

Abstract
A tetra(aniline)-based cationic amphiphile, TANI-NHC(O)C5H10N(CH3)3+Br– (TANI-PTAB) was synthesized, and its emeraldine base (EB) state was found to self-assemble into nanowires in aqueous solution. The observed self-assembly is described by an isodesmic model, as shown by temperature-dependent UV–vis investigations. Linear dichroism (LD) studies, combined with computational modeling using time-dependent density functional theory (TD-DFT), suggests that TANI-PTAB molecules are ordered in an antiparallel arrangement within nanowires, with the long axis of TANI-PTAB arranged perpendicular to the nanowire long axis. Addition of either S- or R- camphorsulfonic acid (CSA) to TANI-PTAB converted TANI to the emeraldine salt (ES), which retained the ability to form nanowires. Acid doping of TANI-PTAB had a profound effect on the nanowire morphology, as the CSA counterions’ chirality translated into helical twisting of the nanowires, as observed by circular dichroism (CD). Finally, the electrical conductivity of CSA-doped helical nanowire thin films processed from aqueous solution was 2.7 mS cm–1. The conductivity, control over self-assembled 1D structure and water-solubility demonstrate these materials’ promise as processable and addressable functional materials for molecular electronics, redox-controlled materials and sensing.
Introduction
Self-assembly1 provides facile, bottom-up access to complex ordered morphologies that can improve material properties, such as charge carrier mobility (μ) and conductivity (σ), and lead to novel emergent properties. Much work has traditionally been focused on self-assembly and processing of materials from organic solvents; however, water-soluble materials present considerable benefits. To this end there has been increasing interest in water-soluble “functional amphiphiles”, typically oligomers or small organic molecules that are covalently connected to hydrophilic head groups causing water solubility and that can undergo self-assembly.2−4 Water solubility, while facilitating routes to biocompatible and sustainable processability, is also advantageous for self-assembly of functional organic materials, where water can promote structure formation5−7 and boost the strength of morphology-directing intermolecular π–π interactions.8 Self-assemblies in aqueous environments of interest for applications include anisotropic 1D fiber- or wire-like structures (to improve charge transport properties of organic semiconductors),9−12 and 3D gel networks, to modify rheological or mechanical properties.13
One class of conjugated oligomeric materials for which aqueous self-assembly is nearly unknown is oligo(aniline)s:14 short (4-, 8- or 16-mer) analogues of the well-known conducting polymer poly(aniline) (PANI).15,16 Oligo(aniline)s, and especially the synthetically accessible tetramer, TANI, have been investigated for their interesting and varied self-assemblies in the contexts of thin films,17,18 crystalline microstructures19−21 and block-like structures.22 The acid/base- and redox-switchable properties of TANI, as well as its switchable transition to a conductive salt state, echoes that of PANI.23 In addition, the improved processability and control over molecular structure inherent to oligomers and small molecules make these materials attractive for further exploitation. However, very few reports of self-assembly exist for oligo(aniline)s compared with other conjugated oligomers such as oligo(thiophene), indicative of the challenges posed by aniline-based materials.
