

Abstract
During development, hematopoietic cells arise from a specialized subset of endothelial cells, hemogenic endothelium (HE). Modeling HE development in vitro is essential for mechanistic studies of the endothelial-hematopoietic transition and hematopoietic specification. Here, we describe a method for the efficient induction of HE from human pluripotent stem cells (hPSCs) by way of overexpression of different sets of transcription factors. The combination of ETV2 and GATA1 or GATA2 TFs is used to induce HE with pan-myeloid potential, while a combination of GATA2 and TAL1 transcription factors allows for the production of HE with erythroid and megakaryocytic potential. The addition of LMO2 to GATA2 and TAL1 combination substantially accelerates differentiation and increases erythroid and megakaryocytic cells production. This method provides an efficient and rapid means of HE induction from hPSCs and allows for the observation of the endothelial-hematopoietic transition in a culture dish. The protocol includes hPSCs transduction procedures and post-transduction analysis of HE and blood progenitors.
Keywords: Developmental Biology, Issue 106, Human embryonic stem cells (hESCs), human induced pluripotent stem cells (hiPSCs), hematopoietic progenitors, hemogenic endothelium, gain-of-function, GATA1, GATA2, ETV2, TAL1, LMO2
Introduction
The unique ability of human pluripotent stem cells (hPSCs) to self-renew and to differentiate into cells of the three germ layers, including blood, make them a valuable tool for the mechanistic studies of hematopoietic development, modeling of blood diseases, drug screening, toxicity studies, and the development of cellular therapies. Because blood formation in the embryo proceeds from hemogenic endothelium (HE) through an endothelial hematopoietic transition1,2, the generation of HE in cultures would be essential to study the molecular mechanisms regulating the endothelial to hematopoietic transition and hematopoietic specification. Current methods for studies of HE are based on the induction of hematoendothelial differentiation in aggregates (EBs) with the addition of hematopoietic cytokines3-5, and coculture of hPSCs with hematopoiesis-supportive stromal cells6,7 or in two-dimensional cultures with extracellular matrices and cytokines8,9. These classical differentiation methods are based on the introduction of external signals acting at the cell surface and initiating cascades of molecular pathways that eventually lead to the activation of transcriptional program guiding hematoendothelial development. Thus, the efficiency of hPSCs differentiations in these systems relies on an effective induction of those signals, signal transduction to the nucleus, and the resulting activation of specific transcriptional regulators. In addition, the study of HE in conventional differentiation cultures requires the additional step of isolating HE cells using cell sorting. Here, we describe a simple protocol for the direct induction of HE and blood by overexpression of hematopoietic transcription factors. This method allows for the efficient induction of HE in a dish and direct observation of the endothelial to hematopoietic transition without the need for isolation of HE using a cumbersome cell sorting procedure.
Formation of HE and blood from human pluripotent stem cells can be efficiently induced by overexpressing just a few transcription factors (TFs). The optimal combination of TFs capable of inducing robust pan-myeloid hematopoiesis from hPSCs includes ETV2 and GATA1 or GATA2. In contrast, combination of GATA2 and TAL1 induces erythromegakaryocytopoiesis10. Programming hPSCs through overexpression of these factors differentiates hPSCs directly to the VE-cad+CD43-CD73- HE cells that gradually acquire the hematopoietic phenotype defined by the expression of the early hematopoietic marker CD437. This lentiviral-based method for the direct programming of human pluripotent stem cells method is applicable for the generation of HE and blood cells for mechanistic studies, studies of endothelial to hematopoietic transition, and transcriptional regulation of hematopoietic development and specification. Although the current protocol describes blood production using constitutive expression of the transgenes, similar results could be obtained using modified mRNA10.
Protocol
1. Virus Preparations and Transcription Factor Combinations
Prepare solutions with pSIN-EF1α lentiviral expression plasmid containing protein-coding DNA for ETV2, GATA1, GATA2, TAL1 and LMO2 (Table 1).
Measure the concentration and purity of plasmid preparations used for lentiviral production by recording UV absorption with a spectrophotometer at 230 nm, 260 nm, and 280 nm. Note: DNA preparations demonstrating A260/280 and A260/230 values greater than 1.8 are generally considered good quality. Lower A260/280 values may indicate protein contamination, while lower A260/230 values indicate impurities with salts or some solvent such as phenol. Recommended plasmid concentration is 1-3 µg/µl.
