

Abstract
The aim of this study was to investigate the relationship between chronic cerebral hypoperfusion and the occurrence and development of Alzheimer’s disease (AD). A cerebral hypoperfusion rat model was established by two vessels occlusion (2VO). The cognitive function of the rats with chronic cerebral hypoperfusion and the expression of p-Tau protein in the hippocampus were observed dynamically. Before the operation, no differences were observed in the cognitive functions of the control and 2VO group (P > 0.05). However, a significant difference was found at 2, 4, 8, and 12 weeks after the operation. The shock number required to reach the “learned” standard in the 2VO group increased remarkably compared with that of the control group (P < 0.01). With the passage of time, the shock number in the model group increased gradually. The p-Tau-positive cells in the CA1 region of the hippocampus also increased markedly in the model group in a time-dependent manner as compared with that in the control group (P < 0.01). Cerebral hypoperfusion can cause and aggravate the phosphorylation of Tau protein in the brain, leading to cognitive dysfunction. Therefore, this protein is an important initiating and promoting factor involved in the development of AD.
Keywords: Cerebral hypoperfusion, phosphorylated Tau protein, cognitive dysfunction, Alzheimer’s disease
Introduction
Alzheimer’s disease (AD) is the most common type of dementia, which has become a common mental disorder currently prevalent in the world [1]. However, no effective prevention and treatment of AD has been found till date. Thus, studies on the etiology and pathogenesis of AD are of great significance. Recent studies suggest that chronic cerebral hypoperfusion may occur according to the common pathological process in various diseases, such as vascular dementia, AD, and Binswanger disease, which may be involved in the occurrence and development of such diseases [2,3].
In the animal experiments on chronic cerebral hypoperfusion, cerebral hypoperfusion seems to initiate cognitive dysfunction in the model [4,5]. The findings of recent animal experiments indicate that cognitive dysfunction is a complicated process, involving various pathophysiological processes [6,7]. Till date, many preliminary studies have been conducted on the loss of cortical neurons, cholinergic neuronal damage, cerebral white matter lesions, abnormal oxygen free radicals, abnormal immune-regulating participation, and energy metabolism disorders [8-10]. However, no studies have been conducted on the correlation between cognitive dysfunction caused by chronic cerebral hypoperfusion and abnormal expression of Tau protein, which is an important limitation in the studies on the role of chronic cerebral hypoperfusion on the pathophysiological mechanisms of cognitive dysfunction and AD.
In recent years, with an increase in the number of studies on the pathogenesis of AD, circulatory disorders were found to play an important role in the pathogenesis and progress of AD [11]. However, most of the current studies focus on the general observation of clinical symptoms or epidemiology investigation. There are few studies focusing on the change in the clinical pathophysiology of the patients, especially the abnormal change law of Tau protein during cerebral hypoperfusion. It has been shown that phosphorylated Tau protein (p-Tau protein) is the key component in AD senile plaque [12]. Thus, in this study, the expression of p-Tau protein in the rat hippocampus was investigated in a two vessels occlusion (2VO) cerebral hypoperfusion rat model. We also observed the dynamic change in cognitive function under the chronic cerebral hypoperfusion (CCH) condition, so as to explore the relationship between CCH and the occurrence and development of AD as well as the circulation disorder mechanism in the process of AD development.
Materials and methods
Animals
Fifty healthy male Sprague-Dawley rats, weighing 250±30 g, were fed a normal diet and housed in a room at a temperature of 18-25°C and humidity of 60-70%. The rats were allowed to drink freely but were subjected to 12 h fasting prior to the operation.
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The animal use protocol has been reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Shandong University.
Establishment of an animal model and grouping
Rats were randomly divided into the 2VO group (n = 25) and sham operation group (n = 25). The 2VO rat model was established according to the DE la Torre method, described previously [13]. Briefly, after the rats were anesthetized with 0.4% pentobarbital sodium (1 mL/100 g) via a intraperitoneal injection, bilateral common carotid arteries was exposed by blunt separation from the anterior midline incision, and care was taken not to damage the vagus nerve. The bilateral common carotid artery was double ligated with silk and was cut off between the two ligation points. The anterior midline incision was sewed up step by step. During the operation, the rectal temperature was monitored, and the body temperature was maintained at 37°C using a roast lamp. Animals in the sham operation group only received the corresponding operation but no ligation or bloof flow blockage was performed. After the operation, each rat was kept in a cage alone to prevent choking.
