

Summary
Chronic granulomatous disease (CGD) is an inherited immunodeficiency linked with mutations in the multi‐subunit leucocyte NADPH oxidase. Myeloid‐derived phagocytic cells deficient in NADPH oxidase fail to produce sufficient levels of reactive oxygen species to clear engulfed pathogens. In this study we show that oxidase also influences B‐cell functions, including responses to single‐stranded RNA or unmethylated DNA by endosomal Toll‐like receptors (TLRs) 7 and 9. In response to TLR7/9 ligands, B‐cell lines derived from patients with CGD with mutations in either the NADPH oxidase p40phox or p47phox subunits produced only low levels of reactive oxygen species. Remarkably, cytokine secretion and p38 mitogen‐activated protein kinase activation by these oxidase‐deficient B cells was significantly increased upon TLR7/9 activation when compared with oxidase‐sufficient B cells. Increased TLR responsiveness was also detected in B cells from oxidase‐deficient mice. NADPH oxidase‐deficient patient‐derived B cells also expressed enhanced levels of TLR7 and TLR9 mRNA and protein compared with the same cells reconstituted to restore oxidase activity. These data demonstrate that the loss of oxidase function associated with CGD can significantly impact B‐cell TLR signalling in response to nucleic acids with potential repercussions for auto‐reactivity in patients.
Keywords: B cell, immunodeficiency diseases, inflammation, reactive oxygen species, Toll‐like receptors
Abbreviations
- (c)DNA
(complementary) deoxyribonucleic acid
- (m)RNA
(message) ribonucleic acid
- CD
cluster of differentiation
- CGD
chronic granulomatous disease
- CpG
cytosine‐phosphate‐guanine
- DPI
diphenyleneiodonium
- EDTA
ethylenediaminetetraacetic acid
- ELISA
enzyme‐linked immunosorbent assay
- FITC
fluorescein isothiocyanate
- GAPDH
glyceraldehyde‐3‐phosphate dehydrogenase
- GT
guanine thymidine
- GTP
guanosine triphosphate
- H2DCFDA
2′7′‐dichlorodihydrofluorescein diacetate
- H2O2
hydrogen peroxide
- IFNα
interferon alpha
- IL
interleukin
- JNK
c‐Jun N‐terminal kinase
- kDa
kilo‐dalton
- MAPK
mitogen‐activated protein kinase
- MyD88
myeloid differentiation primary response gene 88
- PCR
polymerase chain reaction
- PMA
phorbol 12‐myristate 13‐acetate
- ROS
reactive oxygen species
- SDS‐PAGE
sodium dodecyl sulfate‐polyacrylamide gel electrophoresis
- TLR
toll‐like receptor
- TNFα
tumor necrosis factor alpha
Introduction
The NADPH oxidase complex is responsible for the production of antimicrobial oxidants by phagocytic cells including neutrophils and macrophages in response to microbial pathogens. Upon physiological stimulation of these phagocytes, cytoplasmic components of the oxidase, p40phox, p47phox, p67phox, and a small GTPase Rac2, translocate to the catalytic membrane‐bound flavocytochrome, a heterodimer composed of gp91phox (Nox2) and p22phox. The now active NADPH oxidase transfers electrons from cytoplasmic NADPH to molecular oxygen, forming a series of toxic reactive oxygen species (ROS) that targets pathogens for destruction within vacuolar compartments. Mutations in NADPH oxidase subunits result in the development of chronic granulomatous disease (CGD), an immunodeficiency marked by insufficient phagocyte ROS production, hallmark recurrent bacterial and fungal infections, and the development of granulomas.1 Less well understood from a molecular perspective, patients with CGD often experience sterile granulomas and have higher incidences of autoimmune disorders independent of infection.1, 2, 3, 4 These latter observations may suggest a role for the oxidase and its by‐product ROS in modulating other important aspects of immune cell function.
Though expressed on a smaller scale than in phagocytes, NADPH oxidase is also present and functional in B and T lymphocytes, producing ROS upon B‐cell or T‐cell receptor activation.5, 6, 7 Several studies have shown a role for the oxidase in B‐cell and T‐cell function and activation.8 A subset of peritoneal B cells is capable of phagocytosis and oxidase‐derived ROS production linked to bacterial killing.7 However, other B‐cell functions are also regulated by the multi‐subunit oxidase. Disruption of oxidase function in murine B cells led to an increase in cell proliferation and altered antibody production.6, 9 Other defects in peripheral blood B cells were identified in patients with CGD, including low frequencies of memory CD27+ B cells and reduced B‐cell activating factor receptor expression on CD19+ B lymphocytes.10, 11, 12 Additionally, alterations in MHC class II‐restricted antigen presentation were seen in CGD B lymphoblasts.13 Furthermore, T cells from oxidase‐deficient mice also produce increased levels of T helper type 1 and type 17 associated cytokines following activation.14 Intracellular ROS has been implicated in some intracellular signalling pathways,15 although mechanistically how oxidase loss impacts B‐cell activation remains unclear.
Toll‐like receptors (TLRs) recognize a wide array of evolutionarily conserved patterns uniquely found in microbial pathogens and some self‐antigens16, 17 and the activation of these receptors leads to oxidase ROS production. NADPH oxidase assembly and ROS production in murine macrophages and neutrophils are dependent on MyD88 and phospho‐p38, essential components of the TLR signalling pathway.18 Additionally, dendritic cells matured by TLR2 or TLR4 ligands express higher levels of p47phox and gp91phox.19
B cells express TLR7 and TLR9, which recognize single‐stranded RNA and CpG‐containing DNA, respectively, within endosomal compartments. Dysregulation of these endosomal TLRs has been linked to several autoimmune disorders including systemic lupus erythematosus.20, 21, 22, 23 Interestingly, genome‐wide association studies analysis has linked select oxidase subunit mutations with lupus susceptibility.24 Lupus‐prone MRL.Fas lpr mice crossbred to promote oxidase‐deficiency generated offspring with exacerbated lupus and elevated autoantibody production.25
In the current study, B lymphoblasts from patients with CGD with mutations in p40phox or p47phox of the oxidase displayed hyper‐responsiveness to TLR7 and TLR9 ligands as detected by secretion of pro‐inflammatory and anti‐inflammatory cytokines interleukin‐6 (IL‐6), IL‐10 and tumour necrosis‐α (TNF‐α). Restoration of wild‐type oxidase subunits in these cells significantly diminished cytokine production. Reduced oxidase function in these human B cells was associated with higher levels of TLR7 and TLR9 mRNA and protein expression as well as hyper‐activation of p38 mitogen‐activated protein kinase (MAPK). Oxidase‐deficient mouse B cells also exhibited exaggerated cytokine production in response to TLR7/9 ligands and increased phosphorylation of p38 MAPK. Together, these studies suggest an important role of NADPH oxidase in modulating endosomal TLR and innate immune responses to nucleic acids.
Materials and methods
Human and murine B cells
Human B lymphoblastoid cell lines AR and KS were generated from whole blood of two patients diagnosed with CGD by Epstein–Barr virus‐transformation. The AR cell line has defined mutations mapped in each allele of p40phox. The AR paternal p40phox allele has a frame shift mutation and is not expressed, whereas the maternal allele has a point mutation that disrupts normal function.26 AR B cells, previously referred to as AR40.DR4, produce reduced intracellular ROS under resting and PMA‐activated conditions.13 The KS B lymphoblastoid cell line fails to express p47phox because of a GT deletion at base 75 in this subunit and is probably homozygous. Resting KS cells have diminished superoxide production.27 Both AR and KS cells were reconstituted with vectors encoding the appropriate wild‐type oxidase subunit to restore NADPH oxidase ROS production, and termed ARp40 or KSp47, respectively.13, 27 To maintain the expression of the transfected wild‐type subunits, the reconstituted cells were cultured as follows. KSp47 cells were treated with 200 μg/ml hygromycin B (EMD Millipore, Billerica, MA) every 2 weeks to maintain wild‐type selection of p47phox expression. ARp40, previously referred to as AR40.DR4WT, was treated with 200 μg/ml puromycin (Sigma‐Aldrich, St Louis, MO) every 2 weeks to maintain wild‐type p40phox expression. All B cells were cultured in RPMI‐1640, 10% fetal bovine serum and 1% penicillin/streptomycin. Cybb −/− mice, deficient in the gp91phox subunit of NADPH oxidase,28 were analysed alongside age‐ and sex‐matched C57BL/6 wild‐type mice (Jackson Laboratory, Bar Harbor, ME). Mice were treated humanely following institutionally approved protocols. After lysis of red blood cells from crushed murine spleens in ACK buffer (0·15 m NH4Cl, 10 mm KHCO3, 0·1 mm Na2EDTA, pH 7·3), naive murine B cells were isolated in a no‐touch manner by using anti‐CD43+ MACS beads (Miltenyi Biotec, San Diego, CA).