Produce lentiviruses for transductions as described in previously published protocols11. Induction of HE with pan-myeloid potential requires co-expression of ETV2 and GATA1, or ETV2 and GATA2. Induction of HE with erythro- and megakaryocytic potential requires GATA2 and TAL1. The addition of LMO2 significantly improves the yield of erythro-megakaryocytic cells from hPSCs in the presence of GATA2 and TAL110.
2. hPSCs Culture Protocol
Grow pluripotent stem cells in tight colonies with sharp edges to 70-80% confluency before subculturing. Mark and remove spontaneously differentiated colonies before passaging cells.
Dilute commercial extracellular matrix gel (see Table 2) according to the manufacturer’s instructions and coat 6-well plates overnight at 4 °C, or for at least one hour at 37 °C prior to use. Before passaging cells, aspirate the matrix, add 2 ml of fresh commercial complete culturemedium (see Table 2) and keep plates at 37°C, 5% CO2 until the cells are ready for seeding.
- Passage hPSCs
- Aspirate medium from one confluent well of a 6-well plate with hPSCs and add 1.5 ml of pre-warmed Dispase II solution (2 mg/ml in DMEM/F12, sterilized by filtration through 0.22 µm filters). Incubate for 5-7 min at 37 °C, 5% CO2 until the edges of colonies begin to lift from the surface.
- Aspirate Dispase II solution and carefully wash the colonies twice by adding and then aspirating 2 ml of fresh DMEM/F12 medium. Then, using a 5 ml serological glass pipette, dissociate hPSCs with 3 ml of fresh commercial complete culture medium (see Table 2) by pipetting cell suspension up and down several times.
- Add 0.5 ml of cell suspension to each well of a 6-well plate containing 1.5 ml of commercial complete culture medium (see Table 2). Agitate plate back and forth and right to left several times to ensure a uniform cell distribution when situating the plate into the incubator. Keep cells at 37 °C, 5% CO2 for 18-24 hr.
- The next day change medium to fresh commercial complete culture medium (see Table 2) and examine hPSCs morphology. Successful passaging results in small, well-attached colonies retaining hPSC morphology. hPSCs must be fed with 2.5-3 ml freshmedium per well on a daily basis and passaged every 4-5 days at no more than 70-80% confluence. NOTE: If hiPSCs were maintained on mouse embryonic fibroblasts (MEFs), they need to be transferred to feeder-free conditions and passaged 2-3 times before transduction to ensure efficient differentiation.
3. Induction of Hematoendothelial Pprecursors from hPSCs (day 0, Transduction of hPSCs)
- To prepare reaction medium combine 1.3 ml of commercial complete serum-free medium (see Table 2), virus, polybrene, and Y27632 ROCK inhibitor (Table 1): add 1.3 µl of 10 mM ROCK inhibitor to 1.3 ml medium to a final concentration of 10 μM, and polybrene to a final concentration 6 µg/ml.
- Add an appropriate amount of viral concentrate(s) at a final concentration of 0.5-1.0 MOI (multiplicity of infection) of each virus per cell. Keep reaction medium on ice or at 4 °C until the single cell suspension is ready. NOTE: The same amount of MOI for each virus in the GATA1/ETV2, GATA2/TAL1, and GATA2/TAL1/LMO2 reaction mixtures is optimal for induction of differentiation. However, in ETV2/GATA2 transduced cultures doubling MOI for GATA2, relative to ETV2, enhances differentiation. Therefore, for these cultures, ratio 1:2 for MOI of ETV2 and 1 MOI of GATA2 is recommended (e.g. if viral concentrate is estimated approximately 6.8 x 107 particles/ml, add 10 µl of ETV2 and 20 µl of GATA2 viral concentrates to 0.68 x 106 cells per one ETV2/GATA2 reaction in a 35 mm well of a 6-well plate).
- Prepare hPSCs in a single cell suspension.
- Grow hPSCs in feeder-free conditions as described in section 2. On day 4 following the last passage, observe cell density and morphology under an inverted microscope. hPSCs should grow in tight colonies with sharp edges with no spontaneous differentiation and reach ~60-70% confluency on the day of transduction.
- Aspirate medium, add 1.5-2 ml of commercial cell dissociation reagent (Table 1) per well and incubate at 37 °C with 5% CO2 for 5-7 min.
- Collect cells into equal volumes of commercial complete medium (see Table 2) with 10 µM of ROCK inhibitor, count the cells and determine viability. Pellet cells by centrifugation at 200 x g for 5 min and then resuspend cells in commercial complete medium with 10 µM of ROCK inhibitor to a concentration of 3.4-5 x 106 cells per ml. NOTE: Cell viability is critical for successful viral transduction and subsequent differentiation. Typically, cell viability before transduction should exceed 90%.