Ethologic observation
The MG-B electric maze, a Y-electric maze composed of three isometric arms (each 45 cm) and a junctional area, was used to test the rats’ behavior on regaining consciousness. A stimulation signal lamp was installed on the top of each arm to indicate the safety zone (switch off). By randomly changing the location of the safety zone and shock area, the animals’ response to flee from the shock area to the safety zone was observed. A rat escaping from the electric shock area to the safety zone successfully by only one time was defined as “right”. With continuous training, the number of shocks needed for the rat to meet the “learned” standard (nine “right” after ten consecutive tests was defined as “learned”) was recorded as a judge index for evaluating the learning ability and memory of rats. Ethologic tests were carried out between 8:00 am to 11:00 am on the day before the operation and 2, 4, 8, and 12 weeks after the operation.
Sampling and fixation of brain tissues
After the ethologic test, the rats were anesthetized with 0.4% pentobarbital sodium (1 mL/100 g) via an intraperitoneal injection. Then, 100 mL warm physiological saline was perfused via the ascending aorta of the left ventricle, followed by 400 mL precooled 4% paraformaldehyde (PFA), which was initially perfused quickly, but then slowed down. During the perfusion, the rat’s head was kept at a low position to ensure full perfusion in the brain. Forty minutes later, the rat’s brain was dissected and fixed in 4% PFA at 4°C overnight. The brain was dehydrated with 30% sucrose solution until it sank. According to the map of the rat’s brain, the brain tissue was repaired and cut into 5-µm thick continuous coronary slices from the optic chiasma using a AO cryotome. One of every 5 slices was placed on polylysine-coated slides for H&E and immunohistochemical staining.
Immunohistochemistry
SABC method
After incubation with 3% H2O2-methanol solution for 10 min, the slides were treated with 0.01% trypsin for 30 min at room temperature. Slides were incubated with rabbit anti-rat p-Tau polyclonal antibody (Biosource International, Inc. USA) at 4°C overnight, followed by incubation with a goat anti-rabbit secondary antibody at 37°C for 2 h. After that, the slides were incubated with SABC reagent (Wuhan Boster Biological Engineering Co., LTD) at 37°C for 2 h. Finally, a DAB kit was used for chromogenic reaction. After counterstaining with hematoxylin, the slides were dehydrated with gradient alcohol and xylene. Normal rabbit immunoglobulin G (IgG) was substituted for a primary antibody as the negative control.
Evaluation of the immunostaining
Samples were considered to be positive for p-Tau protein if buffy granules were observed in the cytoplasm or nuclei. Three high-power fields (magnification ×400) of the CA1 region of the hippocampus were examined under a light microscope. The mean count of the positive neurons from three fields was calculated.
Statistical analysis
All data were shown as mean ± standard deviation (SD) and analyzed using SPSS version 11.0 for Windows software (SPSS, Chicago, IL). Comparisons among groups were carried out using analysis of variance. The difference between the two groups was compared using t test. P < 0.05 was considered to be statistical significant.
Results
Behavioral observation
Before the operation, no difference in the ethologic test was observed in the rats in both the groups (P > 0.05). However, at 2, 4, 8, and 12 weeks after the operation, a significant difference was found in their behaviors, and the number of electric shocks needed in the 2VO group increased significantly (P < 0.01). Moreover, in the 2VO group, the number of electric shocks increased gradually with increase in time, thereby showing a significant difference (Table 1).
Table 1.
The number of electric shocks needed for rats to meet “learned” standard (n = 5)
| Groups | Preoperative | Postoperative | |||
|---|---|---|---|---|---|
|
| |||||
| 2 w | 4 w | 8 w | 12 w | ||
| Sham operation | 10.25±3.32 | 11.45±3.76 | 10.45±2.96 | 10.85±3.43 | 9.75±2.68 |
| 2VO | 10.75±3.93 | 24.15±3.28* | 32.65±4.14* | 43.05±4.86**,Δ | 51.95±5.16**,Δ |
Data were shown as mean ± SD;
P < 0.01, compared with sham operation group;
P < 0.001, compared with sham operation group;
P < 0.01, compared among the time points.
