

Abstract
Nontypeable Haemophilus influenzae (NTHi) is associated with chronic otitis media (COM). In this study, we generated a murine model of COM by using eustachian tube (ET) obstruction and NTHi (107 CFU) inoculation into the tympanic bulla, and we investigated the relationship between regulatory T cells (Treg) and chronic inflammation in the middle ear. Middle ear effusions (MEEs) and middle ear mucosae (MEM) were collected at days 3 and 14 and at 1 and 2 months after inoculation. Untreated mice served as controls. MEEs were used for bacterial counts and to measure the concentrations of cytokines. MEM were collected for histological evaluation and flow cytometric analysis. Inflammation of the MEM was prolonged throughout this study, and the incidence of NTHi culture-positive MEE was 38% at 2 months after inoculation. The levels of interleukin-1β (IL-β), tumor necrosis factor alpha, IL-10, and transforming growth factor β were increased in the middle ear for up to 2 months after inoculation. CD4+ CD25+ FoxP3+ Treg accumulated in the middle ear, and the percentage of Treg in the MEM increased for up to 2 months after inoculation. Treg depletion induced a 99.9% reduction of bacterial counts in MEEs and also significantly reduced the ratio of NTHi culture-positive MEE. The levels of these cytokines were also reduced in MEEs. In summary, we developed a murine model of COM, and our findings indicate that Treg confer infectious tolerance to NTHi in the middle ear.
INTRODUCTION
Chronic otitis media (COM), including OM with effusion (OME) and recurrent OM, is characterized by clinical evidence of OM and resolution of middle ear effusion (MEE) between episodes. OME represents a spectrum of chronic disease states, ranging from serous to mucoid OM, and is associated with hearing loss, delayed speech development, permanent middle ear damage, and mucosal changes (1). Although COM remains a common problem in pediatric populations, its etiology and pathogenesis are not fully understood. Eustachian tube (ET) dysfunction is considered to be the underlying pathophysiologic event leading to most cases of OME in children. The most frequent causes of ET dysfunction include upper respiratory infections, adenoid tissue hypertrophy, and cleft palate (2, 3). In bacterial infection of COM, the majority of cases are caused by Gram-negative bacteria, whereas in acute OM, Gram-positive bacteria are also frequently isolated (4). Lipopolysaccharides are a component of Gram-negative bacteria that have been detected in human MEEs (5), and their levels are significantly higher in children with chronic OME than in children with acute OME (6). Lipopolysaccharides alone have also been shown to induce mucosal inflammation with the accumulation of effusion in the middle ears of animal models (7, 8). Only 30% of MEEs from COM children yielded an unequivocally positive culture for aerobic bacteria (9). According to previous reports, components from Gram-negative bacteria are thought to induce COM in humans. However, recently, COM was reportedly associated with a persistent bacterial infection, and biofilm structures were identified in 92% of middle ear mucosa (MEM) biopsy specimens from children with COM (10). On the basis of these reports, ET dysfunction and a persistent bacterial infection are closely associated with the pathogenesis of COM. Previously, we established a murine model of COM with effusion by ET blockage and endotoxin inoculation into the bulla (11). COM mice produced by ET blockage showed persistent serous MEE with mild inflammatory responses. COM mice produced by ET blockage and endotoxin inoculation were associated with moderate inflammation and the production of mucoid MEE accompanied by histological changes with inflammatory cell infiltration, especially lymphocytes, and cytokine production in the middle ear cavity. However, this mouse model of COM does not reflect the recent findings of COM in children. Therefore, we need to examine a COM model with ET dysfunction and persistent bacterial infection to investigate the pathogenesis of COM.
Nontypeable Haemophilus influenzae (NTHi) is thought to be the chief pathogen in both acute OM and COM (12). In addition, NTHi is an important pulmonary bacterial pathogen associated with recurrent and persistent lower respiratory tract infections in patients with chronic obstructive pulmonary disease, which affects 16 million people and is the fourth leading cause of death in the United States (13). NTHi is a commensal to opportunistic pathogen that is highly adapted to the human airway (14). NTHi strains can persist within the airway for lengthy periods, during which their carriage is mostly asymptomatic in healthy individuals (15). However, when host mucosal clearance mechanisms are compromised or impaired, NTHi can cause an array of airway infections (14). Recently, biofilm formation by NTHi has been defined for persistence and pathogenicity in chronic airway infections to some degree (16). However, in cases of persisting NTHi infection in the upper and lower airways, little is known about the immunological responses from the viewpoint of the host immune reaction against NTHi.
Regulatory T cells (Treg), also known as suppressor T cells, consist of a specific subpopulation of cells that functionally suppress the activation of the immune system and maintain immune tolerance to self-antigens (17). The gene coding for the Treg cell transcription factor forkhead box P3 (FoxP3) is a master gene for the differentiation and function of Treg (18, 19). Treg consist of 2 major subsets: natural Treg and adaptive or induced Treg. Natural Treg derived from the thymus are considered the classic Treg, while inducible Treg develop in the periphery in response to self or tumor antigens by converting naive CD4+ T cells into Treg (20). Normally, when these two subsets of Treg are depleted, various organ-specific autoimmune diseases occur spontaneously (17). Recent studies, however, indicate that Treg can also be activated and expanded against bacterial, viral, and parasite antigens in vivo (21). Such pathogen-specific Treg are known to prevent infection-induced immunopathology but may also increase the load of infection and prolong pathogen persistence by suppressing protective immune responses. Clarifying whether Treg play a protective or detrimental role in the immune cellular response during upper respiratory tract infection with NTHi could help establish a treatment strategy for COM.
In this study, we explored a murine model of COM with persistent NTHi infection by using ET blockage and NTHi inoculation and investigated the immune cellular response, specifically Treg, and also the effect of the depletion of Treg on chronic middle ear infection.
MATERIALS AND METHODS
Animals.
BALB/c mice were purchased from Kyudo Japan (Fukuoka, Japan). All mice were maintained in a pathogen-free facility until they were 5 weeks old, at which time they were used for the experiments. A total of 136 animals were used in this study. All experiments were approved by the Committee on Animal Experiments of Oita University.
Middle ear challenge with live NTHi and induction of experimental COM.
Strain 76 of NTHi, which was isolated from the nasopharynx of a patient with OME at Oita University, was used for the middle ear challenge, and the bacterial suspension was injected into the right tympanic cavity, as described previously (11). NTHi was grown on chocolate agar at 37°C under 5% CO2 for 16 h. The bacterial concentration was determined by optical density at a wavelength of 600 nm, and then a bacterial suspension was prepared to a concentration of 1.0 × 109 CFU/ml in phosphate-buffered saline (PBS) and stored on ice. The bacterial concentration was confirmed by colony counting after an overnight incubation. In brief, with the animal under anesthesia induced with 0.6 mg/ml ketamine hydrochloride, an incision was made in the submandibular skin, the right inferior bulla was exposed, the pharyngeal part of the ET near the tympanic bulla was cut, and a gelatin sponge was inserted into the ET (ET blockage). Subsequently, 2 microholes were made in the bulla with a 27-gauge needle. A micropipette was inserted into one of the holes, and 10 μl (1.0 × 107 CFU) of live NTHi suspension was inoculated slowly. Mice without both ET blockage and NTHi inoculation served as controls based on our previous study (11). The mice were monitored otomicroscopically to confirm the presence of MEE and tympanic membrane changes. Untreated mice were used as controls according to the same procedure for these experiments. Another set of model mice was made for flow cytometric analysis and real-time reverse transcription-PCR (RT-PCR).