Add 200 µl of the cell suspension, containing 0.68-1 x106 cells, into 1.3 ml of the reaction medium prepared in 3.1.
Aspirate the matrix solution from the overnight coated 6-well plate prepared in 2.2., transfer the reaction mixture to one well of a 6-well plate and distribute cells evenly. Incubate at 37 °C with 5% CO2 for 24 hr. After 24 hr at least 80% of the cells should be attached to the matrix.
4. Induction of Hematoendothelial Precursors from hPSCs (Day 1-7)
24 hr post-transduction, remove virus-containing medium and wash attached cells with commercial incomplete cell culture medium (see Table 1) and then add 3 ml per well of commercial incomplete cell culture medium containing hematopoietic cytokines: SCF 100 ng/ml, TPO 50 ng/ml, bFGF 20 ng/ml (hereafter referred to as 3F-medium).
Replace total medium with fresh 3F-medium on days 2, 3 and day 4 to remove dead cells and debris. After day 4, when floating hematopoietic cells are emerging, replace half of the 3F-medium every other day while maintaining the total volume at 4 ml. NOTE: pSIN-EF1α lentiviral expression plasmids incorporate puromycin resistance gene thereby allowing for positive selection of transduced cells. The 1 μg/ml of puromycin can be added to medium during the first 1-2 days of differentiation to eliminate the any residual undifferentiated hPSCs.
Observe cell morphology on day 4 under an inverted microscope: tight clusters of cells with typical endothelial morphology begin to appear (Figures 2A, 3A). Round blood cells appears from day 5 to day 7 of differentiation, while some areas may retain hPSCs morphology. Analyze HE and hematopoietic differentiation as described below. NOTE: Successful hematoendothelial differentiation results in the production of blood cells that appear as refractile round cells loosely attached to underlying flat endothelial cells. These cells actively proliferate forming small aggregates, and eventually detach and float (Figures 2A and 3A). Live blood cells can be visually distinguished from the dead and dying cells based on their ability to reflect the light and expand in culture conditions.
5. Analysis of Hemogenic Endothelium (HE) Stage of Differentiation.
- Detect the formation of HE between days 3 and 4 of differentiation by microscopic observation of cells with endothelial morphology. There are no floating blood cells in the culture at this stage of differentiation. The presence of HE can be confirmed by immunofluorescent staining and flow cytometric analysis using VE-cadherin, CD73, CD226 and CD43 antibodies.
- To assess HE formation by flow cytometric analysis use cells on day 3 and day 4 from one experiment. Collect cells by incubating of cultures with commercial cell dissociation reagent (see Table 1) for 5-10 min at 37 °C, 5% CO2 and then use up to 1 x 105 cells per staining reaction (flow cytometry staining procedure described in 12). NOTE: VE-cadherin+ HE cells acquire expression of early hematopoietic marker CD226, but lack the expression of CD73 and CD436. In contrast, non-HE cells express CD73 (Figures 2B, 3B).
- Immunofluorescence staining for hPSC-derived HE.
- Wash the attached monolayer of cells with phosphate buffered saline (PBS) and then fix cells in 4% paraformaldehyde from 30 to 60 min at room temperature.
- Wash cells again with PBS and then permeabilize using 0.1% Triton X-100 in PBS for 20 min at room temperature.
- Wash cells in PBS two to three times and then incubate in blocking buffer consisting of PBS with 10% fetal bovine serum (FBS), for 30 to 120 min.
- Prepare staining solution, containing both primary antibodies (1:1,000 dilution) mouse anti-human CD43 and rabbit anti-human VE-cadherin in PBS with 5% FBS, (Table 1).
- Aspirate the blocking buffer from the cells and add 1 ml per well of staining buffer with the added primary antibodies; incubate 3 hr at room temperature, or overnight at 4 °C.
- Wash the cells three times by adding 2 ml of PBS with 5% FBS.
- Prepare a second staining buffer, containing secondary antibodies at 1:500 dilution in PBS with 5% FBS: donkey anti-rabbit conjugated with green fluorescent dye, and anti-mouse conjugated with red fluorescent dye (Table 1). Add 1 ml per well of secondary staining buffer and incubate at room temperature for 1 hr.
- Wash three times with PBS without serum. Stain cells with 300 nM DAPI working solution in dH2O for 10-15 min.
- Wash cells with PBS twice and add 2-3 ml of PBS into well with stained cells. Observe fluorescence with green, red and blue microscope filters.