Oligo(thiophene)24 is the most commonly reported π-conjugated motif on which water-soluble functional amphiphiles are based. Variations in the conjugation length from sexi-,25,26 quinque-,27,28 tetra-,28,29 to terthiophenes,30,31 architecture from bola-25−27 to single-headed amphiphiles28,29 and choice of the polar headgroup from cationic,25 nonionic ethylene oxide26,29 to peptidic27,28,32 are used to tune the solution self-assembly, the resultant morphology, and material properties. Other functional and conjugated organic motifs that have been applied as functional amphiphiles include derivatives of pyrene,33 perylene,4,34 oligo(fluorene)s35−37,24−26 distyrylbenzenes27,28,38,39 and p-phenylenevinylenes.40
Solubility of TANI in water is, as is typical for a π-conjugated oligomer, intrinsically poor. While there are some reports of attempts to improve water-solubility by covalently inducing amphiphilicity, no previous example of a cationic amphiphile exists. Previously, anionic bola-amphiphiles based on penta(aniline) oligomers were reported for their corrosion inhibition properties.41 More frequently, poly(ethylene oxide) blocks have been used to promote water-solubility in adducts with TANI.42,43 We chose a cationic headgroup to decouple the water-solubility from effects of pH and doping of the oligo(aniline). Previously reported anionic oligo(aniline) amphiphiles were self-doped by the acidic sulfonic acid headgroup,41 preventing both study of the native EB state material or the ability to control doping to tune structure, as shown here. Further, where the self-assembly has been investigated, these attempts have only produced zero-dimensional (0D) micelle or vesicle nanostructures. Higher dimensionality is desirable for a host of applications, such as organic electronics, where a 1D self-assembling conducting nanowire, although not often achieved, is an attractive component.44
We report here on the synthesis and self-assembly of the first TANI-based amphiphile bearing a cationic headgroup. TANI was end-functionalized with an amide-linked five-carbon spacer bearing a trimethylammonium headgroup; together, these features endow water-solubility and cause spontaneous self-assembly into high aspect-ratio 1D nanowires. The thermodynamics of self-assembly were studied, with self-assembly found to be strongly enthalpically favored. The highly anisotropic structures could be tuned to form conductive nanowires with induced helicity from addition of an enantiomerically pure chiral sulfonic acid dopant, demonstrating the versatility of the TANI motif as a platform for designing customizable supramolecular structures. Facile processing into thin films of nanowires from aqueous solution was demonstrated, with conductivity measurements of these films showing the formation of 1D conducting nanowires. We believe these attractive features pave the way to further studies on this hitherto under-investigated class of material.
Results and Discussion
Synthesis of Oligo(Aniline) Amphiphile TANI-PTAB
The TANI motif was used here as a functional and conjugated core for the novel amphiphile TANI-PTAB (Figure 1a). TANI was synthesized using the Buchwald-Hartwig amination methodology45−47 combined with an orthogonal protecting-group strategy to yield an asymmetric tetra(aniline) precursor species (Ph/NH2 TANI). The addition of an amide-linked C5 spacer and a quaternary trimethylammonium bromide yielded the water-soluble functional amphiphile TANI-PTAB in the nonconducting emeraldine base (EB) state. Full synthetic details are available in the Supporting Information.
Figure 1.

(a) Molecular structure and DFT-optimized space-filling model of TANI-NHC(O)C5H10N(CH3)3+Br– (TANI-PTAB). (b) Concentration dependent UV–vis spectra of TANI-PTAB in water (arrow indicates change on increasing concentration), inset: linear dependence of absorbance on concentration (values taken at λmax = 580 nm). (c) Critical aggregation concentration for aqueous TANI-PTAB solution, measured by pyrene fluorescence, CAC = 1 × 10–4 M.
Self-Assembly of EB TANI-PTAB
Aqueous solutions of highly water-soluble EB TANI-PTAB were found to show concentration-dependent shifts in UV–vis absorbance (Figure 1b), suggesting aggregation was occurring. Solutions exhibited a shoulder at 490 nm, as well as a 32 nm bathochromic shift in absorbance at high concentrations (λmax = 582 nm for 1 × 10–3 M vs 550 nm for 1 × 10–5 M solutions, in keeping with previous reports).17 The onset of these changes was at 1 × 10–4 M, which was confirmed as a critical aggregation concentration (CAC) by pyrene fluorescence measurements (Figure 1c).48−50 The bathochromic shifts in absorbance observed with increasing concentration were reproduced when the polarity of the solvent was decreased by successive addition of aliquots of THF to a sub-CAC aqueous TANI-PTAB solution; however, no shoulder at 490 nm was seen (Figure S3). This suggests that in water at concentrations above the CAC, the TANI chromophore is sequestered within the less polar interior of aggregates, and aggregation results in vibronic coupling, with emergence of a shoulder peak. The broad absorbance at 550 nm was assigned to the HOMO–LUMO transition of aq. TANI-PTAB based on the main TD-DFT-calculated molecular orbital (MO) contributions to the energy of the transition to the first singlet excited state (2.41 eV, 514 nm; CAM-B3LYP/6-31G*//B3LYP/6-31G* in water PCM (polarizable continuum model), see the Supporting Information for full method and further details, Figure S8).