Collection of samples and treatment.
On day 3, day 14, 1 month, and 2 months after injection, 7 to 10 mice from each group were killed under deep anesthesia by intraperitoneal administration of a pentobarbital solution. Untreated mice were also killed as controls. The mice were monitored otomicroscopically to confirm the presence of MEE and tympanic membrane (TM) changes. The characteristics of MEE were recorded and graded by increasing order of inflammation according to the severity of COM, as follows: 0, gray and translucent TM without MEE (normal); 1, gray and opaque TM with serous MEE or mucoid MEE; 2, yellow and opaque TM with purulent MEE; and 3, opaque TM with pultaceous MEE. These grading scales are referred to and modified from Giebink et al. (22). MEE samples were obtained by myringotomy at the time of decapitation, and the middle ears were washed with 200 μl physiologic saline. MEEs were diluted serially, and the diluted samples were plated on chocolate agar. Bacterial colonies were counted after an overnight incubation to measure the rate of NTHi clearance from the middle ear. NTHi was identified by standard bacteriologic techniques, including Gram staining and determination of their V and X growth factor requirements. MEEs were centrifuged, and the supernatants were collected and then stored at −80°C for cytokine analysis by enzyme-linked immunosorbent assay (ELISA; described below).
Histological preparation.
After the mice were euthanized and MEE samples were collected, they were fixed by intracardiac perfusion with 10% neutral buffered formalin or periodate lysine paraformaldehyde for histologic evaluation. The mice were then decapitated, and their heads were immersed in the same fixative used for perfusion for 6 h and decalcified in 0.12 M EDTA (pH 7.4) for 2 weeks at 4°C. For hematoxylin-eosin (H&E) and immunohistochemical staining, tissues were dehydrated through a graded series of ethanol, cleared in xylene, and embedded in paraffin.
Histological evaluation.
Serial paraffin sections (6 μm) that contained the tympanic bulla were prepared. Mucosal thickness and the number of inflammatory cells in the MEM were measured using ImageJ, a public domain, Java-based image processing program developed at the National Institutes of Health (Bethesda, MD; http://rsbweb.nih.gov/ij/). Sections from the largest middle ear bulla around the tympanic orifice were observed by light microscopy, and histological images (magnification ×400) were captured; mucosal thickness and the number of inflammatory cells in the MEM were measured and analyzed at the anterior, posterior, medial, and lateral parts of the bulla. Three mice were used for this experiment.
Immunohistochemistry for CD4+ and FoxP3+ cells.
Paraffin sections of the middle ear cavity around the tympanic orifice in the ET were used for the detection of CD4+ and FoxP3+ cells. After deparaffinization, the specimens were treated with 3% H2O2 for 20 min at room temperature. Sections were exposed to 5% normal goat serum in PBS for 30 min and then incubated with a rabbit anti-mouse CD4 antibody (Novus Biologicals, Littleton, CO) and a rabbit anti-mouse FoxP3 antibody (Novus Biologicals) diluted 1:500 in 1% bovine serum albumin (BSA)–PBS for 12 h at room temperature. After being rinsed with PBS, the sections were incubated with a biotinylated goat anti-rabbit antibody (1:2,000) in 1% BSA–PBS for 6 h at room temperature. After being rinsed with PBS, the sections were incubated with ABC reagent (Vector Laboratories, Burlingame, CA) for 1 h and developed in 0.05% 3,3′-diaminobenzidine–0.01% H2O2 substrate medium in 0.1 M phosphate buffer for 8 min.
Detection of cytokines in MEE by ELISA.
Assays for tumor necrosis factor alpha (TNF-α), interleukin-1β (IL-1β), IL-10, and transforming growth factor β (TGF-β) were performed with a commercially available mouse enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Inc., Minneapolis, MN), according to the manufacturer's instructions. All samples were run with negative and positive controls. Standard curves were generated from known concentrations of cytokines provided by the manufacturer. Optical densities were read with a microtiter ELISA plate reader (Sanko Junyaku Co., Ltd., Tokyo, Japan).
Flow cytometric analysis of T cells in mononuclear cells infiltrating the MEM.
To analyze the cellular phenotypes of lymphocytes in the middle ear responding to chronic inflammation, mononuclear cells (MNCs) were isolated and stained with antigen-specific antibodies conjugated with a fluorescent label. Another set of COM mice was made, and MNCs from the middle ear were harvested from groups of 5 mice at each time point. For the isolation of MNCs from the MEM, we established a dissociation method (23). Briefly, the skin was removed carefully from the head region, and the bulla was isolated from the temporal bone. Then, the bulla was dissected, and the MEM was detached from the bone under microscopy. MNCs were isolated from the MEM after enzymatic digestion at 37°C for 10 min with 0.5 mg/ml collagenase type IV (Sigma, St. Louis, MO). Approximately 0.5 × 106 to 1.0 × 106 cells were used for flow cytometry. To detect Treg in the MEM, a fluorescein isothiocyanate (FITC)-conjugated anti-CD4 monoclonal antibody (MAb) (17A2; BD Bioscience, San Diego, CA), phycoerythrin (PE)-conjugated anti-CD8 MAb (53-6.7; BD Bioscience), and peridinin chlorophyll protein (PerCP)-conjugated anti-CD3 MAb (145-2C11; BD Bioscience) were used to analyze T cell subsets in 3-color analysis, and an FITC-conjugated anti-CD4 MAb, phycoerythrin-conjugated anti-FoxP3 MAb (Miltenyi Biotec, Auburn, CA), and PerCP-conjugated anti-CD25 MAb (PC61; BD Bioscience) were used to analyze Treg in 3-color analysis. Intracellular FoxP3 staining was performed by using a FoxP3 staining buffer set (Miltenyi Biotec). The corresponding isotype control antibodies were used as negative controls of background staining. The immunofluorescent intensities of Treg were analyzed using CellQuest software (Becton Dickinson, Mountain View, CA), where a lymphocyte gate was defined by forward and side scatter parameters. This experiment was performed independently 3 times, each from a pool of 5 mice.
Detection of FoxP3 mRNA and cytokine mRNA in MNCs from MEM by real-time RT-PCR.