Histopathological examination
H&E staining revealed no significant infarction in the cerebral cortex or hippocampus in both the groups. Pyramidal cells in the CA1 region of the rat hippocampus were arranged closely and were ordered in the control group, with round and big nuclei. The nuclei were weakly stained and the nucleoli were clear. No damage, such as clear concentrated and hyperchromatic cytoplasm, karyopyknosis, and other neuron degeneration were found in the control group. However, in the 2VO group, obvious abnormalities, such as loose arrangement of cells, reduced cell number, and morphological abnormalities, were found in the CA1 region of the rat hippocampus. Some cells also showed reduced volume, hyperchromatic nuclei, and karyopyknosis.
Tau protein in brain tissues
Immunoreactivity of the Tau protein was mainly located in the cytoplasm. The number of p-Tau protein positive cells in the CA1 region of the rat hippocampus increased markedly in the 2VO group compared with that in the control group, and this increased in a time-dependent manner (Figures 1, 2 and 3; Table 2).
Figure 1.
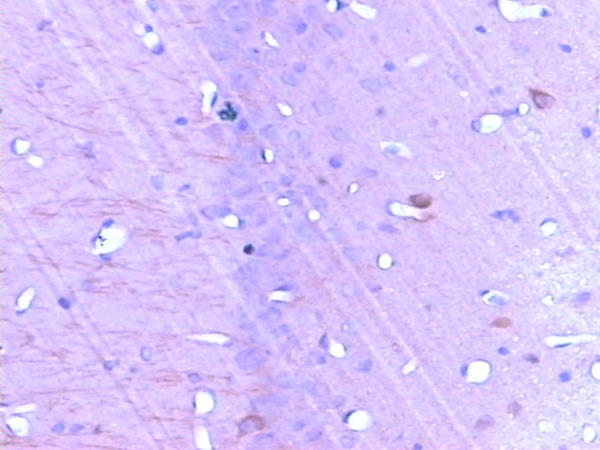
The expression of p-Tau in the CA1 region of hippocampus before operation (SABC, ×200).
Figure 2.
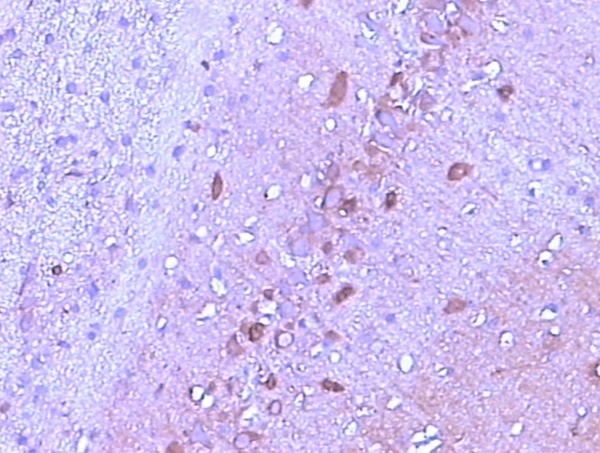
The expression of p-Tau in the CA1 region of hippocampus at 2 w after operation (SABC, ×200).
Figure 3.
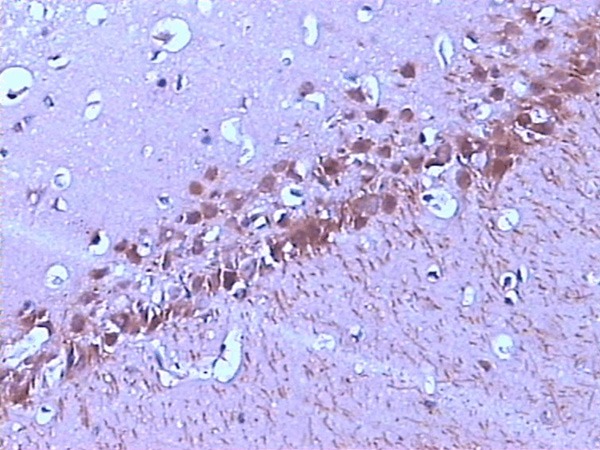
The expression of p-Tau in the CA1 region of hippocampus at 12 w after operation (SABC, ×200).