The aggregate morphology of EB TANI-PTAB was investigated; transmission electron microscopy (TEM) and cryo-TEM (Figure 2) showed that highly anisotropic nanowires were formed with no external treatment. Nanowires with an individual width of 3 nm (Figure 2c) formed fibrous bundles. The nanowire width is comparable to the length calculated for a single TANI-PTAB molecule (2.9 nm), suggesting truly 1D aggregates were formed. Further morphological information on the aggregates in solution was provided by small-angle X-ray scattering (SAXS) experiments (Figure S5a). Fitting the data to a thin plate model (detailed further in the Supporting Information) suggested structures with thickness of approximately 3 nm formed in solution, which is consistent with measurements from analysis of TEM images.
Figure 2.

(a) TEM (stained with 1% uranyl acetate) and (b) cryo-TEM (unstained) of EB TANI-PTAB forming bundles of nanofibers in aqueous solution (4 × 10–3 M and 1 × 10–3 M, respectively). (c) Histogram of the measured widths of nanofibers within bundles, (d) enlarged section of cryo-TEM image showing nanofiber bundles. Scale bars: 50 nm (a, b), 10 nm (d).
The mechanism of self-assembly for TANI-PTAB was investigated by temperature-dependent UV–vis spectroscopy (Figure 3). Heating a 1 × 10–3 M aqueous solution of TANI-PTAB at a rate of 1 K min–1 and sampling spectra at 5 K intervals from 288 K up to 338 K revealed a hypsochromic shift of 32 nm from 582 to 550 nm. The disappearance of a shoulder at 492 nm was also observed, such that the high-temperature spectra resembled those of the molecularly dissolved species in solution at sub-CAC concentrations. The parameter α(T), representing the variation in mole fraction of the aggregated species with temperature, fitted well to a sigmoidal curve (Figure 3, inset), indicative of self-assembly behavior within an isodesmic, or equal-K, model.51
| 1 |
Figure 3.

Temperature-dependent UV–vis for TANI-PTAB (1 × 10–3 M, aqueous solution). Arrow indicates direction of change on increasing temperature. Inset: the mole fraction of aggregated molecules, α(T), as a function of temperature.
From eq 1, A(T) is the measured absorbance at temperature T, Amon and Aagg are the absorbance values of the monomer and fully aggregated states, respectively. Values were taken at λmax = 492 nm, corresponding to the shoulder observed for aggregated species in solution. Thermodynamic parameters were calculated from a Van’t Hoff plot (Figure S4); these values and the melting temperature, Tm, of the aggregates (the temperature, T, at which α(T) = 0.5) are listed in Table 1. Large enthalpic gains, which offset the entropic cost of aggregation, are likely to result from the release of high-energy interfacial water molecules into the bulk5 and the increased interaction between π-conjugated TANI moieties.
Table 1. Thermodynamic Properties of TANI-PTAB Self-Assembly (1 × 10–3 M).
| TANI-PTAB | |
|---|---|
| Tm (K) | 309 |
| ΔH (kJ mol–1) | –180 |
| ΔS (J mol–1 K–1) | –497 |
EB TANI-PTAB solutions above the CAC were highly viscous, likely due to the self-assembled fiber network in solution, and could be shear-aligned. This was advantageous for linear dichroism (LD) studies, where aligned samples were used to investigate the nanowires’ molecular structure in solution. Shear was applied to a TANI-PTAB solution, causing alignment of anisotropic aggregates in the direction of their long axis. Any preference for absorbance of light polarized either parallel (A∥) or perpendicular (A⊥) to the shear direction is indicative of the orientation of electronic transition dipole moments (TDMs) relative to the fiber axis.