ROS production
Cells were treated with or without 10 μm diphenyleneiodonium (DPI) (Sigma‐Aldrich) for 30 min before a 10‐min incubation with 3 μm 2′7′‐dichlorodihydrofluorescein diacetate (H2DCFDA; Thermo Fisher Scientific, Waltham, MA), a dye that fluoresces upon reaction with ROS. Subsequently, cells incubated with the dye only were treated with 10 μg/ml of the TLR7 ligand R848 (Enzo Life Sciences, Farmingdale, NY), 5 μm of the TLR9 ligand human CpG oligodinucleotide 2006 (Invivogen, San Diego, CA), and/or 10 μg/ml PMA (Sigma‐Aldrich) for 30 min. Cellular mean fluorescence intensity was detected by flow cytometry and plotted as a ratio to the mean fluorescence intensity of cells treated with H2DCFDA alone.
B‐cell stimulation
Human B‐cell lines were seeded at 1 × 106/ml and incubated with 10 nm PMA, and/or 2 μg/ml R848 or 50 nm CpG for 24 hr.29 Isolated murine naive B cells were similarly seeded at 1 × 106/ml and incubated for 24 hr with 10 nm PMA, and/or 0·1 μg/ml R848 or 0·5 μm murine CpG oligodinucleotide 1826 (Invivogen). Cell culture supernatants were harvested, cells were removed from wells using 2 mm EDTA (Sigma‐Aldrich), and cell viability was determined via trypan blue exclusion.
ELISA
Cell culture supernatants of B lymphoblasts after 24 hr of TLR and/or PMA activation were analysed using a standard sandwich ELISA to detect human IL‐6 (Thermo Fisher Scientific), human IL‐10 (eBioscience, San Diego, CA), and human TNF‐α (R&D Systems, Minneapolis, MN) or murine IL‐6 (eBioscience) and murine IL‐10 (BioLegend, San Diego, CA) with an adjacent standard curve generated by recombinant cytokines (PeproTech, Rocky Hill, NJ). One hundred microlitres of supernatant was analysed in triplicate for each stimulation condition with replicate independent experiments as indicated. An interferon‐α (IFN‐α) ELISA kit (Quansys Biosciences, Logan, UT) was used to assay supernatants from untreated human cells after a 24‐hr incubation in fresh media.
Western blot
Cells were lysed (10 mm Tris–HCl, 150 mm NaCl, 1% Triton‐X‐100) in the presence of phosphatase inhibitor (Cell Signaling Technology, Danvers, MA) and protease inhibitor cocktails (Sigma‐Aldrich). Fifty to one hundred micrograms of cell lysates was resolved by SDS–PAGE, transferred to a nitrocellulose membrane, and immunoblotted for phospho‐p38 MAPK, phospho‐c‐Jun N‐terminal kinase (JNK), total p38 MAPK, total JNK, and/or MyD88 proteins using rabbit monoclonal antibodies purchased from Cell Signaling Technology. To detect NADPH oxidase subunits and receptor protein levels, untreated cells were similarly lysed and immunoblotted for gp91phox (Santa Cruz Biotechnology, Dallas, TX) p40phox (EMD Millipore), p47phox (BD Biosciences, San Jose, CA), full‐length TLR7 (Abcam, Cambridge, MA), or full‐length TLR9 (Abcam) proteins. GAPDH (EMD Millipore) was detected on all blots as a loading control. Immunoblots were visualized with enhanced chemiluminescent (EMD Millipore) after an incubation with a horseradish peroxidase‐conjugated secondary antibody purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). Due to the detection of two bands close in size when using the antibody for phospho‐p38, a synthetic phospho‐p38 peptide recognized by this antibody was used to verify antibody specificity and that phospho‐p38 species in these B cells migrated on gels as a doublet (Cell Signaling). Protein was quantified by densitometry using imageJ, calculated as a ratio to either GAPDH or the respective total protein to control for sample loading. These protein levels were then normalized to the appropriate untreated reconstituted cell line, which was set to one for each blot.
Quantitative PCR
Total RNA was extracted from B cells using an RNeasy Minikit (Qiagen, Valencia, CA) according to the manufacturer's instructions. Messenger RNA transcripts were reverse transcribed using High‐Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). One hundred nanograms of cDNA was used as a template for PCR amplification of specified genes. Real‐time PCR was performed using commercial TaqMan Gene Expression Assays and TaqMan Fast Universal PCR Master Mix on a 7500 Fast RT‐PCR System machine according to the manufacturer's protocol (Thermo Fisher Scientific). The cDNA template was amplified with the following settings: initial template denaturation for 20 seconds at 95° followed by 40 cycles of 3 seconds at 95° and 30 seconds at 60°. Levels of MYD88, TLR7 and TLR9 mRNA transcripts were calculated using the ΔΔC T method with GAPDH as the housekeeping gene and the untreated reconstituted cell line as the calibrator, which was set to one.
Inhibition of p38 MAPK activity
Human B cells were seeded at 1 × 106/ml and incubated for 24 hr with 0, 1, 5, 10, 50 or 100 μm of SB202190 p38 MAPK activity inhibitor (Sigma‐Aldrich). Cell viability was determined by trypan blue exclusion. Treatment with 10 μm SB202190 was the highest concentration that did not compromise cell viability and was therefore selected for future experiments. Cells were incubated with 10 μm SB202190 for 1 hr before 2 μg/ml of R848, 50 nm CpG, and/or 10 nm PMA were added to the media. After 24 hr, cell culture supernatants were harvested and screened for IL‐6 by sandwich ELISA.
Statistical analysis
P‐values were calculated using either multiple t‐tests or analysis of variance with Bonferroni's correction for multiple comparisons using graphpad prism6 software (La Jolla, CA).
Results
Reduced NADPH oxidase subunit expression and ROS production in B lymphoblasts from patients with CGD was corrected by ectopic expression of the appropriate wild‐type oxidase subunit
The lymphoblast cell line AR was generated from a p40phox‐deficient patient with a paternal truncated allele and a maternal point‐mutated allele of the p40phox gene.26 Retroviral transduction was used to ectopically express the wild‐type p40phox allele in the AR cells yielding the cell line ARp40.13 AR and ARp40 cells were lysed and immunoblotted to detect p40phox protein levels. Expression of p40 was reduced by ~93% in the AR B‐cell line compared with the restored cell line ARp40 (Fig. 1a). A classical GT deletion mutation associated with loss of p47phox function was mapped in the human B lymphoblast cell KS, which is probably homozygous. To restore p47phox function in KS cells, the cDNA for the wild‐type human p47phox allele was inserted into an expression vector and transfected in KS cells generating the cell line KSp47.27 To verify p47phox deficiency and restoration, KS and KSp47 cells were lysed and immunoblotted to detect p47phox (Fig. 1b).
Figure 1.