To elucidate the inflammatory responses in the middle ear after bacterial injection, real-time RT-PCR was performed to assess the expression of FoxP3 and cytokine mRNA in the MNCs that had infiltrated the MEM. These MNCs were isolated from 5 mice at the same time as those used for flow cytometry. For the detection of FoxP3, IL-10, TGF-β, IL-1β, and TNF-α mRNAs, total RNA was extracted from the MNCs that had infiltrated the MEM. The primers and probes for FoxP3 mRNA used in this study were obtained from Applied Biosystems (Foster City, CA). Total cellular RNA extraction and first-strand cDNA synthesis were performed using an RNA extraction and reverse transcription kit (Qiagen, Tokyo, Japan). The total concentration of RNA used for this assay in each group was adjusted to 0.25 μg. Each cDNA sample was then used as a template in a PCR amplification mixture containing forward and reverse primers for FoxP3, IL-10, TGF-β, IL-1β, and TNF-α, forward and reverse primers for 18S rRNA (internal control), and TaqMan Universal PCR master mix (Applied Biosystems). PCR amplifications for the target cytokines and internal control 18S rRNA were performed in a single well of a capped 96-well optical plate. Reaction mixtures were subjected to the following amplification scheme: 1 cycle at 50°C for 2 min and 1 cycle at 95°C for 10 min, followed by 40 cycles at 95°C for 15 s (denaturation) and 60°C for 1 min (annealing and extension). Real-time RT-PCR data were analyzed using a StepOnePlus real-time PCR system (Applied Biosystems). Final mRNA quantities were derived using the comparative threshold cycle (CT) method. These quantities are expressed as the fold difference relative to a calibrator cDNA (from mice that had no treatment as a control) prepared in parallel with the experimental cDNAs. Data are expressed as the arithmetic means of the percentage of positive cells from 3 independent experiments (± standard deviation [SD]), each from a pool of 5 mice.
In vivo depletion of Treg by administration of an anti-CD25 MAb.
In vivo depletion of Treg is a means of studying the role of these subpopulations in the effector phases of particular in vivo immune responses in COM. To deplete Treg, an anti-CD25 MAb (PC61) was used. Onizuka et al. reported that the administration of anti-CD25 MAb (PC61) caused a reduction in the number of CD4+ CD25+ cells in peripheral lymphoid tissues, and they also reported that 125 μg was an effective dose to reduce Treg (24).
Experimental COM mice were made as described above. The mice were injected intraperitoneally with 125 μl anti-CD25 MAb (PC61.5 at 125 μg/mouse; eBioscience, San Diego, CA) or PBS as a control at 2, 4, and 6 weeks after ET blockage and NTHi inoculation. In a preliminary study, we administered the anti-CD25 antibody once, twice, and three times, and administration of the antibody three times was found to be more effective and stable than administration once and twice (data not shown). At 2 months after treatment, 8 mice from each group were killed under deep anesthesia by intraperitoneal administration of a pentobarbital solution. The same evaluations as in our earlier experiment were performed. To investigate the changes of Treg after treatment with the anti-CD25 MAb, the spleen (SPL) and cervical lymph nodes (CLNs) were collected for single-cell suspension by gentle teasing through a stainless steel mesh. MEMs were then dissociated with collagenase type IV to obtain single-cell suspensions as described above. MNCs from these tissues were used for flow cytometry to detect CD4+ CD25+ FoxP3+ Treg. The protocol for flow cytometric analysis of T cells in MNCs from these tissues was the same as described above.
Statistical analysis.
Statistical comparisons between appropriate groups were performed with a Mann-Whitney test and chi-square test. P values of <0.05 were considered significant.
RESULTS
Middle ear findings of COM.
In the control group, no mice had MEE. In the COM group, MEE was induced in all mice that received an NTHi injection and ET obstruction (Fig. 1A). As for the characteristics of MEE (Fig. 1B), 100% (10/10) of effusions were purulent at day 3 after inoculation, and 71% (5/7) were purulent at 2 weeks. At 1 month after inoculation, the ratio of mucoid effusions was increased to 70% (7/10). At 2 months after inoculation, 50% (4/8) of MEEs had become serous, and 38% (3/8) of effusions had become pultaceous. The severity score of COM at all time points examined showed a significant difference compared to the control group (Table 1).
FIG 1.
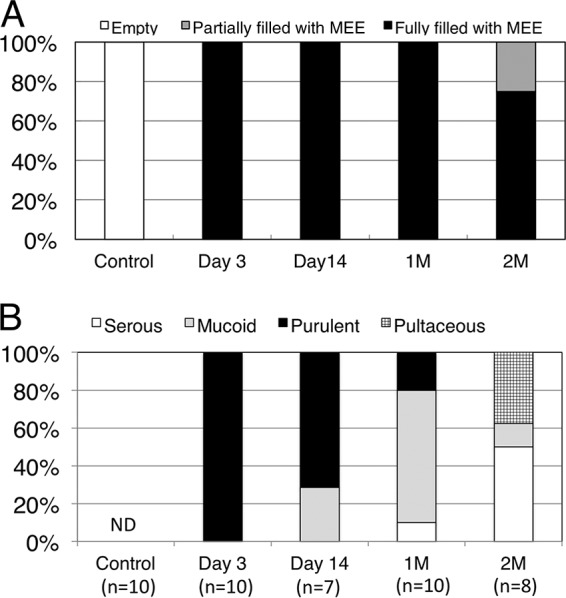
In the control group, no mice had MEE. In the COM group, MEE was induced in all mice that received an NTHi injection and ET obstruction (A). All MEEs were purulent at day 3 after inoculation, and 71% were purulent at 2 weeks. At 1 month (1M) after inoculation, the ratio of mucoid effusions was increased to 70%. At 2 months (2M) after inoculation, 50% of MEEs had become serous and 38% of effusions had become pultaceous (B). ND, not detected.
TABLE 1.
Severity scores of OM
| Groupa | Severity score of OMb |
|---|---|
| Control | 0 ± 0 |
| Day 3 | 2 ± 0* |
| Day 14 | 1.7 ± 0.5* |
| 1 mo | 1.2 ± 0.4* |
| 2 mo | 1.8 ± 1.0* |
Seven to 10 mice from each group were monitored otomicroscopically at day 3, day 14, 1 month, and 2 months after injection with NTHi.
The severity score of OM was measured according to tympanic membrane (TM) color and opacity and the presence of middle ear effusion (MEE), as follows: 0, gray and translucent TM without MEE (normal); 1, gray and opaque TM with serous or mucoid MEE; 2, yellow and opaque TM with purulent MEE; and 3, opaque TM with pultaceous MEE. The severity score is expressed as the mean ± SD. *, P < 0.05 compared to the control by Mann-Whitney test.
Bacterial clearance and inflammatory cell infiltration.
Bacterial counts of NTHi from the middle ear are shown in Fig. 2. The bacterial counts in MEE decreased gradually during the course of COM. NTHi concentrations were highest at day 3 after inoculation (log10 4.9 ± 0.9 CFU/ml) and decreased gradually but were sustained for up to 2 months after inoculation (log10 1.4 ± 2.1 CFU/ml). The incidence of an NTHi culture-positive MEE was 100% at day 3, which was sustained from 57% to 38% from day 14 to 2 months after inoculation (Table 2).