6. Analysis of Induced Hematopoietic Precursors
Maintain differentiation cultures until floating cells expand significantly, up to 10-14 days, changing half of the 3F-medium, as described in 3.1, every two days.
Dissociate and collect differentiating monolayers of cells by treating cells with a commercial cell dissociation reagent (see Table 1) for 5-10 min at 37°C, 5% CO2.
Perform flow cytometry using anti-VE-cadherin, CD43 and CD45 antibodies to evaluate hematopoiesis in cultures12. Note that a large fraction of cells, up to 40% of ETV2 and GATA2 transduced cells, co-expresses VE-cad and CD43. The following antibodies can be used to detect specific cell lineages following expansion or differentiation of induced blood progenitors: CD235a (erythroid cells), CD41a (megakaryocytic), CD32 (all types of myeloid cells), CD66b (neutrophils) and CD163 (macrophages).
- Perform CFC-assay using a commercial clonogenic medium (Table 1) according to the manufacturer’s instructions.
- Transfer 1-2 x 104 cells into 3 ml of commercial clonogenic medium (see Table 3) to assess the number of colony-forming cells and types of hematopoietic colonies. Optimal seeding density allows for the identification and isolation of single colonies that grow separately from each other and do not overlap. Seeding density may vary between experiments depending on differentiation efficiency, but should not exceed 1 x 104 cells per 1 ml of commercial clonogenic medium.
- Mix cells in commercial clonogenic medium by gentle vortexing, and transfer 3 ml of suspension to two 35 mm ultra-low attachment dishes for CFC assay, 1.5 ml of suspension to each dish. Distribute medium evenly and incubate at 37 °C, 5% CO2 for 14 days. Avoid disturbing the dishes; observe colony formation on day 7 and day 10 of assay.
- Identify and count hematopoietic colonies according to their morphologies on day 14.
Representative Results
The schematic diagram of HE and blood induction from hPSCs by overexpression of transcription factors is shown in Figure 1. ETV2 with GATA1 or GATA2 combination induces pan-myeloid hematopoiesis, while a GATA2, TAL1 +/- LMO2 combination induces predominantly erythro-megakaryocytic hematopoiesis. Both TF combinations directly induced HE cells that subsequently transformed into blood progenitors with a distinct spectrum of hematopoietic differentiation. Differentiation of hPSCs from the pluripotent state to blood producing HE takes on average 4 days. Round blood cells first emerge by day 5 of differentiation. Subsequently, numerous floating blood cells appear and expand in cultures. To maximize the number of floating blood cells, post-transduction cultures can be prolonged to 10-14 days. The desired lineage of cells can be expanded in low-adherent conditions with the appropriate combination of hematopoietic cytokines. Figure 2 demonstrates the induction of blood from hPSCs using ETV2 and GATA1 or GATA2 TFs. Following transduction with ETV2 and GATA2, cells acquire a typical endothelial morphology by day 4 of differentiation, and eventually transform into round blood cells (day 6-8 of differentiation (Figure 2A). Cells collected on days 3-4 of differentiation show a typical VE-cadherin+CD226+CD73- HE phenotype (Figure 2B, 3B). By day 7 of differentiation more than 50% of VE-cadherin+ cells acquire the hematopoietic CD43+ phenotype (Figure 2C). CFC-assays of GATA2/ETV2-induced cells reveal that colonies are comprised of high proliferative potential (HPP) CFCs, Erythroid (E) CFCs, macrophages (M) CFCs, and granulocytes (G) CFCs (Figure 2E). The hematoendothelial differentiation in hPSCs following overexpression of GATA2 and TAL1 alone or with LMO2 is shown in Figure 3. HE stage is best identified in cultures with GATA2 and TAL1. The addition of LMO2 accelerates differentiation and markedly contracts HE stage. The GATA2 and TAL1 combination alone or with LMO2 demonstrates differentiation and maturation of blood progenitors to mostly erythroid and megakaryocytic cells Figure 3D and 3E. Figure 4 shows optimization of transduction efficiency in H1 hESCs using GFP lentivirus (A) and cell death and differentiation efficiency in cultures transduced with GATA2/TAL1/LMO2 at high and low MOI.