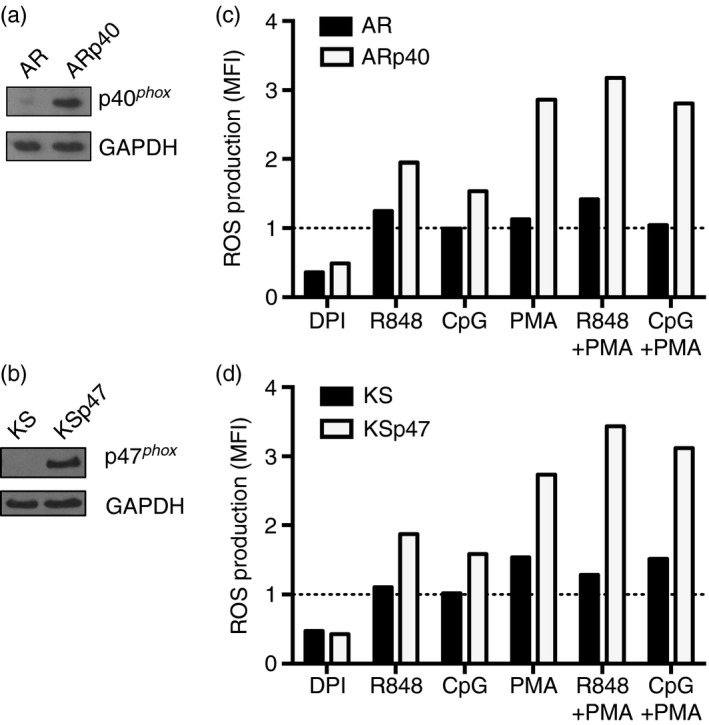
Decreased reactive oxygen species (ROS) production after Toll‐like receptor 7 ( TLR7) or TLR9 stimulation in NADPH oxidase‐deficient B‐cell lines. (a) AR, a B‐cell line derived from a patient with mutations in both paternal and maternal alleles of p40phox and ARp40, the AR cell reconstituted with wild‐type p40phox, were lysed and the lysates were immunoblotted to detect p40phox and GAPDH proteins. (b) Cell lysates from KS, a B‐cell line derived from a patient lacking functional p47phox and KSp47, the KS B‐cell line reconstituted with p47phox, were immunoblotted to detect p47phox and GAPDH proteins. (c,d) ROS production by B cells was monitored after a 30‐min treatment with diphenyleneiodonium (DPI), R848, CpG and/or PMA and a 10‐min incubation with 2′7′‐dichlorodihydrofluorescein diacetate (H2 DCFDA) as described in the Materials and methods. Mean fluorescence intensity (MFI) was detected by flow cytometry and plotted as a ratio to the MFI of cells treated with H2 DCFDA alone which was set to one (dashed line). Data shown in (c) and (d) represent one of three independent experiments.
To verify restoration of NADPH oxidase function in ARp40 and KSp47 cells after wild‐type p40phox or p47phox reconstitution, the original cell lines from each CGD patient and their reconstituted counterparts were pre‐treated with or without DPI, an inhibitor of ROS production, and H2DCFDA, a sensor of intracellular ROS (Fig. 1c,d). Parental CGD and reconstituted cells constitutively produced ROS, which was reduced by treatment with DPI. Splenic B lymphocytes from healthy mice have been shown to produce ROS after TLR4 ligand stimulation.30 Here, treatments using TLR7 or TLR9 ligands in the presence or absence of PMA were similarly able to induce ROS production by the reconstituted B cells, whereas the levels of ROS detected in the parental CGD cell lines increased only slightly or remained similar to unstimulated cells. With TLR stimulation alone, NADPH oxidase‐sufficient B cells, ARp40 and KSp47 produced 30–50% more ROS compared with untreated cells, but the oxidase‐deficient cells had minimal response. Treatment with PMA alone greatly enhanced ROS production in NADPH oxidase‐sufficient cells by two‐ to threefold, whereas a comparatively smaller change was seen in NADPH oxidase‐deficient cells. Together, these results demonstrate that TLR7/9 stimulation in human B cells promotes intracellular ROS production, which is linked to functional NADPH oxidase.
Enhanced cytokine production upon TLR7 or TLR9 stimulation of NADPH oxidase‐deficient B cells
To examine endosomal TLR7 and TLR9 activation further in the context of oxidase deficiency, the CGD‐derived and oxidase‐reconstituted B cells were stimulated with R848 or CpG in the presence or absence of PMA for 24 hr. Cell culture supernatants were assayed for cytokines IL‐6, IL‐10 and TNF‐α by ELISA. In the absence of stimuli, the p40phox‐deficient B‐cell line AR secreted about seven times more IL‐10 compared with ARp40 cells (Fig. 2a–c, left panel). Activation by TLR7 or TLR9 of AR cells resulted in >10‐ to 250‐fold increases in IL‐6 and IL‐10 release compared with ARp40 cells and a more modest 1·5‐fold increase in TNF‐α release. Studies with human B cells indicate that endosomal TLR signalling in response to ligands can be enhanced by PMA treatment in part through increased protein kinase C activation and the expression of intracellular proteases required for TLR functional maturation.31, 32 Treatment of p40phox‐deficient AR human B lymphoblasts with PMA alone led to an ~10‐fold increase of IL‐10 and IL‐6 but slightly less TNF‐α release than ARp40 cells (Fig. 2a–c, right panel). Treating B cells with PMA and TLR7 or TLR9 ligands enhanced IL‐6 and IL‐10 production fivefold to 100‐fold in the p40phox‐deficient cell line compared with the p40phox‐sufficient cell line, a trend not seen for TNF‐α production.
Figure 2.
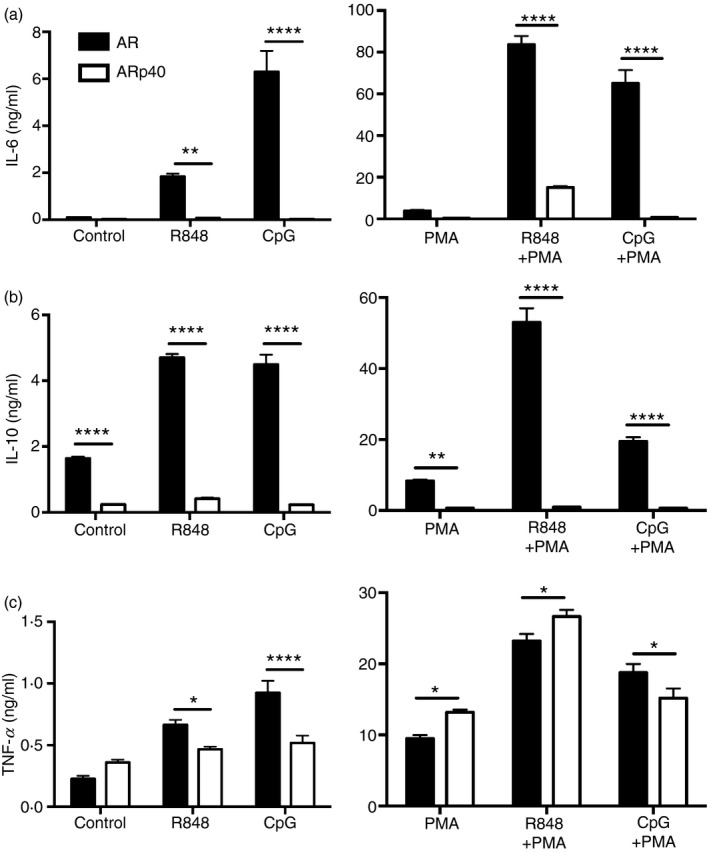
Enhanced secretion of select cytokines by human B lymphoblasts lacking functional p40phox upon Toll‐like receptor 7 (TLR7) or TLR9 activation. AR and ARp40 cell lines were treated with R848, CpG and/or PMA as described in the Materials and methods. After 24 hr, cell culture supernatants were harvested and screened for interleukin‐6 (IL‐6) (a), IL‐10 (b), and tumour necrosis factor‐α (TNF‐α) (c) by ELISA. Data are expressed as mean ± SEM from four independent experiments. Analysed by two‐way analysis of variance with Bonferroni's correction for multiple comparisons. *P ≤ 0·05, **P ≤ 0·01, ***P ≤ 0·001, ****P ≤ 0·0001.
To determine if the observed increases in endosomal TLR responsiveness were associated with oxidase deficiency overall, and were not simply a loss of functional p40phox, a similar analysis was conducted using KS, a p47phox‐deficient B‐cell line. Basal levels of IL‐6, IL‐10 and TNF‐α secretion were 2‐ to 10‐fold higher for KS cells when compared with the oxidase‐sufficient KSp47 cells (Fig. 3a–c, left panel). Cytokine release by KS cells in response to TLR7 ligands was consistently greater than observed with TLR9 ligands, whereas in all cases oxidase‐deficient KS cells secreted more cytokine with TLR stimulation (3‐ to 30‐fold increase) than KSp47 cells. Cytokine release in response to PMA alone was greater for p47phox‐deficient KS cells compared with oxidase‐sufficient KSp47 cells (Fig. 3a–c, right panel). Similarly, exposure of cells lacking functional p47phox to TLR ligands and PMA together resulted in greater cytokine release (3‐ to 60‐fold increase) than cells with wild‐type p47phox.
Figure 3.