A negative LD signal was observed for the HOMO–LUMO45 transition of EB TANI-PTAB, indicating greater A⊥ (Figure 4a). To relate LD data to the molecular structure of TANI-PTAB, TD-DFT modeling of the excited state determined the orientation of the TDM for the HOMO–LUMO transition is orientated along the long axis of the TANI moiety (exact Cartesian coordinates for the TDM can be found in the Supporting Information). For a strong absorbance, this TDM must be aligned in the same direction as the polarization of light. The calculated orientation of the TDM, along with LD results, suggests that within nanowires TANI-PTAB molecules are arranged normal to the long axis of the nanowires.
Figure 4.
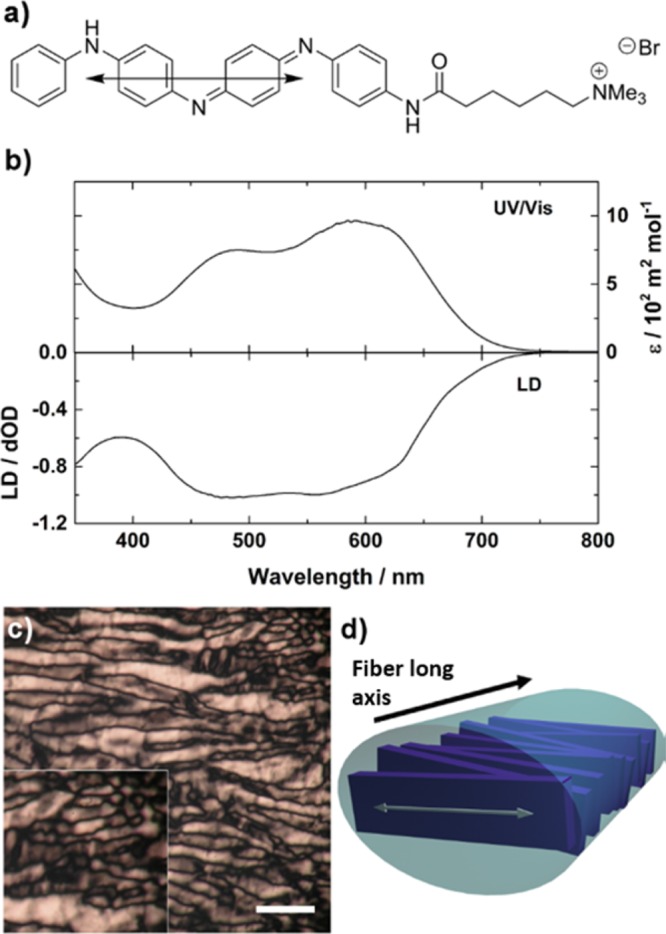
(a) Calculated orientation of the transition dipole moment relative to the EB TANI-PTAB molecular structure. (b) LD and UV–vis spectra of the HOMO–LUMO spectral region of TANI-PTAB (4 × 10–3 M). (c) Birefringent optical textures observed under crossed polars for TANI-PTAB between glass plates (4 × 10–3 M), scale bar: 100 μm. (d) Proposed alignment of TANI chromophores within nanofibers, with transition dipole moment (white arrow) overlaid, relative to fiber axis (black arrow).
Polarized optical microscopy (POM) investigations revealed that these solutions displayed birefringent optical textures typical of chromonic lyotropic nematic phases (Figure 4c).52,53 Chromonic LC phases consist of cofacial stacks of water-soluble aromatic molecules that assemble via an isodesmic mechanism. Based on LD, TD-DFT and POM, and by analogy to other chromonic dyes, amphiphiles based on perylene diimides54 and oligo(aniline) in the solid state,19 we suggest the TANI chromophores are stacked cofacially, normal to the fiber axis. A disordered, loosely cylindrical stack that allows maximal shielding of the hydrophobic TANI from water by the cationic headgroups is likely, though further investigation by X-ray diffraction would be required for a definitive picture of the cationic headgroup positions within nanowires.