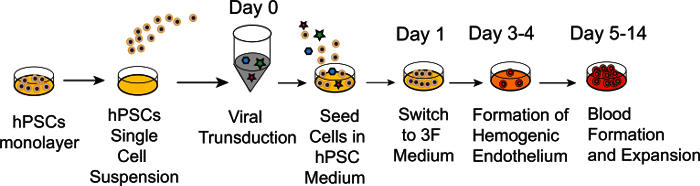 Figure 1: Schematic diagram of protocol for induction of hemogenic endothelium and blood cells via the forced expression of TFs. Cartoon outlines major experimental steps and timelines. Following preparation of single cell suspension, hPSCs are transduced with TFs and seeded on matrix in complete commercial serum-free medium. Next day, medium is replaced with growth factor free commercial medium supplemented with bFGF, TPO and SCF (3F Medium). Following 3-4 days of culture, HE is formed. Subsequently, HE undergoes endothelial to hematopoietic transition with formation of multiple blood cells.
Figure 1: Schematic diagram of protocol for induction of hemogenic endothelium and blood cells via the forced expression of TFs. Cartoon outlines major experimental steps and timelines. Following preparation of single cell suspension, hPSCs are transduced with TFs and seeded on matrix in complete commercial serum-free medium. Next day, medium is replaced with growth factor free commercial medium supplemented with bFGF, TPO and SCF (3F Medium). Following 3-4 days of culture, HE is formed. Subsequently, HE undergoes endothelial to hematopoietic transition with formation of multiple blood cells.
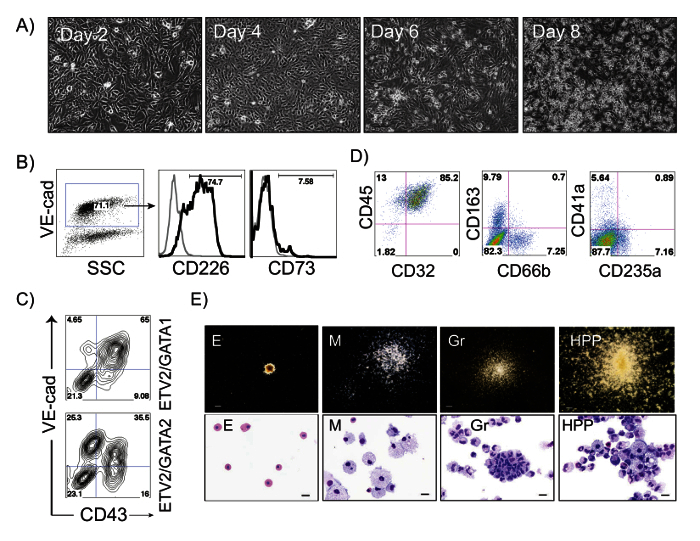 Figure 2: ETV2/GATA2-induced differentiation of hPSCs. (A) Phase contrast images of ETV2/GATA2-induced differentiation in H1 hESCs at days 2, 4, 6 and 8 after transduction; Scale bar, 100 µm. (B) Flow cytometric analysis of ETV2/GATA2-induced cells undergoing endothelial to hematopoietic transition at day 3 of differentiation: VE-cadherin+ HE cells express CD226 and lack expression of CD73. A small percentage of endothelial cells express CD73 (non-HE). (C) Flow cytometric analysis of ETV2/GATA1- and ETV2/GATA2-induced H1 hPSCs on day 7 post-transduction. (D) Flow cytometric analysis of ETV2/GATA2-induced hematopoietic cells expanded in medium supplemented with FBS and SCF, IL3, IL6, GM-CSF, G-CSF and EPO for 14 days. (E) Types of hematopoietic colonies formed from ETV2/GATA2 transduced cells in CFC-assay and corresponding Wright-stained cytospins representing Erythroid (E), Macrophages (M), Granulocytes (Gr) and High Proliferative Potential colonies (HPPs). Scale bars for CFC-assay, 250 µm, for cytospins, 20 µm. Please click here to view a larger version of this figure.
Figure 2: ETV2/GATA2-induced differentiation of hPSCs. (A) Phase contrast images of ETV2/GATA2-induced differentiation in H1 hESCs at days 2, 4, 6 and 8 after transduction; Scale bar, 100 µm. (B) Flow cytometric analysis of ETV2/GATA2-induced cells undergoing endothelial to hematopoietic transition at day 3 of differentiation: VE-cadherin+ HE cells express CD226 and lack expression of CD73. A small percentage of endothelial cells express CD73 (non-HE). (C) Flow cytometric analysis of ETV2/GATA1- and ETV2/GATA2-induced H1 hPSCs on day 7 post-transduction. (D) Flow cytometric analysis of ETV2/GATA2-induced hematopoietic cells expanded in medium supplemented with FBS and SCF, IL3, IL6, GM-CSF, G-CSF and EPO for 14 days. (E) Types of hematopoietic colonies formed from ETV2/GATA2 transduced cells in CFC-assay and corresponding Wright-stained cytospins representing Erythroid (E), Macrophages (M), Granulocytes (Gr) and High Proliferative Potential colonies (HPPs). Scale bars for CFC-assay, 250 µm, for cytospins, 20 µm. Please click here to view a larger version of this figure.