Table 2.
Comparisons of the number of p-Tau protein positive cells in the CA1 region of rat hippocampus (n = 5)
| Groups | Preoperative | Postoperative | |||
|---|---|---|---|---|---|
|
| |||||
| 2 w | 4 w | 8 w | 12 w | ||
| Sham operation | 12.34±3.56 | 13.25±4.16 | 12.21±3.06 | 14.06±3.23 | 13.81±3.78 |
| 2VO | 13.15±3.63 | 31.25±4.08* | 45.65±4.94* | 58.63±5.26**,Δ | 71.95±7.08**,Δ |
Data were shown as mean ± SD;
P < 0.01, compared with sham operation group;
P < 0.001, compared with sham operation group;
P < 0.01, compared among the time points.
Discussion
In recent years, many of the epidemiological, clinical, and pathological studies have shown that long-term cerebral blood supply shortage and free radical damage induced by chronic hypoperfusion may be the causes of AD [14,15]. Till date, a majority if reports in the risk factors of AD include vascular factors that reduce cerebral blood flow perfusion [16]. Epidemiological investigations show that many risk factors associated with vascular factors or hemodynamics, such as hypertension, diabetes, hypercholesterolemia, cerebral thrombosis, high level of fibrinogen, high serum homocysteine, arteriosclerosis, fibrillation atrial, smoking, and alcohol poisoning, can increase the risk of AD. All of these factors can reduce or damage normal cerebral perfusion. AD is a progressive degenerative disease of the central nervous system with clinical presentations of progressive cognitive hypofunction. It is pathologically characterized by the presence of beta-amyloid in the cerebral cortex and hippocampus, formation of neural plaques or age spots, and accumulation of abnormally phosphorylated Tau protein causing nerve fiber entanglement in cranial nerve cells.
Tau is a microtubule-related protein, associated with mitosis, intracellular transport, and other functions. Normally, Tau protein exists in a balance of phosphorylation and dephosphorylation in the human brain [17]. Excessive phosphorylation of Tau protein induces change in its physiological function and is thought to play an important role in the pathogenesis of AD. However, the initiating factors are still unclear, especially the relationship between a blood circulation disorder and Tau protein as well as its phosphorylated state.
In this study, we found that the expression of p-Tau protein increased in the CA1 region of the hippocampus in a cerebral hypoperfusion rat model, and the cognitive function of rats was disturbed. This may be associated with a change in the phosphorylation in cerebral hypoperfusion. Such a phenomenon can also be found in focal cerebral ischemia [18]. The pathophysiological mechanism of cerebral hypoperfusion status influencing cognitive function is complicated and may be associated with various factors. The first factor is the presence of energy metabolic disorders. Results of animal experiments on chronic cerebral ischemia [19,20] suggest that chronic reduction in the cerebral blood flow plays an initiating role in the process of cognitive decline. Energy metabolic disorders began when cerebral blood flow was reduced to a critical threshold, i.e., 60-75% of the normal level, or the nutrients transported to neurons and glial cells were decreased to a certain concentration. This resulted in a series of reactions including ion pump failure and the inhibition of protein synthesis, transcription, translation, glutamate uptake, axoplasmic transportation, neural transmission, and synaptic activity. Dissemination of information in the brain cells is blocked and eventually leads to cognitive impairment. The second factor is protein metabolism abnormality in the brain. It has been reported that chronic cerebral hypoperfusion can cause abnormal expression of amyloid β-protein and microvascular JRP-1 protein, which are closely related to cognitive function [21-23]. The third factor is oxidative stress. Chronic cerebral hypoperfusion has been proven to activate oxidative stress pathways and produce a large amount of oxygen-derived free radicals, which may cause neuron damage in the striatum and hippocampus, as well as acetylcholine system dysfunction, thereby leading to cognitive dysfunction [16,24,25]. The fourth factor is inflammatory response. Activity of immune cells can be elevated by chronic cerebral hypoperfusion, which is associated with the degree of brain damage and cognitive impairment [26]. With increase in immune cell activity, brain damage and cognitive impairment aggravate gradually, thereby suggesting that chronic cerebral hypoperfusion can promote the occurrence and development of cognitive impairment. In the 2VO rat model, the brain was in a persistent low perfusion state and excessive phosphorylation of the Tau protein was found in the hippocampus. Excessively phosphorylated Tau protein not only competes with normal microtubule-associated protein and influences the formation of microtubules but also promotes the separation of the normal microtubule-associated protein from microtubules, leading microtubule disintegration. It also leads to sediment formation in the neurons, thereby leading to neuron damage [27]. These processes may be important in the pathophysiology of AD. Excessively phosphorylated Tau protein aggregates in neurons and forms nerve fiber tangles, which is a classic pathological characteristic of AD. From the above results, it can be seen that cerebral hypoperfusion may lead to excessive expression of Tau protein in the brain, thereby affecting the pathophysiological process of AD as an initiating factor of excessive phosphorylation of the Tau protein.