FIG 2.
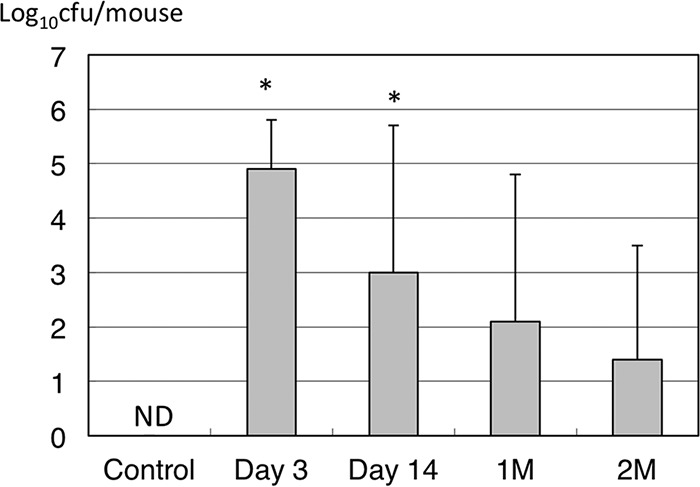
The bacterial counts in MEEs decreased gradually during the course of COM. NTHi concentrations were highest at day 3 after inoculation and decreased gradually but were sustained for up to 2 months after inoculation. ND, not detected; 1M, 1 month; 2M, 2 months. *, P < 0.05 compared to the control by Mann-Whitney test.
TABLE 2.
Range of NTHi infection and rate of MEE
| Groupa | NTHi infection, CFU/mouse | Rate of culture positive MEE, no. positive/total (%) |
|---|---|---|
| Control | 0 | 0/10 (0) |
| Day 3 | 2.4 × 103–1.1 × 106 | 10/10 (100) |
| Day 14 | 0 × 106–1.3 × 106 | 4/7 (57) |
| 1 mo | 0 × 106–1.6 × 106 | 4/10 (40) |
| 2 mo | 0 × 104–5.4 × 104 | 3/8 (38) |
MEEs were collected from 7 to 10 mice at day 3, day 14, 1 month, and 2 months after injection with NTHi and ET blockage.
Histological evaluation.
In terms of H&E staining of the middle ear (Fig. 3A), few inflammatory cells were seen in the middle ear of the control animals. At day 3 after inoculation, the MEM showed thickening and inflammatory cell infiltration containing polymorphonuclear cells, and these inflammatory responses were strong up to day 14. From 1 to 2 months after inoculation, mucosal thickening and inflammatory cell infiltration continued with lymphocyte migration in the MEM. Mucosal thickness (Fig. 3B) and the number of inflammatory cells (Fig. 3C) in the middle ear were analyzed using ImageJ software. Compared to the control group, mucosal thickness of the middle ear increased significantly throughout the experiments (mean thickness of MEM ± SD in control versus day 3, day 14, 1 month, and 2 months, 15 ± 9 versus 35 ± 11, 59 ± 20, 48 ± 31, and 41 ± 8 nm, respectively; P < 0.05), peaking at day 14. Inflammatory cell infiltration also increased (mean number of inflammatory cells in the MEM ± SD in control versus day 3, day 14, 1 month, and 2 months, 19 ± 11 versus 161 ± 58, 104 ± 54, 124 ± 93, and 72 ± 27 cells/10,000 nm2, respectively; P < 0.05). The inflammatory responses due to NTHi inoculation continued for up to 2 months in the middle ear.
FIG 3.

(A) H&E image of the middle ear. In the control mice, few inflammatory cells were seen in the middle ear. At day 3 after inoculation, the MEM showed thickening and inflammatory cell infiltration containing polymorphonuclear cells, and these inflammatory responses were strong up to day 14. From 1 month (1M) to 2 months (2M) after inoculation, mucosal thickening and inflammatory cell infiltration continued with lymphocyte migration in the MEM. (B) Mucosal thickness of the MEM. (C) Number of inflammatory cells in the MEM. Compared to the control group, mucosal thickness of the middle ear increased significantly throughout the experiments. Inflammatory cell infiltration also increased throughout the experiments. *, P < 0.05 compared to the control by Mann-Whitney test.
Distribution of CD4+ T cells in the middle ear.
In immunohistochemistry for CD4+ cells (Fig. 4A), CD4+ cells in the MEM were increased at day 14 after inoculation. The distribution of positive cells increased and peaked at 1 month after inoculation; however, there were large numbers of positive cells at 1 and 2 months after inoculation. Figure 4B shows flow cytometric analysis for T cell subsets in the MEM. The percentage of CD4− CD8− T cells increased in the middle ear from day 3 to 2 months after inoculation. However, the percentage of CD4+ T cells was always greater than that of CD8+ T cells in the middle ear throughout the experiment, and the CD4/CD8 ratio increased from day 3, 1 and 2 months after inoculation compared to the control group (Fig. 4B). Table 3 shows the average frequency of CD4 and CD8 cells in the middle ear, and there was no statistical difference between the control and other groups (average CD4/CD8 ratio for control versus day 3, day 14, 1 month, and 2 months, 2.7 versus 4.0, 2.4, 4.6, and 4.8, respectively).
FIG 4.

(A) Immunohistochemistry for CD4+ cells in the MEM. A rabbit polyclonal anti-mouse CD4 MAb was used. CD4+ cells in the MEM were increased at day 14 after inoculation, and there were a large number of positive cells at 1 and 2 months after inoculation (1 M and 2 M, respectively). (B) Flow cytometric analysis for T cell subsets in the MEM. The percentage of CD4− CD8− T cells was increased in the middle ear from day 3 to 2 months after inoculation. However, the percentage of CD4+ T cells was always greater than that of CD8+ T cells in the middle ear throughout the experiment.
TABLE 3.
Average frequencies of CD4, CD8, and regulatory T cells in the middle ear
| Groupa | Frequency of cells: |
||||
|---|---|---|---|---|---|
| CD3+ |
CD4+b |
||||
| % CD4 | % CD8 | CD4/CD8 ratio | % FoxP3+ | % CD25+ FoxP3+ | |
| Control | 42.8 ± 15.6 | 17.7 ± 11 | 2.7 ± 1.2 | 18.5 ± 1.3 | 12.2 ± 2.4 |
| Day 3 | 40.2 ± 8.3 | 10.0 ± 2.1 | 4.0 ± 0.2 | 15.4 ± 4.5 | 10.6 ± 1.8 |
| Day 14 | 30.8 ± 8.6 | 12.8 ± 1.5 | 2.4 ± 0.6 | 45.0 ± 4.0* | 38.3 ± 4.9* |
| 1 mo | 33.7 ± 11.0 | 8.4 ± 4.7 | 4.6 ± 1.6 | 40.3 ± 1.8* | 34.7 ± 2.0* |
| 2 mo | 33.6 ± 8.6 | 7.3 ± 2.3 | 4.8 ± 1.2 | 34.4 ± 2.5* | 27.8 ± 2.3* |
MNCs were collected from 5 mice. This experiment was performed independently 3 times.