 Figure 3: GATA2/TAL1 and GATA2/TAL1/LMO2-induced differentiation of hPSCs. (A) Phase contrast images of GATA2/TAL1/LMO2-induced differentiation in H1 hESCs at days 2, 4, 6 and 8; Scale bar, 100 µm. (B) Flow cytometric analysis of GATA2/TAL1-induced cells undergoing endothelial to hematopoietic transition on day 3 of differentiation: VE-cadherin+ cells acquire expression of HE marker CD226. A small percentage of endothelial cells express CD73 (non-HE). (C) Immunofluorescent staining of GATA2/TAL1-induced differentiation in H1 hESC, day 5; VE-cadherin+ cells (green) acquire expression of hematopoietic marker CD43 (red). (D) Flow cytometric analysis of GATA2/TAL1/LMO2-inducedhematopoietic cultures in suspension on day 14 post-transduction following expansion in serum-free medium supplemented with SCF, TPO, EPO and bFGF; (E) Types of hematopoietic colonies formed in CFC-assay by GATA2/TAL1/LMO2 differentiated cellsand corresponding Wright-stained cytospins representing Erythroid (E), Macrophages (M), and Megakaryocytes (Mk). Scale bars for CFC-assay, 250 µm, for cytospins, 20 µm. Please click here to view a larger version of this figure.
Figure 3: GATA2/TAL1 and GATA2/TAL1/LMO2-induced differentiation of hPSCs. (A) Phase contrast images of GATA2/TAL1/LMO2-induced differentiation in H1 hESCs at days 2, 4, 6 and 8; Scale bar, 100 µm. (B) Flow cytometric analysis of GATA2/TAL1-induced cells undergoing endothelial to hematopoietic transition on day 3 of differentiation: VE-cadherin+ cells acquire expression of HE marker CD226. A small percentage of endothelial cells express CD73 (non-HE). (C) Immunofluorescent staining of GATA2/TAL1-induced differentiation in H1 hESC, day 5; VE-cadherin+ cells (green) acquire expression of hematopoietic marker CD43 (red). (D) Flow cytometric analysis of GATA2/TAL1/LMO2-inducedhematopoietic cultures in suspension on day 14 post-transduction following expansion in serum-free medium supplemented with SCF, TPO, EPO and bFGF; (E) Types of hematopoietic colonies formed in CFC-assay by GATA2/TAL1/LMO2 differentiated cellsand corresponding Wright-stained cytospins representing Erythroid (E), Macrophages (M), and Megakaryocytes (Mk). Scale bars for CFC-assay, 250 µm, for cytospins, 20 µm. Please click here to view a larger version of this figure.
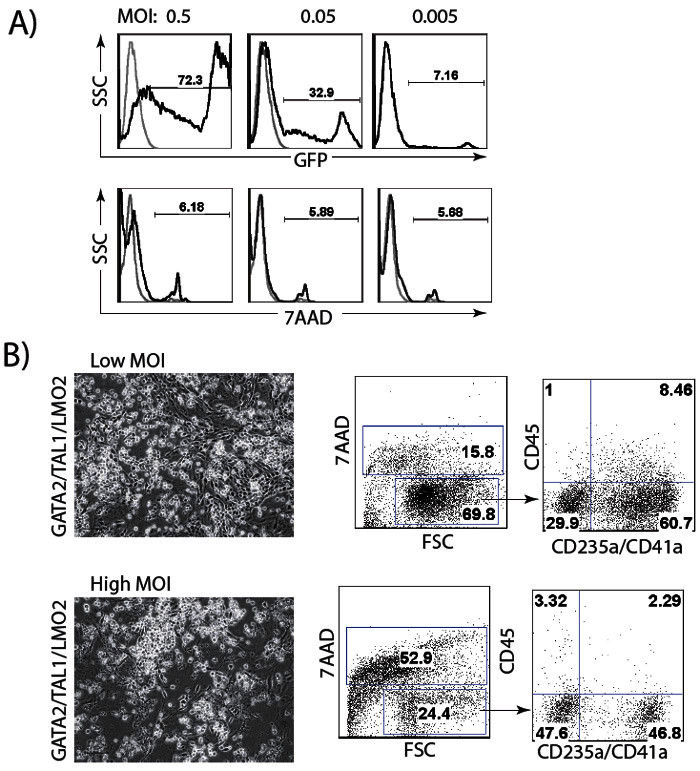 Figure 4: Optimization of lentiviral transduction procedure. (A) Transduction of H1 hESCs with different MOI of GFP lentivirus. (B) Cell viability and differentiation efficiency in cultures transduced with GATA2/TAL1/LMO2 at low (1 for each virus) and high (2 for each virus) MOI.