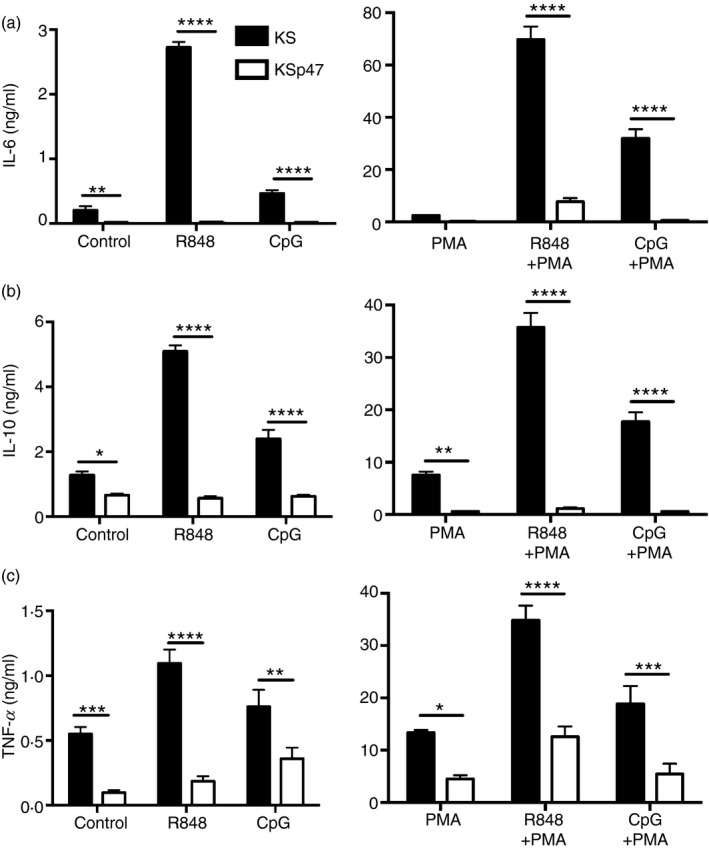
Absence of p47phox expression in human B lymphoblasts favoured higher cytokine secretion upon Toll‐like receptor 7 (TLR7)/TLR9 activation. KS and KSp47 cell lines were treated with R848, CpG and/or PMA as described in the Materials and methods. After 24 hr, cell culture supernatants were harvested and screened for interleukin‐6 (IL‐6) (a), IL‐10 (b), and tumour necrosis factor‐α (TNF‐α) (c) by ELISA. Data expressed as mean ± SEM from at least three independent experiments. Analysed by two‐way analysis of variance with Bonferroni's correction for multiple comparisons. *P ≤ 0·05, **P ≤ 0·01, ***P ≤ 0·001, ****P ≤ 0·0001.
Furthermore, analysis of both p40phox‐ and p47phox‐deficient B cells revealed higher basal levels of IFN‐α production compared with B cells with wild‐type oxidase expression (see Supplementary material, Fig. S1). Higher secretion of both pro‐inflammatory and anti‐inflammatory cytokines by p40phox‐deficient and p47phox‐deficient B cells in response to TLR7 or TLR9 ligands suggests that the reduced oxidase function of these cells may bias them towards hyperactivity to TLR‐induced signals overall. The detected increased cytokine production by the oxidase‐deficient CGD B‐cell lines was not the result of increased cell proliferation after TLR stimulation as determined by trypan blue exclusion (see Supplementary material, Fig. S2).
Enhanced TLR7 and TLR9 mRNA and protein expression in NADPH oxidase‐deficient B cells
Increased expression of TLR7 and TLR9 mRNA associated with oxidase deficiency could contribute to the heightened cytokine production detected in NADPH oxidase‐deficient cells upon TLR7 or TLR9 stimulation. Both AR and KS cells displayed ~1·5‐fold higher TLR7 mRNA and ~3‐fold higher levels of TLR9 mRNA than their reconstituted counterpart cells (Fig. 4a,b). To evaluate whether these changes in mRNA levels translated to higher levels of TLR proteins, cells were screened by Western blotting to detect full‐length (~120 000 MW) TLR7 and TLR9 (Fig. 4c–f) proteins. NADPH oxidase‐deficient cell lines expressed approximately three to four times more TLR7 and TLR9 protein than NADPH oxidase‐sufficient cell lines, as determined by imageJ analysis. These results demonstrated higher TLR7 and TLR9 mRNA and protein levels detected in NADPH oxidase‐deficient B cells compared with oxidase‐sufficient B cells.
Figure 4.
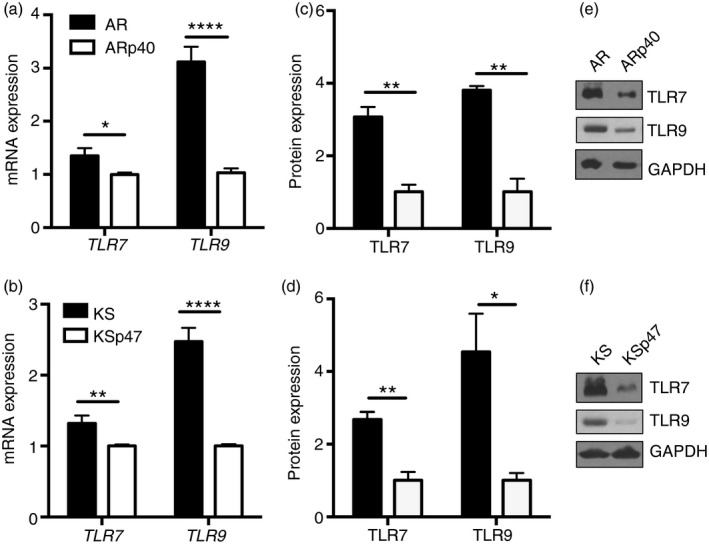
Elevated Toll‐like receptor 7 (TLR7) and TLR9 mRNA and protein expression in human B cells lacking functional NADPH oxidase. TLR7 and TLR9 transcript expression were measured by quantitative PCR. (a) AR mRNA expression is presented as a fold change relative to ARp40 mRNA expression for each respective gene (n = 3). (b) KS mRNA expression is presented as a fold change relative to KSp47 mRNA expression for each respective gene (n = 6). AR and KS cell lysates were immunoblotted to detect full‐length TLR7, full‐length TLR9, and GAPDH proteins (e,f) followed by densitometry to quantify protein expression. TLR protein levels were expressed as a ratio to GAPDH and then normalized to protein levels of ARp40 (c) and KSp47 (d), which were set to one (n = 3). Data expressed as mean ± SEM. Analysed by multiple t‐tests. *P ≤ 0·05, **P ≤ 0·01, ****P ≤ 0·0001.
MyD88 mRNA and protein expression in B cells was not altered by NADPH oxidase deficiency
To see whether downstream effectors impact endosomal TLR hyper‐activation in NADPH oxidase‐deficient human B cells, mRNA and protein levels of the common adaptor for TLR7 and TLR9, MyD88 were examined. There was a slight overall increase in both mRNA and protein expression of MyD88 in ARp40 compared with oxidase‐deficient AR cells (Fig. 5a,b). In contrast, NADPH oxidase‐deficient KS expressed slightly higher mRNA and protein expression of MyD88 than KSp47 cells (Fig. 5c,d). Levels of MyD88 expression did not change with TLR7 or PMA stimulation. These results suggest that changes in MyD88 mRNA and protein expression were not linked to loss of oxidase function in CGD B cells.
Figure 5.
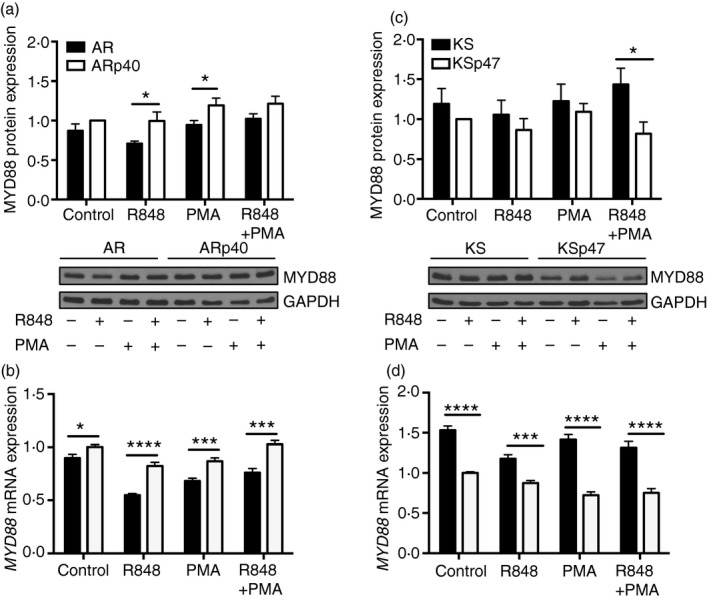
MyD88 mRNA and protein expression was independent of NADPH oxidase function in human B lymphoblasts. Cells were treated with R848 and/or PMA for 24 hr. AR and KS cell lysates were immunoblotted to detect MyD88 and GAPDH proteins followed by densitometry to quantify protein expression. MyD88 protein levels were expressed as a ratio to GAPDH and then normalized to protein levels of untreated ARp40 (a) and KSp47 (c) for each blot. Data expressed as mean ± SEM from at least five independent experiments. *P ≤ 0·05. MYD88 mRNA expression was measured by quantitative PCR after cells were similarly treated with Toll‐like receptor 7 (TLR7) ligand and PMA. MYD88 mRNA expression is presented as a fold change of mRNA expression of untreated ARp40 (b) or untreated KSp47 (d) (n = 3). Data expressed as mean ± SEM. Analysed by multiple t‐tests. ***P ≤ 0·001, ****P ≤ 0·0001.