*, P < 0.05 compared to control by Mann-Whitney test.
Distribution of FoxP3+ cells in the middle ear.
In immunohistochemistry for FoxP3+ cells (Fig. 5A), FoxP3+ cells were also seen at day 14. The distribution of positive cells was sustained at 1 and 2 months. Figure 5B shows flow cytometric analysis for CD4+ FoxP3+ Treg in the MEM. The percentage of CD4+ FoxP3+ T cells increased in the middle ear from day 14 to 2 months after inoculation compared to the control group. Table 3 shows the significant differences between the control group and at day 14 and 1 and 2 months after inoculation (average frequency of CD4+ FoxP3+ T cells for control versus day 14, 1 month, and 2 months, 18.5% versus 45.0%, 40.3%, and 34.4%, respectively). In the cell fraction of FoxP3+ T cells (Fig. 5C), the number of CD25+ T cells also increased after inoculation, and the number of CD25+ FoxP3+ T cells in the MEM was greater than that of CD25− FoxP3+ T cells among the CD4+ T cells at all time points examined. The distribution of CD4+ CD25+ FoxP3+ T cells increased from day 14 to 2 months. Table 3 shows the significant differences between the control group and at day 14 and 1 and 2 months after inoculation (average frequency of CD4+ CD25+ FoxP3+ T cells for control versus day 14, 1 month, and 2 months, 12.2% versus 38.3%, 34.7%, and 27.8%, respectively). CD4+ CD25+ FoxP3+ T cells accounted for approximately 80% of CD4+ FoxP3+ T cells from day 14 to 2 months.
FIG 5.

(A) Immunohistochemistry for FoxP3+ cells in MEM. A rabbit polyclonal anti-mouse FoxP3 MAb was used. FoxP3+ cells were also seen at day 14. The distribution of positive cells was sustained at 1 and 2 months (1 M and 2 M, respectively). (B) Percentage of CD4+ FoxP3+ T cells in the MEM. The percentage of CD4+ FoxP3+ T cells increased in the middle ear from day 14 to 2 months after inoculation compared to in the control group. (C) Cell fraction of FoxP3+ T cells. The number of CD25+ T cells also increased after inoculation, and the number of CD25+ FoxP3+ T cells in the MEM was greater than that of CD25− FoxP3+ T cells among the CD4+ T cells at all time points examined. One representative experiment out of 3 is shown.
Cytokine levels in MEE and cytokine mRNA expression in MNCs in the middle ear.
Figure 6 shows the cytokine levels in MEE. According to ELISA, in the MEE samples, the levels of IL-1β and TNF-α in the COM group were higher than in the control group. The differences in IL-1β concentration reached statistical significance throughout the experiments (mean concentration of IL-1β ± SD in control versus day 3, day 14, 1 month, and 2 months, 4.5 ± 10 versus 297.6 ± 163.8, 118.9 ± 59.5, 106.3 ± 79.6, and 75.6 ± 87.7 pg/ml, respectively; P < 0.05). The TNF-α concentration was also significantly higher in the COM group at day 3, day 14, and 1 month after inoculation (mean concentration of TNF-α ± SD in control versus day 3, day 14, and 1 month, 0 ± 0 versus 143.7 ± 79.6, 52.3 ± 64.6, and 32.7 ± 43.3 pg/ml, respectively; P < 0.05). Similarly, the IL-10 concentration in the MEE samples was significantly higher in the COM group at day 3, day 14, and 2 months after inoculation (mean concentration of IL-10 ± SD in control versus day 3, day 14, and 2 months, 0 ± 0 versus 30 ± 26.5, 21 ± 15.2, and 16 ± 10 pg/ml, respectively; P < 0.05), as was the TGF-β concentration at day 3, day 14, 1 month, and 2 months after inoculation (mean concentration of TGF-β ± SD in control versus day 3, day 14, 1 month, and 2 months, 0 ± 0 versus 186.5 ± 260.5, 457.6 ± 421, 418.6 ± 367.2, and 359.3 ± 333.8 pg/ml, respectively; P < 0.05).
FIG 6.
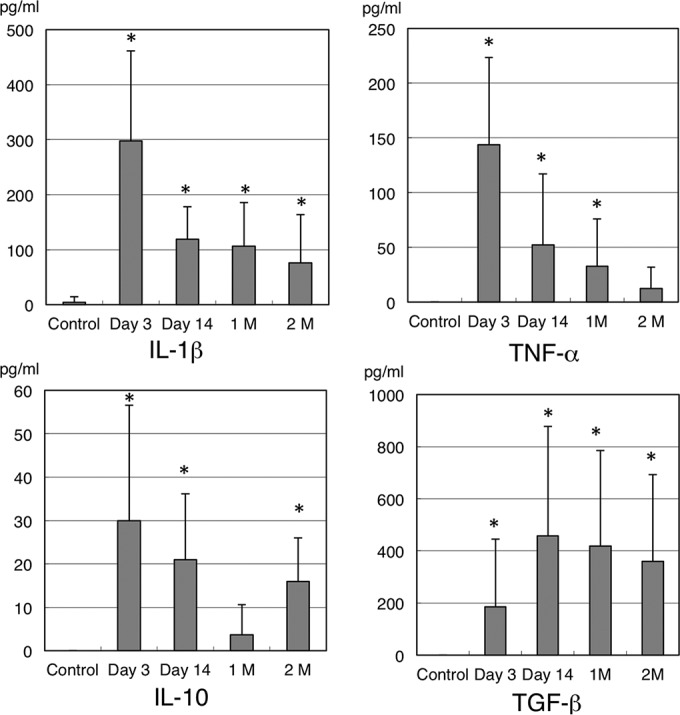
Levels of cytokines in MEE. In the MEE samples, the levels of IL-1β and TNF-α were higher in the COM group than in the control group. The differences in the concentration of IL-1β were statistically significant throughout the experiments. The TNF-α concentration was also significantly higher in the COM group at day 3, day 14, and 1 month (1 M) after inoculation. Similarly, the concentration of IL-10 and TGF-β in the MEE samples increased significantly in the COM group for up to 2 months (2 M) after inoculation. *, P < 0.05 compared to the control by Mann-Whitney test.
To define the induction of cytokine transcription in MNCs from the MEM, the expression of the IL-1β, TNF-α, IL-10, and TGF-β genes was evaluated by real-time RT-PCR analysis (Fig. 7). The FoxP3, IL-1β, TNF-α, IL-10, and TGF-β genes were significantly more induced by inoculation with NTHi than in the controls.
FIG 7.