In this study, we dynamically observed the expression of p-Tau protein expression and cognitive function in experimental rats with different periods of cerebral hypoperfusion. The results showed that the expression of p-Tau protein increased with an increase in the hypoperfusion time and the degree of cognitive dysfunction aggravated as well. Studies have shown that the p-Tau protein is the main element of neurofibrillary tangles. Excessive p-Tau protein accumulates in the cell body of degenerated neurons and is positively correlated with the degree of clinical dementia in patients with AD [28]. The pathological changes of Tau protein occur before the onset of dementia symptoms and are independent of β-amyloid anomalies [29]. This indicates that excessive phosphorylation of the Tau protein plays an important role in promoting nerve cell degeneration. Sustained hypoperfusion causes excessive Tau phosphorylation that aggregates in neurons, leading to progressive exacerbation of cognitive dysfunction seen in AD [30]. Therefore, chronic cerebral hypoperfusion plays an important role in the occurrence and development of AD and excessive p-Tau protein may play an important role in this mechanism.
In conclusion, cerebral hypoperfusion can cause excessive phosphorylation of the Tau protein in the brain, thereby resulting in cognitive dysfunction. Cerebral hypoperfusion is involved in the development of AD and may be an important initiating and promoting factor. Thus, further studies are warranted to determine the relationship between cerebral hypoperfusion and AD, the results of which can aid in the discovery of a new effective method to determine the occurrence and development of AD.
Acknowledgements
This study are supported by the grant from the Science and Technology Development Program of Shandong province (No. 2014GSF118106).
Disclosure of conflict of interest
None.
References
- 1.Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, Hall K, Hasegawa K, Hendrie H, Huang Y, Jorm A, Mathers C, Menezes PR, Rimmer E, Scazufca M. Alzheimer’s Disease International. Global prevalence of dementia: a Delphi consensus study. Lancet. 2005;366:2112–2117. doi: 10.1016/S0140-6736(05)67889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang R, Liu J, Khan A, Tseng B, Tarumi T, Armstrong K, Hill C, Martin-Cook K, Weiner M, Cullum M. Brain hypoperfusion and neurovascular decoupling in amnestic mild cognitive impairment. Alzheimers Dement. 2013;9:775. [Google Scholar]
- 3.Ferreira ED, Romanini CV, Mori MA, de Oliveira RM, Milani H. Middle-aged, but not young, rats develop cognitive impairment and cortical neurodegeneration following the four-vessel occlusion/internal carotid artery model of chronic cerebral hypoperfusion. Eur J Neurosci. 2011;34:1131–1140. doi: 10.1111/j.1460-9568.2011.07824.x. [DOI] [PubMed] [Google Scholar]
- 4.De la Torre JC. Is Alzheimer’s disease preceded by neurodegeneration or cerebral hypoperfusion? Ann Neurol. 2005;57:783–784. doi: 10.1002/ana.20516. [DOI] [PubMed] [Google Scholar]
- 5.Vemuri P, Przybelski SA, Gunter JL, Senjem ML, Boeve BF, Knopman DS, Petersen RC, Jack CR. Quantitative measurement of cerebral hypoperfusion and atrophy in Alzheimer’s disease using MRI. Alzheimers Dement. 2009;5:78. [Google Scholar]
