.pone.0090676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair PA, Ramesh P. Electrospun biodegradable calcium containing poly(ester-urethane)urea: synthesis, fabrication, in vitro degradation, and biocompatibility evaluation. J. Biomed. Mater. Res., Part A. 2013;101:1876–1887. doi: 10.1002/jbm.a.34490. [DOI] [PubMed] [Google Scholar]
- Caracciolo P, Thomas V, Vohra Y, Buffa F, Abraham G. Electrospinning of novel biodegradable poly(ester urethane)s and poly(ester urethane urea)s for soft tissue-engineering applications. J. Mater. Sci.: Mater. Med. 2009;20:2129–2137. doi: 10.1007/s10856-009-3768-3. [DOI] [PubMed] [Google Scholar]
- Hong Y, et al. A small diameter, fibrous vascular conduit generated from a poly(ester urethane)urea and phospholipid polymer blend. Biomaterials. 2009;30:2457–2467. doi: 10.1016/j.biomaterials.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pego AP, et al. Preparation of degradable porous structures based on 1,3-trimethylene carbonate and D,L-lactide (co)polymers for heart tissue engineering. Tissue Eng. 2003;9:981–994. doi: 10.1089/107632703322495628. [DOI] [PubMed] [Google Scholar]
- Niu H, Wang H, Zhou H, Lin T. Ultrafine PDMS fibers: preparation from in situ curing-electrospinning and mechanical characterization. RSC Adv. 2014;4:11782–11787. [Google Scholar]
- Kim YB, Cho D, Park WH. Electrospinning of poly(dimethyl siloxane) by sol–gel method. J. Appl. Polym. Sci. 2009;114:3870–3874. [Google Scholar]
- Kenawy E-R, et al. Release of tetracycline hydrochloride from electrospun poly(ethylene-co-vinylacetate), poly(lactic acid), and a blend. J. Control. Release. 2002;81:57–64. doi: 10.1016/s0168-3659(02)00041-x. [DOI] [PubMed] [Google Scholar]
- Uykun N, et al. Electrospun antibacterial nanofibrous polyvinylpyrrolidone/cetyltrimethylammonium bromide membranes for biomedical applications. J. Bioact. Compat. Polym. 2014;29:382–397. [Google Scholar]
- Panthi G, et al. Preparation and characterization of nylon-6/gelatin composite nanofibers via electrospinning for biomedical applications. Fibers Polym. 2013;14:718–723. [Google Scholar]
- Pant HR, et al. Chitin butyrate coated electrospun nylon-6 fibers for biomedical applications. Appl. Surf. Sci., Part B. 2013;285:538–544. [Google Scholar]
- Pant HR, Kim CS. Electrospun gelatin/nylon-6 composite nanofibers for biomedical applications. Polym. Int. 2013;62:1008–1013. [Google Scholar]
- Correia DM, et al. Influence of electrospinning parameters on poly(hydroxybutyrate) electrospun membranes fiber size and distribution. Polym. Eng. Sci. 2014;54:1608–1617. [Google Scholar]
- Tong H-W, Wang M. Electrospinning of poly(hydroxybutyrate-co-hydroxyvalerate) fibrous tissue engineering scaffolds in two different electric fields. Polym. Eng. Sci. 2011;51:1325–1338. [Google Scholar]
- Carampin P, et al. Electrospun polyphosphazene nanofibers for in vitro rat endothelial cells proliferation. J. Biomed. Mater. Res., Part A. 2007;80:661–668. doi: 10.1002/jbm.a.30999. [DOI] [PubMed] [Google Scholar]
- Lin Y-J, et al. Effect of solvent on surface wettability of electrospun polyphosphazene nanofibers. J. Appl. Polym. Sci. 2010;115:3393–3400. [Google Scholar]
- Zhang J, et al. Engineering of vascular grafts with genetically modified bone marrow mesenchymal stem cells on poly (propylene carbonate) graft. Artif. Organs. 2006;30:898–905. doi: 10.1111/j.1525-1594.2006.00322.x. [DOI] [PubMed] [Google Scholar]
- Nagiah N, Sivagnanam UT, Mohan R, Srinivasan NT, Sehgal PK. Development and characterization of electropsun poly(propylene carbonate) ultrathin fibers as tissue engineering scaffolds. Adv. Eng. Mater. 2012;14:B138–B148. [Google Scholar]
- Welle A, et al. Electrospun aliphatic polycarbonates as tailored tissue scaffold materials. Biomaterials. 2007;28:2211–2219. doi: 10.1016/j.biomaterials.2007.01.024. [DOI] [PubMed] [Google Scholar]
- Khanam N, Mikoryak C, Draper RK, Balkus KJ. Electrospun linear polyethyleneimine scaffolds for cell growth. Acta Biomater. 2007;3:1050–1059. doi: 10.1016/j.actbio.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Xu X, Zhang J-F, Fan Y. Fabrication of cross-linked polyethyleneimine microfibers by reactive electrospinning with in situ photo-cross-linking by UV radiation. Biomacromolecules. 2010;11:2283–2289. doi: 10.1021/bm1003509. [DOI] [PubMed] [Google Scholar]
- Wang S, et al. Fabrication and morphology control of electrospun poly(Γ-glutamic acid) nanofibers for biomedical applications. Colloids Surf. B. 2012;89:254–264. doi: 10.1016/j.colsurfb.2011.09.029. [DOI] [PubMed] [Google Scholar]
- Sakai S, Yamada Y, Yamaguchi T, Kawakami K. Prospective use of electrospun ultra-fine silicate fibers for bone tissue engineering. Biotechnol. J. 2006;1:958–962. doi: 10.1002/biot.200600054. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Sakai S, Kawakami K. Application of silicate electrospun nanofibers for cell culture. J. Sol-Gel Sci. Technol. 2008;48:350–355. [Google Scholar]
- Vazquez G, Alvarez E, Navaza JM. Surface-tension of alcohol plus water from 20-degrees C to 50-degrees. C. J. Chem. Eng. Data. 1995;40:611–614. [Google Scholar]
- Hoke BC, Patton EF. Surface tensions of propylene glycol water. J. Chem. Eng. Data. 1992;37:331–333. [Google Scholar]
- Azizian S, Hemmati M. Surface tension of binary mixtures of ethanol + ethylene glycol from 20 to 50. C. J. Chem. Eng. Data. 2003;48:662–663. [Google Scholar]
- Nayak BK, Caffrey PO, Speck CR, Gupta MC. Superhydrophobic surfaces by replication of micro/nano-structures fabricated by ultrafast-laser-microtexturing. Appl. Surf. Sci. 2013;266:27–32. [Google Scholar]