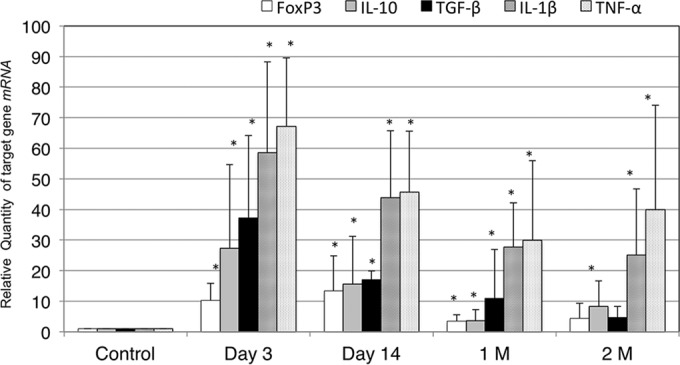
Quantification of the levels of mRNA transcripts. The FoxP3, IL-1β, TNF-α, IL-10, and TGF-β genes were significantly more induced by inoculation with NTHi than the controls throughout the experiment. 1 M, 1 month; 2 M, 2 months; *, P < 0.05 compared to the control by Mann-Whitney test.
Effect of Treg depletion on immune responses in COM.
Figure 8A shows the results of flow cytometry for CD4+ CD25+ FoxP3+ T cells with anti-CD25 MAb or PBS treatment. Compared with the PBS-treated mice, in which the percentages of Treg were 7.0% in the SPL, 8.5% in the CLNs, and 32.4% in the MEM, anti-CD25 MAb-treated mice had a lower percentage of Treg in each tissue (1.3% in the SPL, 1.3% in the CLNs, and 18.2% in the MEM). In terms of bacterial counts in MEE, PBS-treated mice had higher counts than the anti-CD25 MAb-treated mice; a 99.9% reduction was seen in the anti-CD25 MAb-treated mice (Fig. 8B). Figure 8B also shows that there was a significantly higher percentage of NTHi culture-positive MEE in the PBS-treated mice than in the anti-CD25 MAb-treated mice (rate of NTHi culture-positive MEE in PBS-treated versus anti-CD25 MAb-treated mice, 62.5% [5/8] versus 12.5% [1/8], respectively; chi-square test, P < 0.05). For the cytokine levels in MEE, the levels of all cytokines tested, including proinflammatory and immunosuppressive cytokines, were reduced by anti-CD25 MAb treatment. Especially, IL-1β was reduced significantly between the two groups (Fig. 8C).
FIG 8.

(A) Population of CD4+ CD25+ FoxP3+ T cells in MEM, CLNs, and SPL. Compared with PBS-treated mice, the anti-CD25 MAb-treated mice had a lower percentage of Treg in each tissue type. (B) Bacterial counts in MEEs. The PBS-treated mice had higher counts than the anti-CD25 MAb-treated mice; the numbers show the arithmetic means of CFU/mouse, and a 99.9% reduction was seen in the anti-CD25 MAb-treated mice. (C) Cytokine levels in MEEs. The levels of all cytokines tested were reduced by anti-CD25 MAb treatment. Especially, IL-1β was reduced significantly between the two groups. MEM, middle ear mucosa; CLNs, cervical lymph nodes; SPL, spleen; anti-CD25(−), PBS-treated mice; anti-CD25(+), anti-CD25 MAb-treated mice. †, P < 0.05 compared to anti-CD25(−) by chi-square test; *, P < 0.05 compared to anti-CD25(−) by Mann-Whitney test.
DISCUSSION
In this study, we developed an experimental mouse model of COM by inoculating NTHi into the middle ear bulla and using ET blockage and then studied the local immune responses of the middle ear against NTHi. We found that middle ear inflammation, such as mucosal thickening and inflammatory cell infiltration, continued for at least 2 months after inoculation of the middle ear, and NTHi was detected using a bacterial culture method in the middle ear cavity in 40% of our COM model mice for up to 2 months after the challenge. This is the first report showing chronic inflammation can be induced in the middle ear with NTHi culture-positive MEEs. To our knowledge, these are also the first detailed findings regarding the inflammatory responses of the middle ear in a murine COM model. This is in contrast to other animal models using NTHi, such as the chinchilla and rat. Green et al. (25) reported that inoculation of viable NTHi induced acute OM in the chinchilla and that the bacterial cells existed for 14 days of the challenge. Melhus et al. (26) found that middle ear infections were resolved within 8 days in a rat model. These animal models were not treated with an ET block, and middle ear infections were limited to several weeks. On the basis of these findings and compared with previous studies, this mouse model treated with both an ET block and NTHi inoculation in the middle ear may make a useful model for studying the role of chronic inflammation in a persistent NTHi infection.
Recently, the characterization of T cell subsets has accelerated the understanding of inflammatory and humoral immune responses in part by unveiling the enormous plasticity within these subsets. The balance in T helper subsets as observed in healthy mucosa is changed in inflamed mucosa. According to flow cytometric analysis of the middle ear in our model, the CD4/CD8 ratio was increased from day 3 to 2 months after inoculation compared to the control group, as Derycke et al. (27) reported the CD4/CD8 ratio was increased depending on the severity of inflammation in chronic rhinosinusitis. This result showed that chronic inflammation occurred in the middle ear for a period of 2 months. Furthermore, the number of CD4+ FoxP3+ Treg was significantly increased in the COM group from 2 weeks to 2 months after inoculation compared with the control group. In the CD4+ FoxP3+ Treg, CD4+ CD25+ FoxP3+ Treg were mainly increased in the MEM. The immunohistochemistry findings also supported the results of these flow cytometric analyses. CD4+ CD25+ FoxP3+ Treg have a primary effect on T cells and/or dendritic cells by the inhibition of IL-2 gene expression, modulation of costimulatory molecules on antigen-presenting cells, interaction of lymphocyte activation gene 3 with major histocompatibility complex (MHC) class II molecules, secretion of immunosuppressive cytokines such as IL-10 and TGF-β, induction of tryptophan catabolism through cytotoxic-T lymphocyte-associated antigen 4 (CTLA-4), and cytotoxicity (28). Previously, CD4+ CD25+ FoxP3+ Treg were considered natural Treg, which are physiologically produced by the normal thymus as a functionally mature and distinct population, and their development and function depend on the expression of the transcription factor FoxP3 (17, 18, 28). However, CD4+ CD25− naive T cells can be converted to CD4+ CD25+ Treg by the TGF-β-induced expression of FoxP3 (29). Adaptive or induced CD4+ Treg include two types of cells: T regulatory type 1 (Tr1) cells and Th3 Treg. Tr1 cells suppress antigen-specific immune responses by producing high levels of IL-10, and Tr1 cells can transiently express FoxP3 upon activation (30, 31). Th3 cells secrete TGF-β and IL-10 and express FoxP3 (31, 32). Th3 cells play a crucial role in inducing and maintaining peripheral tolerance by driving the differentiation of antigen-specific FoxP3 regulatory cells in the periphery by producing high levels of TGF-β (33). In this study, not only inflammatory cytokines (IL-1β and TNF-α) but also immunosuppressive cytokines (IL-10 and TGF-β) increased in the middle ear at the protein and mRNA level after inoculation compared to the control group. Although we did not distinguish natural Treg from adaptive or induced Treg in the CD4+ FoxP3+ Treg, these Treg are considered to be related to the persistent inflammation in NTHi infection.