- 6.Aliev G, Shenk JC, Fischbach K, Cobb CJ, Pacheco GJ, Gasimov E, Perry G. Cerebral hypoperfusion-induced oxidative stress and mitochondrial failure as initiators of aging and Alzheimer’s disease pathology. Alzheimers Dement. 2008;4:T721. [Google Scholar]
- 7.Cai Z, Yan Y, Sun S, Zhang J, Huang L, Yan L, Li J. Upregulation of BACE1 and β-amyloid protein mediated by chronic cerebral hypoperfusion contributes to cognitive impairment and pathogenesis of Alzheimer’s Disease. Neur Res. 2009;34:1226. doi: 10.1007/s11064-008-9899-y. [DOI] [PubMed] [Google Scholar]
- 8.Jack CR, Barrio JR, Kepe V. Cerebral amyloid PET imaging in Alzheimer’s disease. Acta Neuropathologica. 2013;126:643–657. doi: 10.1007/s00401-013-1185-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herholz K. Cerebral glucose metabolism in preclinical and prodromal Alzheimer’s disease. Expert Rev Neurother. 2010;10:1667–1673. doi: 10.1586/ern.10.136. [DOI] [PubMed] [Google Scholar]
- 10.Marmarelis VZ, Shin DC, Orme ME, Zhang R. Model-based quantification of cerebral hemodynamics as a physiomarker for alzheimer’s disease? Ann Biomed Eng. 2013;41:2296–2317. doi: 10.1007/s10439-013-0837-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gurol ME. Cerebral hypoperfusion and white matter disease in healthy elderly and patients with Alzheimer’s disease. Eur J Neurol. 2013;20:214–215. doi: 10.1111/j.1468-1331.2012.03865.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sokolowemail S, Luu SH, Vinters HV, Miller CA, Gylys KH. Glutamatergic (VGLUT1) synapses are not lost in Alzheimer’s disease cerebral cortex, but preferentially accumulate amyloid-beta and p-Tau pathology. Alzheimers Dement. 2010;6:S243. [Google Scholar]
- 13.De la Torre JC, Pappas BA, Prevot V, Emmerling MR, Mantione K, Fortin T, Watson MD, Stefano GB. Hippocampal nitric oxide upregulation precedes memory loss and A beta 1-40 accumulation after chronic brain hypoperfusion in rats. Neurol Res. 2003;25:635–641. doi: 10.1179/016164103101201931. [DOI] [PubMed] [Google Scholar]
- 14.De la Torre JC. Alzheimerps disease is a vasocognopathy: a newterm to describe its nature. Neurol Res. 2004;26:517–524. doi: 10.1179/016164104225016254. [DOI] [PubMed] [Google Scholar]
- 15.Koistinaho M, Koistinaho J. Interactions between Alzheimer’s disease and cerebral ischemia--focus on inflammation. Brain Res Brain Res Rev. 2005;48:240–250. doi: 10.1016/j.brainresrev.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 16.Liu J, Zhu YS, Khan MA, Brunk E, Martin-Cook K, Weiner MF, Cullum CM, Lu H, Levine BD, Diaz-Arrastia R, Zhang R. Global brain hypoperfusion and oxygenation in amnestic mild cognitive impairment. Alzheimers Dement. 2014;10:162–170. doi: 10.1016/j.jalz.2013.04.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gong CX, Iqbal K. Hyperphosphorylation of microtubule-associated protein Tau: a promising therapeutic target for Alzheimer disease. Curr Med Chem. 2008;15:2321–2328. doi: 10.2174/092986708785909111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wen Y, Yang S, Liu R, Simpkins JW. Transient cerebral ischemia induces site-specific hyperphosphorylation of tau protein. Brain Res. 2004;1022:30–38. doi: 10.1016/j.brainres.2004.05.106. [DOI] [PubMed] [Google Scholar]
- 19.Otori T, Katsumata T, Muramatsu H, Kashiwagi F, Katayama Y, Terashi A. Long-term measurement of cerebral blood flow and metabolism in a rat chronic hypoperfusion model. Clin Exp Pharmacol Physiol. 2003;30:266–272. doi: 10.1046/j.1440-1681.2003.03825.x. [DOI] [PubMed] [Google Scholar]