Downstream TLR signalling p38 MAPK was hyper‐phosphorylated in NADPH oxidase‐deficient B cells
TLR7 and TLR9 signalling in B cells can lead to the phosphorylation of p38 MAPK, which promotes cytokine expression and secretion. Phospho‐p38 protein levels were increased in the oxidase‐deficient cells compared with the B cells reconstituted with wild‐type oxidase subunits, whereas total p38 MAPK protein expression in cells was consistent regardless of the functional status of the oxidase (Fig. 6). Interestingly, phospho‐p38 protein was detected even in unstimulated B cells from the patients with CGD, consistent with hyperactivity of the p38 pathway in these cells. Overall, p38 phosphorylation in the oxidase‐deficient cells appeared to be ~1·5‐ to 2‐fold more than in the reconstituted cells with a greater difference seen between the p47phox‐deficient cells.
Figure 6.
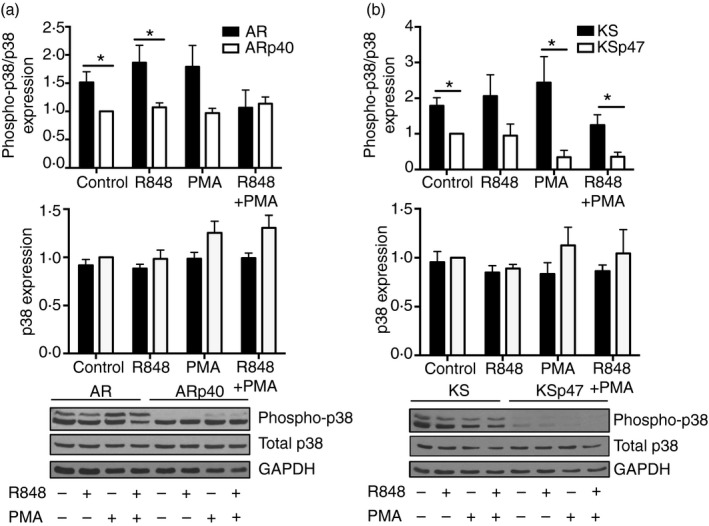
Human B lymphoblasts lacking functional oxidase expressed higher levels of phospho‐p38. Cells were treated with R848 and/or PMA for 24 hr. AR and KS cell lysates were immunoblotted to detect phospho‐p38, total p38 and GAPDH proteins followed by densitometry to quantify protein expression. Phospho‐p38 protein levels were expressed as a ratio to total p38. Total p38 protein levels were expressed as a ratio to GAPDH. Phospho‐p38 and total p38 protein ratios were then normalized to protein levels of untreated ARp40 (a) and KSp47 (b) for each blot. Data expressed as mean ± SEM from at least three independent experiments. Analysed by multiple t‐tests. *P ≤ 0·05.
Similar hyper‐phosphorylation of p38 MAPK was seen upon TLR activation of B cells from oxidase‐deficient mice, consistent with the results using B cells from patients with CGD. Naive murine B cells from wild‐type C57BL/6 mice and animals with targeted deletion of the oxidase catalytic subunit gp91phox (see Supplementary material, Fig. S3a) were examined for their response to TLR7 and TLR9 ligands. Secretion of IL‐6 and IL‐10 following TLR ligand exposure was up to twice as high from the oxidase‐deficient murine B cells than from wild‐type murine B cells (see Supplementary material, Fig. S3b, c), similar to results using human B lymphoblasts from patients with CGD. Unlike in human B‐cell lines, loss of NADPH oxidase expression in mice did not correlate with increased TLR7 and TLR9 mRNA expression (see Supplementary material, Fig. S3d). Oxidase‐deficient murine B cells expressed higher levels of phosphorylated p38 MAPK, but this was only seen upon TLR and PMA stimulation (see Supplementary material, Fig. S3e).
To see whether other downstream kinases were also constitutively activated in NADPH oxidase‐deficient B cells, human B‐cell lysates were monitored for phospho‐JNK and total JNK protein expression with or without TLR7 and PMA treatment. There was no consistent trend between JNK phosphorylation and NADPH oxidase deficiency (data not shown). To further evaluate whether phospho‐p38 MAPK was involved in hyper‐secretion of cytokines by NADPH oxidase‐deficient B cells, cells were treated with p38 MAPK activity inhibitor SB202190 before incubation with TLR7 or TLR9 ligands. Interleukin‐6 release by NADPH oxidase‐deficient B cells decreased ~5‐ to 20‐fold in response to TLR ligands and/or PMA when pre‐treated with the p38 MAPK activity inhibitor (Fig. 7). The inhibition of p38 activity also decreased TLR7/9 and/or PMA‐induced IL‐6 production by NAPDH oxidase‐sufficient B cells about twofold to sevenfold. These results suggest that in human B lymphoblasts, p38 MAPK signalling plays an important role in TLR7/9‐induced cytokine release, and that the hyper‐activation of this kinase in oxidase‐deficient B cells contributes to excessive cytokine production.
Figure 7.
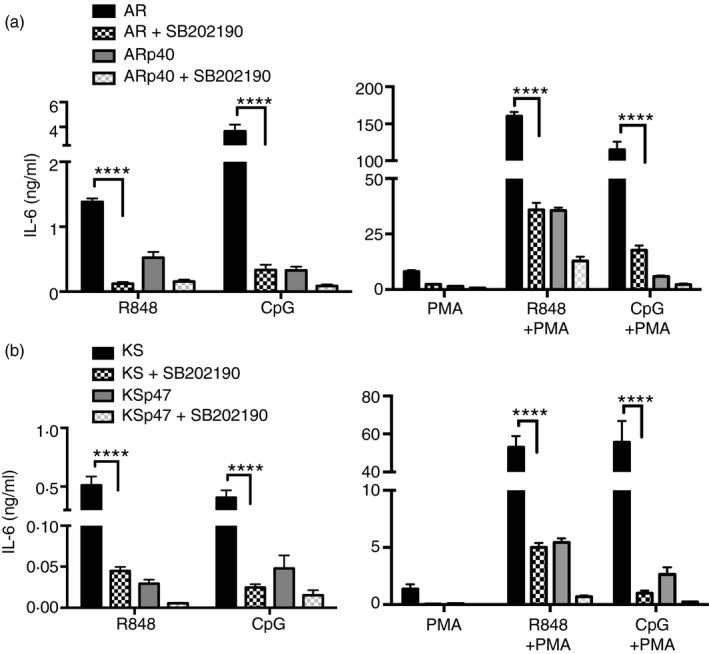
Inhibition of p38 mitogen‐activated protein kinase (MAPK) activity reduced cytokine secretion by NADPH oxidase‐deficient B cells. Cells were treated with SB202190 p38 MAPK activity inhibitor for 1 hr before 24 hr incubation with R848, CpG and/or PMA. Cell culture supernatants of AR (a) and KS (b) were screened for interleukin‐6 (IL‐6) by ELISA. Data expressed as mean ± SEM from three independent experiments. Analysed by two‐way analysis of variance with Bonferroni's correction for multiple comparisons. ****P ≤ 0·0001.