Figure 4: Optimization of lentiviral transduction procedure. (A) Transduction of H1 hESCs with different MOI of GFP lentivirus. (B) Cell viability and differentiation efficiency in cultures transduced with GATA2/TAL1/LMO2 at low (1 for each virus) and high (2 for each virus) MOI.
Discussion
The above-described method for hematopoietic differentiation of hPSCs by overexpression of TFs, represents a rapid and efficient approach for the generation of HE and myeloid and erytho-magakaryocytic progenitors from hESCs and iPSCs, thereby allowing the production of up to 30 millions blood cells from one million pluripotent stem cells10. This method exhibited consistent differentiation in multiple hESC and iPSCs lines10. During differentiation by ETV2 and GATA2, GATA1 factors as well as GATA2 and TAL1 factors, cells form blood producing endothelium, HE and undergo an endothelium to hematopoietic transition. Formation of HE is usually observed between days 3 and 4 of differentiation. Addition of LMO2, a transcriptional co-factor of TAL1, to the GATA2 and TAL1 combination, significantly increased the robustness of differentiation, while the HE stage became markedly contracted.
The success of directed differentiation of hPSCs by transcription factors mainly depends on (i) an effective delivery of nucleic acids into cells, (ii) cell viability following genetic manipulations, and (iii) an effective translation of exogenous transcripts inside the cell.
One of the key steps of the described method is the production of effective lentiviral units, which depends on a precise buffer pH for HEK 293T cell transfection, and the absence of endotoxin contaminants at all steps of viral production and transduction experiments. The integrity of all constructs, including packaging and envelope plasmids, must be verified if no signs of viral production in HEK 293T cells is observed13.
The differentiation protocol design using combinations of transcription factors should consider several variables. High efficiency of viral transduction is important for successful differentiation. Since transduction efficiency depends on the method used for virus production and purification, optimize hPSC transduction using a GFP-lentiviral vector is recommended (Figure 4A). Viral integration following transduction should be verified using genomic PCR with transgene-specific primers (Table 3). Differentiation optimization should also include adjustment of lentiviral concentration in the reaction mixture. A relatively low amount of virus, MOI of 0.5-1, is capable of inducing hPSC differentiation. While a higher MOI increases the efficiency of differentiation, it also increases cell death (Figure 4B). Additional optimization step may include MOI ratio adjustment for each virus in the reaction mixture. In GATA2/ETV2 transduced cultures, doubling the MOI for GATA2, relative to ETV2, increases the efficacy of differentiation. For GATA1/ETV2, GATA2/TAL1 and GATA2/TAL1/LMO2 equal MOI for each virus is optimal.
Following transduction with TFs, a vast majority of cells underwent angiohematopoietic differentiation while only a very few cells retained an undifferentiated morphology. Since pSIN-EF1α lentiviral vectors incorporate antibiotic resistance, treatment of cultures with puromycin can be used to eliminate residual undifferentiated cells.
The formation of cells with characteristic endothelial morphologies, and subsequent round blood cells, indicate a successful differentiation. On average, the GATA2/ETV2 combination produces 40-50% of CD43+VE-cadherin+/- blood cells by day 9-10 of differentiation with up to 30% cells remaining VE-cadherin+CD43-. GATA1/ETV2 combination induces CD43 expression more quickly and generates a greater number of CD43+ cells as compared to the GATA2/ETV2 combination. In these cultures, the majority of CD43+ cells coexpress VE-cadherin. In cultures transduced with GATA2/TAL1, the number of CD43+ cells usually reaches 40-50%. The addition of LMO2 to GATA2/TAL1 substantially accelerates and enhances the blood production, and markedly contracts the endothelial stage of development.