- 20.Staffen W, Bergmann J, Schönauer U, Zauner H, Kronbichler M, Golaszewski S, Ladurner G. Cerbral perfusion( HMPAO-SPECT) in patients with depression with cognitive impairment versus those with mild cognitive impairment and dementia of Alzheimer’s type: a semiquantitative and automated evalution. Eur J Nucl Med Mol Imaging. 2009;36:801–810. doi: 10.1007/s00259-008-1028-2. [DOI] [PubMed] [Google Scholar]
- 21.Zhao J, Fu Y, Yasvoina M, Shao P, Hitt B, O’Connor T, Logan S, Maus E, Citron M, Berry R, Binder L, Vassar R. Beta-site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer’s disease pathogenesis. J Neurosci. 2007;27:3639–3649. doi: 10.1523/JNEUROSCI.4396-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamada M, Ihara M, Okamoto Y, Maki T, Washida K, Kitamura A, Hase Y, Ito H, Takao K, Miyakawa T, Kalaria RN, Tomimoto H, Takahashi R. The Influence of Chronic Cerebral Hypoperfusion on Cognitive Function and Amyloid β Metabolism in APP Overexpressing Mice. PLoS One. 2011;6:e16567. doi: 10.1371/journal.pone.0016567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mancuso M, Orsucci D, Siciliano G, Murri L. Mitochondria, mitochondrial DNA and Alzheimer’s disease. What comes first? Curr Alzheimer Res. 2008;5:457–468. doi: 10.2174/156720508785908946. [DOI] [PubMed] [Google Scholar]
- 24.Liu H, Zhang J. Cerebral Hypoperfusion and Cognitive Impairment: The Pathogenic Role of Vascular Oxidative Stress. Int J Neurosci. 2012;122:494–499. doi: 10.3109/00207454.2012.686543. [DOI] [PubMed] [Google Scholar]
- 25.Kim HA, Miller AA, Drummond GR, Thrift AG, Arumugam TV, Phan TG, Srikanth VK, Sobey CG. Vascular cognitive impairment and Alzheimer’s disease: role of cerebral hypoperfusion and oxidative stress. Naunyn Schmiedebergs Arch Pharmacol. 2012;385:953–959. doi: 10.1007/s00210-012-0790-7. [DOI] [PubMed] [Google Scholar]
- 26.Vicente E, Degerone D, Bohn L. Astroglial and cognitive effects of chronic cerebral hypoperfusion in the rat. Brain Res. 2009;1251:204–212. doi: 10.1016/j.brainres.2008.11.032. [DOI] [PubMed] [Google Scholar]
- 27.Amniai L, Barbier P, Sillen A, Wieruszeski JM, Peyrot V, Lippens G, Landrieu I. Alzheimer disease specific phosphoepitopes of Tau interfere with assembly of tubulin but not binding to microtubules. FASEB J. 2009;23:1146–1152. doi: 10.1096/fj.08-121590. [DOI] [PubMed] [Google Scholar]
- 28.Iqbal K, Alonso Adel C, El-Akkad E, Gong CX, Haque N, Khatoon S, Pei JJ, Tanimukai H, Tsujio I, Wang JZ, Grundke-Iqba I. Alzheimer neurofibrillary degeneration: therapeutic targets and high-throughput assays. J Mol Neurosci. 2003;20:425–429. doi: 10.1385/jmn:20:3:425. [DOI] [PubMed] [Google Scholar]
- 29.Delacourte A, Sergeant N, Champain D, Wattez A, Maurage CA, Lebert F, Pasquier F, David JP. Nonoverlapping but synergetic Tau and APP pathologies in sporadic Alzheimer’s disease. Neurology. 2002;59:398–407. doi: 10.1212/wnl.59.3.398. [DOI] [PubMed] [Google Scholar]
- 30.Du AT, Jahng GH, Hayasaka S, Kramer JH, Rosen HJ, Gorno-Tempini ML, Rankin KP, Miller BL, Weiner MW, Schuff N. Hypoperfusion in frontotemporal dementia and Alzheimer disease by arterial spin labeling MRI. Neurology. 2006;67:1215–1220. doi: 10.1212/01.wnl.0000238163.71349.78. [DOI] [PMC free article] [PubMed] [Google Scholar]