Treg are also known to play an important role in controlling infectious disease, including viruses, parasites, and bacteria, and Treg play two different key immunological roles—protective or detrimental—in host defense.
Treg play protective roles against certain viral pathogens. Lund et al. (34) showed an accelerated fatal infection with increased herpes simplex virus loads in the vaginal mucosa and central nervous system after ablation of Treg. Similarly, FoxP3+ Treg ablation before West Nile virus infection in mice led to an increase of disease severity, which was associated with higher viral loads in the brain and spinal cord (35). As for bacterial infections, Treg reportedly continue to play a detrimental role in host defense. To investigate the relationship between Treg and pregnancy-associated infection susceptibility, Rowe et al. (36) demonstrated that expansion of immune-suppressive FoxP3+ Treg, which occurs physiologically during pregnancy or when experimentally induced in transgenic mice, caused enhanced susceptibility to prenatal pathogens, including Listeria and Salmonella species. Reciprocally, infection susceptibility was uniformly reduced with Treg ablation. Johanns et al. (37) also demonstrated that FoxP3+ Treg dictated the natural progression of persistent virulent Salmonella infection in mice. In human cases, Helicobacter pylori-positive patients revealed a 19- to 25-fold induction of FoxP3 transcript levels in the antrum and cardia, and naturally occurring Treg (CD4+ CD25high FoxP3+) are associated with higher H. pylori colonization and increased TGF-β1 gene expression (38). In addition, CD4+ FoxP3+ Treg suppressed host responses to Haemophilus ducreyi during experimental skin infection of human volunteers (39). On the basis of these reports, Treg are associated with the pathogenesis of a persistent bacterial infection, but not viral infection, in both animal models and humans. In this study, Treg depletion induced a 99.9% reduction of bacterial counts in MEE and also significantly reduced the ratio of NTHi culture-positive MEE. The levels of proinflammatory cytokines were also reduced in MEE in our model. Kaur et al. (40) reported that the elevation of proinflammatory cytokines in MEEs in children with acute OM is dependent on the number of bacterial species identified, and this supports our findings of the cytokine levels in MEE. On the basis of our observations, Treg may also suppress immune responses and cause persistent infection in the middle ear of a mouse model of COM induced by NTHi inoculation and ET blockade.
In conclusion, we developed a murine model of COM with persistent NTHi infection by using an ET block for 2 months. Treg gathered in the middle ear, and especially CD4+ CD25+ FoxP3+ Treg are assumed to confer infectious tolerance to NTHi in the middle ear. These phenomena may also be at work in the pathogenesis of COM with NTHi infection in other upper and lower airway diseases. A new therapeutic strategy may be developed against chronic inflammatory diseases with NTHi infection by using our mice model.
ACKNOWLEDGMENTS
This work was supported by a Grant-in-Aid for Scientific Research (C) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (25462648).
We thank A. Iwamoto and K. Maeda in our department for invaluable assistance. We appreciate the late G. Mogi for guiding us in our experiments.
REFERENCES
- 1.Bennett KE, Haggard MP, Silva PA, Stewart IA. 2001. Behaviour and developmental effects of otitis media with effusion into the teens. Arch Dis Child 85:91–95. doi: 10.1136/adc.85.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuo CL, Lien CF, Chu CH, Shiao AS. 2013. Otitis media with effusion in children with cleft lip and palate: a narrative review. Int J Pediatr Otorhinolaryngol 77:1403–1409. doi: 10.1016/j.ijporl.2013.07.015. [DOI] [PubMed] [Google Scholar]
- 3.Wright ED, Pearl AJ. 1998. Laterally hypertrophic adenoids as a contributing factor in otitis media. Int J Pediatr Otorhinolaryngol 45:208–214. [DOI] [PubMed] [Google Scholar]
- 4.Bluestone CD, Stephenson JS, Martin LM. 1992Ten-year review of otitis media pathogenesis. Pediatr Infect Dis J 11:S7–S11. doi: 10.1097/00006454-199208001-00002. [DOI] [PubMed] [Google Scholar]
- 5.DeMaria TF, Prior RB, Briggs BR, Lim DJ, Birck HG. 1984. Endotoxin in middle-ear effusions from patients with chronic otitis media with effusion. J Clin Microbiol 20:15–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nell MJ, Grote JJ. 1999. Endotoxin and TNF-alpha in middle ear effusions: in relation with upper airway infection. Laryngoscope 109:1815–1819. doi: 10.1097/00005537-199911000-00017. [DOI] [PubMed] [Google Scholar]
- 7.DeMaria TF, Briggs BR, Lim DJ, Okazaki N. 1984. Experimental otitis media with effusion following middle ear inoculation of nonviable H. influenzae. Ann Otol Rhinol Laryngol 93:52–56. doi: 10.1177/000348948409300113. [DOI] [PubMed] [Google Scholar]
- 8.Nonomura N, Nakano Y, Satoh Y, Fujioka O, Niijima H, Fujita M. 1986. Otitis media with effusion following inoculation of Haemophilus influenzae type b endotoxin. Arch Otorhinolaryngol 243:31–35. doi: 10.1007/BF00457904. [DOI] [PubMed] [Google Scholar]
- 9.Giebink GS, Mills EL, Huff JS, Edelman CK, Weber ML, Juhn SK, Quie PG. 1979. The microbiology of serous and mucoid otitis media. Pediatrics 63:915–919. [PubMed] [Google Scholar]
- 10.Hall-Stoodley L, Hu FZ, Gieseke A, Nistico L, Nguyen D, Hayes J, Forbes M, Greenberg DP, Dice B, Burrows A, Wackym PA, Stoodley P, Post JC, Ehrlich GD, Kerschner JE. 2006. Direct detection of bacterial biofilms on the middle-ear mucosa of children with chronic otitis media. JAMA 296:202–211. doi: 10.1001/jama.296.2.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maeda K, Hirano T, Ichimiya I, Kurono Y, Suzuki M, Mogi G. 2004. Cytokine expression in experimental chronic otitis media with effusion in mice. Laryngoscope 114:1967–1972. doi: 10.1097/01.mlg.0000147930.29261.51. [DOI] [PubMed] [Google Scholar]
- 12.van Alphen L. 1992. Epidemiology and prevention of respiratory tract infections due to nonencapsulated Haemophilus influenzae. J Infect Dis 165:S177–S180. doi: 10.1093/infdis/165-Supplement_1-S177. [DOI] [PubMed] [Google Scholar]
- 13.Sethi S, Murphy TF. 2001. Bacterial infection in chronic obstructive pulmonary disease in 2000: a state-of-the-art review. Clin Microbiol Rev 14:336–363. doi: 10.1128/CMR.14.2.336-363.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erwin AL, Smith AL. 2007. Review. Nontypeable Haemophilus influenzae: understanding virulence and commensal behavior. Trends Microbiol 15:355–362. [DOI] [PubMed] [Google Scholar]