Colony-forming assays should be used to determine the frequency and type of induced hematopoietic progenitors. The nature of colony-forming cells can be confirmed by preparing Wright-stained cytospins from individual colonies. hPSCs transduced with ETV2 and GATA2 produce mostly large (greater than 0.5 mm in diameter) high proliferative potential (HPP) myeloid colonies composed of immature granulocytic and monocytic cells, with some mature myeloid cells and occasional erythroid and megakaryocytic cells. On average more than 80% of CFCs in GATA2/ETV2 transduced cultures are myeloid HPPs. In addition, these cultures generate erythroid and macrophage colonies (Figure 2E), which usually comprise less than 20% of total CFCs. hPSC transduction with ETV2 and GATA1 significantly increases the proportion of CFC-E to up to 50%. TAL1 and GATA2 transduced cultures produce mostly erythroid (more than 80% of total CFCs) and megakaryocytic colonies (more than 10% of total CFCs) with very few macrophage colonies (less than 5% of total CFCs). The addition of LMO2 to the TAL1 and GATA2 combination significantly increases the frequency of colony-forming cells with erythro-megakaryocytic potential10. The pattern of CFC activity following transduction with the described combinations of transcription factors is consistent among different hESC and hiPSC lines10.
In summary, lentiviral-mediated overexpression in hPSCs is a relatively quick and efficient tool for the induction of HE and blood using TFs. This system can also be used for assessing the differentiation capacity of other TFs and identify those that are required for endothelial and hematopoietic specification. However, the current protocol has a limited utility for studies of extracellular signaling involved in the induction of HE, as it uses TFs to bypass the surface receptor-mediated signaling. Although the current protocol describes blood production using the constitutive expression of a transgene, similar results could be obtained using modified mRNAs10.
Disclosures
I.S. is a founding shareholder and consultant for Cynata.
Acknowledgments
We thank Matt Raymond for editorial assistance. This work was supported by funds from the National Institute of Health (U01HL099773, R01HL116221, and P51 RR000167) and The Charlotte Geyer Foundation.
References
- Zape JP, Zovein AC. Hemogenic endothelium: origins, regulation, and implications for vascular biology. Semin Cell Dev Biol. 2011;22:1036–1047. doi: 10.1016/j.semcdb.2011.10.003. [DOI] [PubMed] [Google Scholar]
- Swiers G, Rode C, Azzoni E, de Bruijn MF. A short history of hemogenic endothelium. Blood Cells, Mol & Dis. 2013;51:206–212. doi: 10.1016/j.bcmd.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy M, et al. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Rep. 2012;2:1722–1735. doi: 10.1016/j.celrep.2012.11.003. [DOI] [PubMed] [Google Scholar]
- Wang L, et al. Endothelial and hematopoietic cell fate of human embryonic stem cells originates from primitive endothelium with hemangioblastic properties. Immunity. 2004;21:31–41. doi: 10.1016/j.immuni.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Rafii S, et al. Human ESC-derived hemogenic endothelial cells undergo distinct waves of endothelial to hematopoietic transition. Blood. 2012;121(5):770–780. doi: 10.1182/blood-2012-07-444208. [DOI] [PubMed] [Google Scholar]
- Choi KD, et al. Identification of the hemogenic endothelial progenitor and its direct precursor in human pluripotent stem cell differentiation cultures. Cell Rep. 2012;2:553–567. doi: 10.1016/j.celrep.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodyanik MA, Thomson JA, Slukvin II. Leukosialin (CD43) defines hematopoietic progenitors in human embryonic stem cell differentiation cultures. Blood. 2006;108:2095–2105. doi: 10.1182/blood-2006-02-003327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, et al. TGFbeta inhibition enhances the generation of hematopoietic progenitors from human ES cell-derived hemogenic endothelial cells using a stepwise strategy. Cell Res. 2012;22:194–207. doi: 10.1038/cr.2011.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uenishi G, et al. Tenascin C promotes hematoendothelial development and T lymphoid commitment from human pluripotent stem cells in chemically defined conditions. Stem Cell Rep. 2014;3:1073–1084. doi: 10.1016/j.stemcr.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elcheva I, et al. Direct induction of haematoendothelial programs in human pluripotent stem cells by transcriptional regulators. Nat Commun. 2014;5:4372. doi: 10.1038/ncomms5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiscornia G, Singer O, Verma IM. Production and purification of lentiviral vectors. Nat Protoc. 2006;1:241–245. doi: 10.1038/nprot.2006.37. [DOI] [PubMed] [Google Scholar]
- Vodyanik MA, Slukvin II. Hematoendothelial differentiation of human embryonic stem cells. Curr Protoc Cell Biol. 2007;Chapter 23:Unit 23.6. doi: 10.1002/0471143030.cb2306s36. [DOI] [PubMed] [Google Scholar]
- Cao F, et al. Comparison of gene-transfer efficiency in human embryonic stem cells. Mol Imgn and Biol. 2010;12:15–24. doi: 10.1007/s11307-009-0236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]