Discussion
Exposure of human B lymphoblasts to TLR7 or TLR9 ligands triggered NADPH oxidase‐derived intracellular ROS, and the impairment of this oxidase modulated B‐cell TLR expression and signal transduction. B cells from patients with CGD with dysfunctional p40phox or p47phox subunits of the oxidase displayed impaired ROS production when stimulated with endosomal TLR ligands or the phorbol ester PMA, an established promoter of oxidase activation. Production of ROS was restored in B cells from patients with CGD following cellular reconstitution with the appropriate wild‐type oxidase subunit. B cells derived from patients with CGD produced elevated levels of pro‐inflammatory and anti‐inflammatory cytokines upon TLR7 or TLR9 stimulation. This hyper‐response to TLR ligands was explained in part by the up‐regulation of TLR7 and TLR9 mRNA and protein expression in the oxidase‐deficient human B cells. Additionally, B cells with defects in oxidase function displayed higher phosphorylation of the downstream MAPK p38 compared with oxidase‐sufficient B cells. Exaggerated IL‐6 and IL‐10 production along with enhanced p38 MAPK phosphorylation were also seen upon TLR7 or TLR9 activation of naive B cells from gp91phox‐deficient mice compared with B cells from wild‐type mice. Signalling via this kinase contributed to cytokine release by the oxidase‐sufficient B cells, but was even more critical for the hyper‐response observed in the oxidase‐deficient human B cells.
Even though NADPH oxidase is recognized for its role in microbial killing following pathogen phagocytosis, loss of this oxidase also impacts multiple functions of B lymphocytes. Alterations in MHC class II antigen presentation were detected in B cells from patients with CGD and studies using oxidase‐deficient mice revealed changes in cell proliferation and antibody production in response to B‐cell receptor cross‐linking.6, 9, 13 In this study, upon TLR7 or TLR9 activation, both pro‐inflammatory and anti‐inflammatory cytokine secretion by B cells was greatly enhanced when NADPH oxidase was functionally impaired.
The patient‐derived B‐cell lines examined here have mutations in either the p40phox or p47phox subunits of the oxidase, each of which translocate to the membrane‐bound oxidase subunits during activation. The AR cell line expresses low levels of a single allele of p40phox with reduced function and lipid association.26 In this patient's neutrophils, mutations in p40phox altered oxidase activation by different stimuli and resulted in hyper‐inflammatory disease colitis. By contrast, mutations in the KS cell line disrupt expression of the p47phox protein, which is required for oxidase activation.27 Rodents with mutations and deficiencies in p47phox display a predisposition to autoimmune disorders and inflammation.33 Patients deficient in the catalytic gp91phox subunit of the oxidase as well as some of their first‐degree relatives are also more susceptible to autoimmunity.4, 34
Although each patient‐derived B‐cell line exhibited reduced ROS production, there were some differences in their responses to oxidase activation. The AR B‐cell line, which has mutations in p40phox, displayed significantly greater TLR or PMA‐induced production of IL‐6 and IL‐10 compared with AR cells reconstituted with wild‐type p40phox. TLR7/9 activation of these cells also promoted increased TNF‐α production but the addition of PMA led to cytokine release in the p40phox‐deficient cells compared with the oxidase‐sufficient ARp40 cells. By contrast, KS cells exposed to TLR7/9 ligands and/or PMA consistently produced higher levels of IL‐6, IL‐10 and TNF‐α. In addition to elevated cytokine release upon TLR7/9 stimulation, TLR7 and TLR9 gene and full‐length protein expression were also elevated in these oxidase‐deficient human B cells. Activation of these TLRs is dependent in part on a cleavage event within endosomal compartments. Detection of these cleaved receptors in B cells has proven difficult,31 although the increased levels of the full‐length receptor proteins detected here are consistent with the higher mRNA levels of these receptors in oxidase‐deficient human B cells.
For the current study, peripheral blood B cells from patients were transformed with Epstein–Barr virus to immortalize the cells. This transformation did not alter the loss of oxidase ROS production, or restoration of oxidase function by reconstitution of each cell with the appropriate wild‐type oxidase subunit. Reports are conflicting as to whether Epstein–Barr virus infection can impact TLR7 and TLR9 gene expression and activation in human B cells.35, 36 To examine the effects of oxidase loss on B‐cell function in primary cells without virus, TLR ligand activation of naive B cells from oxidase‐deficient gp91phox knock‐out and wild‐type mice was analysed. Exaggerated production of IL‐6 and IL‐10 was detected upon TLR7 or TLR9 stimulation of the oxidase‐deficient murine B cells compared with their wild‐type counterparts. The enhanced secretion of both pro‐inflammatory and anti‐inflammatory cytokines is consistent with dysregulated cell signalling and cytokine release. Yet, unlike human B lymphoblasts from patients with CGD, the increased expression of TLR7 and TLR9 mRNA was not detected when comparing oxidase‐deficient and oxidase‐sufficient mouse B cells. This may reflect differences in the regulation of human and mouse TLR gene expression, as has been seen with TLR4.37 Alternatively, viral transformation of the B‐cell lines derived from patients with CGD may increase TLR7 and TLR9 gene expression, while restoration of oxidase promotes down‐regulation of these receptors.
Parallel to heightened secretion of cytokines by NADPH oxidase‐deficient B cells, p38 MAPK, which plays a pivotal role downstream of TLR7 and TLR9 signalling, was hyper‐phosphorylated in patient B cells compared with cells with restored oxidase function. The importance of hyper‐activation of the p38 kinase in oxidase‐deficient human B cells was demonstrated using a p38 MAPK activity inhibitor. Inhibition of p38 MAPK activity disrupted IL‐6 production by B cells after TLR7 or TLR9 ligand treatment of both oxidase‐deficient and oxidase‐sufficient human B cells. These results indicate that p38 MAPK signalling is an important contributor to cytokine production by B cells in response to endosomal TLR7 and TLR9 receptors. Inhibitor treatment of oxidase‐deficient B cells often reduced cytokine release below that seen in oxidase‐sufficient cells. Together, these data suggest that p38 MAPK signalling is probably the dominant pathway leading to cytokine overproduction in oxidase‐deficient B cells.
Although CGD is classified as an inherited immunodeficiency with patients suffering from frequent and recurring microbial infections, reports have documented dysregulated cellular immune response and increased autoimmune susceptibility in a percentage of patients with CGD as well as oxidase‐deficient mice.4, 24, 25, 33, 38 Total peripheral blood mononuclear cells isolated from gp91phox‐deficient mice and patients produce increased levels of cytokines upon TLR2, TLR4 or TLR9 stimulation.39 The release of pro‐inflammatory and anti‐inflammatory cytokines was more elevated in whole blood from patients with CGD than from healthy donors after Aspergillus fumigatus exposure.40 Neutrophils isolated from gp91phox‐deficient patients display up‐regulated expression of several pro‐inflammatory genes constitutively and have an elevated release of pro‐inflammatory cytokines upon TLR4 activation.41, 42 Monocytes isolated from patients with CGD also had up‐regulation of many inflammatory genes constitutively and upon TLR2 or TLR4 stimulation.43 Similarly, dendritic cells from NADPH oxidase‐deficient mice secrete heightened levels of cytokine in response to TLR4 stimulation or microbial activation.14, 44 The current study is among the first to describe a hyper‐inflammatory phenotype in CGD B cells, which is further enhanced by endosomal TLR activation. Remarkably, elevated p38 phosphorylation and cytokine production was seen here even in unstimulated oxidase‐deficient B cells, consistent with the observation that many CGD patients display constitutive hyper‐inflammation and granuloma development even in the absence of infection.1, 2
The molecular basis responsible for the hyper‐inflammation observed in some patients with CGD and animal models has been difficult to pinpoint. Humans and mice with NADPH oxidase deficiency frequently show increased susceptibility to autoimmune disorders. However, the hyper‐inflammation associated with oxidase inactivation can confer some benefit as gp91phox‐deficient mice more effectively clear and recover from influenza virus infections.45, 46 It has been proposed that oxidase‐derived H2O2 potentially has an inhibitory role in intracellular signalling, including the inhibition of p38 MAPK activation in dendritic cells.44 The concept that ROS intermediates act as negative regulators of p38 kinase may be consistent with the increased phosphorylation of this kinase in B cells from patients with CGD observed in this study.
Patients with CGD suffer from recurring microbial infections during their lifetime with many fatalities attributed to species in the Aspergillus genus.1, 2 In Aspergillus infections, TLR7 and TLR9 activation have been reported in neutrophils.47, 48 Patients with CGD, and some of their first‐degree relatives, are more susceptible to systemic lupus erythematosus and inflammatory bowel disease, in which dysregulation of TLR7 and TLR9 signalling has been observed.21, 22, 23, 24, 49 Furthermore, elevated IFN‐α is frequently seen in the serum of patients with systemic lupus erythematosus,50 which parallels the hyper‐secretion of IFN‐α detected here with oxidase‐deficient B cells. Together, these results further suggest an important role for B cells and TLR7/9 signalling in the hyper‐inflammatory response and increased autoimmune susceptibility detected in some patients with CGD.