- 15.Mukundan D, Ecevit Z, Patel M, Marrs CF, Gilsdorf JR. 2007. Pharyngeal colonization dynamics of Haemophilus influenzae and Haemophilus haemolyticus in healthy adult carriers. J Clin Microbiol 45:3207–3217. doi: 10.1128/JCM.00492-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swords WE. 2012. Nontypeable Hemophilus influenza biofilm: role in chronic airway infections. Front Cell Infect Microbiol 2:97. doi: 10.3389/fcimb.2012.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. 1995. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor α-chains (CD25): breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 155:1151–1164. [PubMed] [Google Scholar]
- 18.Hori S, Nomura T, Sakaguchi S. 2003. Control of regulatory T cell development by the transcription factor Foxp3. Science 299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 19.Fontenot JD, Gavin MA, Rudensky AY. 2003. FOXP3 programs the development and function of CD4+ CD25+ regulatory T-cells. Nat Immunol 4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 20.Curotto de Lafaille MA, Lafaille JJ. 2009. Natural and adaptive Foxp3+ regulatory T cells: more of the same or a division of labor? Immunity 30:626–635. doi: 10.1016/j.immuni.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 21.Rowe JH, Ertelt JM, Way SS. 2012. Foxp3+ regulatory T cells, immune stimulation and host defence against infection. Immunology 136:1–10. doi: 10.1111/j.1365-2567.2011.03551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giebink GS, Wright PF. 1983. Different virulence of influenza A virus strains and susceptibility to pneumococcal otitis media in chinchillas. Infect Immun 41:913–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kodama S, Suenaga S, Hirano T, Suzuki M, Mogi G. 2000. Induction of specific immunoglobulin A and Th2 immune responses to P6 outer membrane protein of nontypeable Haemophilus influenzae in middle ear mucosa by intranasal immunization. Infect Immun 68:2294–2300. doi: 10.1128/IAI.68.4.2294-2300.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. 1999. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor α) monoclonal antibody. Cancer Res 59:3128–3133. [PubMed] [Google Scholar]
- 25.Green BA, Vazquez ME, Zlotnick GW, Quigley-Reape G, Swarts JD, Green I, Cowell JL, Bluestone CD, Doyle WJ. 1993. Evaluation of mixtures of purified Haemophilus influenzae outer membrane proteins in protection against challenge with nontypeable H. influenzae in the chinchilla otitis media model. Infect Immun 61:1950–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Melhus A, Ryan AF. 2000Expression of cytokine genes during pneumococcal and nontypeable Haemophilus influenzae acute otitis media in the rat. Infect Immun 68:4024–4031. doi: 10.1128/IAI.68.7.4024-4031.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Derycke L, Eyerich S, Van Crombruggen K, Pérez-Novo C, Holtappels G, Deruyck N, Gevaert P, Bachert C. 2014. Mixec T helper cell signatures in chronic rhinosinusitis with and without polyps. PLoS One 9:e97581. doi: 10.1371/journal.pone.0097581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyara M, Sakaguchi S. 2007. Natural regulatory T cells: mechanisms of suppression. Trends Mol Med 13:108–116. doi: 10.1016/j.molmed.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 29.Chen WJ, Jin W, Hardegen N, Lei K, Marinos N, McGrady G, Wahl SM. 2003. Conversion of peripheral CD4+ CD25− naïve T cells to CD4+ CD25+ regulatory T cells by TGF-β induction of transcription factor FOXP3. J Exp Med 198:1875–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Groux H, O'Garra A, Bigler M, Rouleau M, Antonenko S, de Vries JE, Roncarolo MG. 1997. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 389:737–742. doi: 10.1038/39614. [DOI] [PubMed] [Google Scholar]
- 31.Beissert S, Schwartz A, Schwartz T. 2006. Regulatory T cells. J Investig Dermatol 126:15–24. doi: 10.1038/sj.jid.5700004. [DOI] [PubMed] [Google Scholar]
- 32.Chen Y, Kuchroo VK, Inobe J, Hafler DA, Weiner HL. 1994. Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science 265:1237–1240. doi: 10.1126/science.7520605. [DOI] [PubMed] [Google Scholar]
- 33.Carrier YW, Yuan J, Kuchroo VK, Howard L. 2007. Th3 cells in peripheral tolerance, induction of Foxp3-positive regulatory T cells by Th3 cells derived from TGF-beta T cell-transgenic mice. J Immunol 178:179–185. doi: 10.4049/jimmunol.178.1.179. [DOI] [PubMed] [Google Scholar]
- 34.Lund JM, Hsing L, Pham TT, Rudensky AY. 2008. Coordination of early protective immunity to viral infection by regulatory T cells. Science 320:1220–1224. doi: 10.1126/science.1155209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lanteri MC, O'Brien KM, Purtha WE, Cameron MJ, Lund JM, Owen RE, Heitman JW, Custer B, Hirschkorn DF, Tobler LH, Kiely N, Prince HE, Ndhlovu LC, Nixon DF, Kamel HT, Kelvin DJ, Busch MP, Rudensky AY, Diamond MS, Norris PJ. 2009. T regs control the development of symptomatic West Nile virus infection in humans and mice. J Clin Invest 119:3266–3277. doi: 10.1172/JCI39387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rowe JH, Ertelt JM, Aguilera MN, Farrar MA, Way SS. 2011. Foxp3+ regulatory T cell expansion required for sustaining pregnancy compromises host defense against prenatal bacterial pathogens. Cell Host Microbe 10:54–64. doi: 10.1016/j.chom.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johanns TM, Ertelt JM, Rowe JH, Way SS. 2010. Regulatory T cell suppressive potency dictates the balance between bacterial proliferation and clearance during persistent Salmonella infection. PLoS Pathog 6:e1001043. doi: 10.1371/journal.ppat.1001043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kandulski A, Wex T, Kuester D, Peitz U, Gebert I, Roessner A, Malfertheiner P. 2008. Naturally occurring regulatory T cells (CD4+, CD25high, FOXP3+) in the antrum and cardia are associated with higher H. pylori colonization and increased gene expression of TGF-beta. Helicobacter 13:295–303. doi: 10.1111/j.1523-5378.2008.00612.x. [DOI] [PubMed] [Google Scholar]
- 39.Li W, Tenner-Racz K, Racz P, Janowicz DM, Fortney KR, Katz BP, Spinola SM. 2010. Role played by CD4+ FOXP3+ regulatory T cells in suppression of host responses to Haemophilus ducreyi during experimental infection of human volunteers. J Infect Dis 201:1839–1848. doi: 10.1086/652781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaur R, Casey J, Pichichero M. 2014. Cytokine, chemokine, and Toll-like receptor expression in middle ear fluids of children with acute otitis media. Laryngoscope 125:E39–E44. doi: 10.1002/lary.24920. [DOI] [PMC free article] [PubMed] [Google Scholar]