Disclosures
The authors have no financial or commercial conflicts of interest.
Supporting information
Figure S1. NADPH oxidase deficiency in B lymphoblasts led to more interferon‐α (IFN‐α) secretion. Cell culture supernatants were collected after a 24‐hr incubation in fresh media and screened for IFN‐α in duplicate by ELISA. Data are expressed as a mean ± SEM from two independent experiments.
Figure S2. Cell survival after Toll‐like receptor 7 (TLR7) or TLR9 activation was independent of NADPH oxidase function. Cells were seeded at 1 × 106/ml (dashed line) and treated with of R848, CpG and/or PMA as described in the Materials and methods. After 24 hr, cell number was determined by trypan blue exclusion. Data are expressed as mean ± SEM and represent at least three independent experiments.
Figure S3. Absence of gp91phox in murine splenic naive B cells led to an increased response to Toll‐like receptor 7/9 (TLR7/9) ligands. (a) Naive B cells were isolated from the spleens of age‐ and sex‐matched Cybb −/− and C57BL/6 mice, lysed and immunoblotted for the heavily glycosylated gp91phox and GAPDH proteins. Supernatants of splenic murine B cells were screened for interleukin‐6 (IL‐6) (b) and IL‐10 (c) after a 24‐hr treatment with R848, CpG and/or PMA as described in the Materials and methods. Data are expressed as a mean ± SEM from at least three independent experiments and analysed by two‐way analysis of variance with Bonferroni's correction for multiple comparisons. ****P ≤ 0·0001. (d) Cybb −/− tlr7 and tlr9 gene expression presented as a fold change relative to the respective C57BL/6 gene expression. Data expressed as a mean ± SEM from six independent experiments. (e) Murine splenic B cells stimulated for 24 hr with both PMA and TLR7 or TLR9 ligand were lysed and immunoblotted to detect phospho‐p38, total p38 and GAPDH proteins followed by densitometry to quantify protein expression. Phospho‐p38 protein levels were expressed as a ratio to total p38. Total p38 protein levels were expressed as a ratio to GAPDH. Phospho‐p37 and total p38 protein ratios were normalized to protein levels of B cells from C57BL/6 mice treated with PMA alone. Data shown are representative of at least two independent experiments using at least two pooled mice in each group.
Acknowledgements
JSB and SM designed the study; MD and BV provided cell lines and edited the manuscript; SM performed the experiments and analysed the data; JSB and SM wrote the paper. We would like to thank Lynette Guindon, Liliana Pérez, Gail Gardiner, Sarah Deffit, Jean Liu and Crystal Walline for sharing techniques, reagents, guiding experimentation and providing useful ideas and suggestions. This work was supported by NIH grant RO1AI07965 and Children's Discovery Institute at Washington University and St. Louis Children's Hospital.
References
- 1. Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Baltimore 2000; 79:170–200. [DOI] [PubMed] [Google Scholar]
- 2. Bylund J, Goldblatt D, Speert DP. Chronic granulomatous disease: from genetic defect to clinical presentation. Adv Exp Med Biol 2005; 568:67–87. [DOI] [PubMed] [Google Scholar]
- 3. Rosenzweig SD. Inflammatory manifestations in chronic granulomatous disease (CGD). J Clin Immunol 2008; 28:67–72. [DOI] [PubMed] [Google Scholar]
- 4. Gardiner GJ, Deffit SN, McLetchie S, Perez L, Walline CC, Blum JS. A role for NADPH oxidase in antigen presentation. Front Immunol 2013; 4:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jackson SH, Devadas S, Kwon J, Pinto LA, Williams MS. T cells express a phagocyte‐type NADPH oxidase that is activated after T cell receptor stimulation. Nat Immunol 2004; 5:818–27. [DOI] [PubMed] [Google Scholar]
- 6. Richards SM, Clark EA. BCR‐induced superoxide negatively regulates B‐cell proliferation and T‐cell‐independent type 2 Ab responses. Eur J Immunol 2009; 39:3395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kovacs I, Horvath M, Lanyi A, Petheo GL, Geiszt M. Reactive oxygen species‐mediated bacterial killing by B lymphocytes. J Leukoc Biol 2015; 97:1–5. [DOI] [PubMed] [Google Scholar]
- 8. Cachat J, Deffert C, Hugues S, Krause KH. Phagocyte NADPH oxidase and specific immunity. Clin Sci (Lond) 2015; 128:635–48. [DOI] [PubMed] [Google Scholar]
- 9. Wheeler ML, Defranco AL. Prolonged production of reactive oxygen species in response to B cell receptor stimulation promotes B cell activation and proliferation. J Immunol 2012; 189:4405–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moir S, De Ravin SS, Santich BH, Kim JY, Posada JG, Ho J, et al Humans with chronic granulomatous disease maintain humoral immunologic memory despite low frequencies of circulating memory B cells. Blood 2012; 120:4850–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bleesing JJ, Souto‐Carneiro MM, Savage WJ, Brown MR, Martinez C, Yavuz S, et al Patients with chronic granulomatous disease have a reduced peripheral blood memory B cell compartment. J Immunol 2006; 176:7096–103. [DOI] [PubMed] [Google Scholar]
- 12. Matharu K, Zarember KA, Marciano BE, Kuhns DB, Spalding C, Garofalo M, et al B‐cell activating factor (BAFF) is elevated in chronic granulomatous disease. Clin Immunol 2013; 148:258–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Crotzer VL, Matute JD, Arias AA, Zhao H, Quilliam LA, Dinauer MC, et al Cutting edge: NADPH oxidase modulates MHC class II antigen presentation by B cells. J Immunol 2012; 189:3800–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. George‐Chandy A, Nordstrom I, Nygren E, Jonsson IM, Postigo J, Collins LV, et al Th17 development and autoimmune arthritis in the absence of reactive oxygen species. Eur J Immunol 2008; 38:1118–26. [DOI] [PubMed] [Google Scholar]
- 15. Davis Volk AP, Moreland JG. ROS‐containing endosomal compartments: implications for signaling. Methods Enzymol 2014; 535:201–24. [DOI] [PubMed] [Google Scholar]
- 16. Armant MA, Fenton MJ. Toll‐like receptors: a family of pattern‐recognition receptors in mammals. Genome Biol 2002; 3:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brencicova E, Diebold SS. Nucleic acids and endosomal pattern recognition: how to tell friend from foe? Front Cell Infect Microbiol 2013; 3:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Laroux FS, Romero X, Wetzler L, Engel P, Terhorst C. Cutting edge: MyD88 controls phagocyte NADPH oxidase function and killing of gram‐negative bacteria. J Immunol 2005; 175:5596–600. [DOI] [PubMed] [Google Scholar]
- 19. Vulcano M, Dusi S, Lissandrini D, Badolato R, Mazzi P, Riboldi E, et al Toll receptor‐mediated regulation of NADPH oxidase in human dendritic cells. J Immunol 2004; 173:5749–56. [DOI] [PubMed] [Google Scholar]
- 20. Marshak‐Rothstein A. Toll‐like receptors in systemic autoimmune disease. Nat Rev Immunol 2006; 6:823–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chauhan SK, Singh VV, Rai R, Rai M, Rai G. Distinct autoantibody profiles in systemic lupus erythematosus patients are selectively associated with TLR7 and TLR9 upregulation. J Clin Immunol 2013; 33:954–64. [DOI] [PubMed] [Google Scholar]
- 22. Celhar T, Magalhaes R, Fairhurst AM. TLR7 and TLR9 in SLE: when sensing self goes wrong. Immunol Res 2012; 53:58–77. [DOI] [PubMed] [Google Scholar]
- 23. Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, et al Control of toll‐like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity 2007; 27:801–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Winkelstein JA, Marino MC, Johnston RB Jr, Boyle J, Curnutte J, Gallin JI, et al Chronic granulomatous disease. Report on a national registry of 368 patients. Baltimore 2000; 79:155–69. [DOI] [PubMed] [Google Scholar]
- 25. Campbell AM, Kashgarian M, Shlomchik MJ. NADPH oxidase inhibits the pathogenesis of systemic lupus erythematosus. Sci Transl Med 2012; 4:157ra41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Matute JD, Arias AA, Wright NA, Wrobel I, Waterhouse CC, Li XJ, et al A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40 phox and selective defects in neutrophil NADPH oxidase activity. Blood 2009; 114:3309–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Volpp BD, Lin Y. In vitro molecular reconstitution of the respiratory burst in B lymphoblasts from p47‐phox‐deficient chronic granulomatous disease. J Clin Invest 1993; 91:201–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bjorgvinsdottir H, Ding C, Pech N, Gifford MA, Li LL, Dinauer MC. Retroviral‐mediated gene transfer of gp91phox into bone marrow cells rescues defect in host defense against Aspergillus fumigatus in murine X‐linked chronic granulomatous disease. Blood 1997; 89:41–8. [PubMed] [Google Scholar]
- 29. Poovassery JS, Vanden Bush TJ, Bishop GA. Antigen receptor signals rescue B cells from TLR tolerance. J Immunol 2009; 183:2974–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vene R, Delfino L, Castellani P, Balza E, Bertolotti M, Sitia R, et al Redox remodeling allows and controls B‐cell activation and differentiation. Antioxid Redox Signal 2010; 13:1145–55. [DOI] [PubMed] [Google Scholar]
- 31. Hipp MM, Shepherd D, Gileadi U, Aichinger MC, Kessler BM, Edelmann MJ, et al Processing of human toll‐like receptor 7 by furin‐like proprotein convertases is required for its accumulation and activity in endosomes. Immunity 2013; 39:711–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Loegering DJ, Lennartz MR. Protein kinase C and toll‐like receptor signaling. Enzyme Res 2011; 2011:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hultqvist M, Olofsson P, Holmberg J, Backstrom BT, Tordsson J, Holmdahl R. Enhanced autoimmunity, arthritis, and encephalomyelitis in mice with a reduced oxidative burst due to a mutation in the Ncf1 gene. Proc Natl Acad Sci USA 2004; 101:12646–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Battersby AC, Cale AM, Goldblatt D, Gennery AR. Clinical manifestations of disease in X‐linked carriers of chronic granulomatous disease. J Clin Immunol 2013; 33:1276–84. [DOI] [PubMed] [Google Scholar]
- 35. Younesi V, Nikzamir H, Yousefi M, Khoshnoodi J, Arjmand M, Rabbani H, et al Epstein Barr virus inhibits the stimulatory effect of TLR7/8 and TLR9 agonists but not CD40 ligand in human B lymphocytes. Microbiol Immunol 2010; 54:534–41. [DOI] [PubMed] [Google Scholar]
- 36. Martin HJ, Lee JM, Walls D, Hayward SD. Manipulation of the toll‐like receptor 7 signaling pathway by Epstein–Barr virus. J Virol 2007; 81:9748–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bekeredjian‐Ding I, Jego G. Toll‐like receptors – sentries in the B‐cell response. Immunology 2009; 128:311–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rieber N, Hector A, Kuijpers T, Roos D, Hartl D. Current concepts of hyperinflammation in chronic granulomatous disease. Clin Dev Immunol 2012; 2012:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bylund J, MacDonald KL, Brown KL, Mydel P, Collins LV, Hancock RE, et al Enhanced inflammatory responses of chronic granulomatous disease leukocytes involve ROS‐independent activation of NF‐κB. Eur J Immunol 2007; 37:1087–96. [DOI] [PubMed] [Google Scholar]
- 40. Warris A, Netea MG, Wang JE, Gaustad P, Kullberg BJ, Verweij PE, et al Cytokine release in healthy donors and patients with chronic granulomatous disease upon stimulation with Aspergillus fumigatus . Scand J Infect Dis 2003; 35:482–7. [DOI] [PubMed] [Google Scholar]
- 41. Kobayashi SD, Voyich JM, Braughton KR, Whitney AR, Nauseef WM, Malech HL, et al Gene expression profiling provides insight into the pathophysiology of chronic granulomatous disease. J Immunol 2004; 172:636–43. [DOI] [PubMed] [Google Scholar]
- 42. Hatanaka E, Carvalho BT, Condino‐Neto A, Campa A. Hyperresponsiveness of neutrophils from gp 91phox deficient patients to lipopolysaccharide and serum amyloid A. Immunol Lett 2004; 94:43–6. [DOI] [PubMed] [Google Scholar]
- 43. Brown KL, Bylund J, MacDonald KL, Song‐Zhao GX, Elliott MR, Falsafi R, et al ROS‐deficient monocytes have aberrant gene expression that correlates with inflammatory disorders of chronic granulomatous disease. Clin Immunol 2008; 129:90–102. [DOI] [PubMed] [Google Scholar]
- 44. Jendrysik MA, Vasilevsky S, Yi L, Wood A, Zhu N, Zhao Y, et al NADPH oxidase‐2 derived ROS dictates murine DC cytokine‐mediated cell fate decisions during CD4 T helper‐cell commitment. PLoS One 2011; 6:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. To EE, Broughton BR, Hendricks KS, Vlahos R, Selemidis S. Influenza A virus and TLR7 activation potentiate NOX2 oxidase‐dependent ROS production in macrophages. Free Radic Res 2014; 48:940–7. [DOI] [PubMed] [Google Scholar]
- 46. Vlahos R, Stambas J, Bozinovski S, Broughton BR, Drummond GR, Selemidis S. Inhibition of Nox2 oxidase activity ameliorates influenza A virus‐induced lung inflammation. PLoS Pathog 2011; 7:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bellocchio S, Moretti S, Perruccio K, Fallarino F, Bozza S, Montagnoli C, et al TLRs govern neutrophil activity in aspergillosis. J Immunol 2004; 173:7406–15. [DOI] [PubMed] [Google Scholar]
- 48. Mansour MK, Tam JM, Vyas JM. The cell biology of the innate immune response to Aspergillus fumigatus . Ann NY Acad Sci 2012; 1273:78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Berkowitz D, Peri R, Lavy A, Kessel A. Increased Toll‐like receptor 9 expression by B cells from inflammatory bowel disease patients. Hum Immunol 2013; 74:1519–23. [DOI] [PubMed] [Google Scholar]
- 50. Pascual V, Farkas L, Banchereau J. Systemic lupus erythematosus: all roads lead to type I interferons. Curr Opin Immunol 2006; 18:676–82. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. NADPH oxidase deficiency in B lymphoblasts led to more interferon‐α (IFN‐α) secretion. Cell culture supernatants were collected after a 24‐hr incubation in fresh media and screened for IFN‐α in duplicate by ELISA. Data are expressed as a mean ± SEM from two independent experiments.
Figure S2. Cell survival after Toll‐like receptor 7 (TLR7) or TLR9 activation was independent of NADPH oxidase function. Cells were seeded at 1 × 106/ml (dashed line) and treated with of R848, CpG and/or PMA as described in the Materials and methods. After 24 hr, cell number was determined by trypan blue exclusion. Data are expressed as mean ± SEM and represent at least three independent experiments.
Figure S3. Absence of gp91phox in murine splenic naive B cells led to an increased response to Toll‐like receptor 7/9 (TLR7/9) ligands. (a) Naive B cells were isolated from the spleens of age‐ and sex‐matched Cybb −/− and C57BL/6 mice, lysed and immunoblotted for the heavily glycosylated gp91phox and GAPDH proteins. Supernatants of splenic murine B cells were screened for interleukin‐6 (IL‐6) (b) and IL‐10 (c) after a 24‐hr treatment with R848, CpG and/or PMA as described in the Materials and methods. Data are expressed as a mean ± SEM from at least three independent experiments and analysed by two‐way analysis of variance with Bonferroni's correction for multiple comparisons. ****P ≤ 0·0001. (d) Cybb −/− tlr7 and tlr9 gene expression presented as a fold change relative to the respective C57BL/6 gene expression. Data expressed as a mean ± SEM from six independent experiments. (e) Murine splenic B cells stimulated for 24 hr with both PMA and TLR7 or TLR9 ligand were lysed and immunoblotted to detect phospho‐p38, total p38 and GAPDH proteins followed by densitometry to quantify protein expression. Phospho‐p38 protein levels were expressed as a ratio to total p38. Total p38 protein levels were expressed as a ratio to GAPDH. Phospho‐p37 and total p38 protein ratios were normalized to protein levels of B cells from C57BL/6 mice treated with PMA alone. Data shown are representative of at least two independent experiments using at least two pooled mice in each group.