Acid Doping: Controlling Self-Assembly in Solution
An appealing feature of oligo(aniline)-based materials is their reversible protonic doping, allowing switching between EB and emeraldine salt (ES) states via protonation of quinoid N atoms of the EB state. The acid counterion is included in the doped material,55,56 enabling customization of the morphology of self-assembled structures by selection of the acid.19,57 Control over self-assembly in an aqueous environment, with no requirement for solvent mixtures, solvent exchange or slow crystallization processes, is an advantage that we demonstrate here with TANI-PTAB.
Camphorsulfonic acid is a typical dopant for PANI that enables high conductivity.58 Addition of enantiomerically pure camphorsulfonic acid (S-CSA) to EB TANI-PTAB in aqueous solution caused a color change from blue to bright green. This change is associated with formation of a polaron species containing cation radicals, typified in PANI systems by absorbances at 440 nm and, for compact coil conformations at 780 nm.59 As TANI is much shorter than PANI, and thus unable to form extended coil conformations, no true “free-carrier tail” absorbance band was observed. However, the observed absorbance bands suggest polaron formation (Figure 5) and the absence of absorbance in the HOMO–LUMO transition region of EB TANI-PTAB (λmax = 550 nm) indicated that EB TANI-PTAB was fully doped by CSA (to yield ES-state TANI(CSA)2-PTAB).
Figure 5.

UV–vis spectra showing the transition from doped ES TANI(CSA)2-PTAB to dedoped EB TANI-PTAB with dilution. Arrows indicate change on diluting TANI(CSA)2-PTAB (1 × 10–4 M).
TEM and cryo-TEM (Figure 6) confirmed the presence of nanowires even after protonation. Image analysis showed that the width of individual wires within the bundles increased to approximately 6 nm when CSA was added, suggesting incorporation of the CSA counterions into the fiber structure (Figure 6c).
Figure 6.
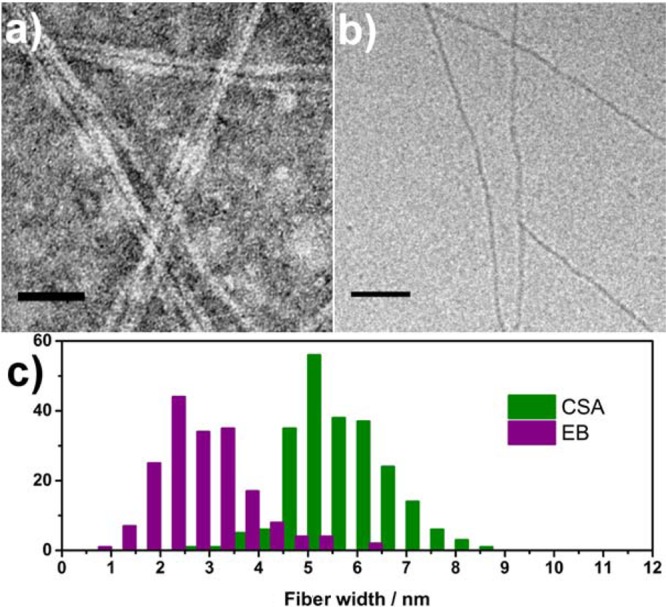
(a) TEM (stained with 1% uranyl acetate) and (b) cryo-TEM (unstained) of TANI(CSA)2-PTAB (4 × 10–3 M). (c) Histogram showing comparison of measured nanowire widths for EB and CSA-doped TANI-PTAB. Scale bars: 50 nm.
SAXS confirmed the presence of aggregates of TANI(CSA)2-PTAB in solution (Figure S5b). However, the intensity gradient tended to be flat at low Q suggesting a significant structure factor resulting from interaggregate interactions. A precise thickness determination is therefore not possible but the thin plate model fit is consistent with a thickness on the nanometer scale.
Investigation of the doped TANI(CSA)2-PTAB by CD spectroscopy (Figure 7) showed exciton-coupled bisignate spectra indicative of helices in solution. A positive Cotton effect was observed for TANI(S-CSA)2-PTAB, indicating right-handed helicity. TANI(R-CSA)2-PTAB produced a negative Cotton effect, suggesting left-handed helices, while racemically doped TANI(±-CSA)2-PTAB showed no ellipticity by CD. EB TANI-PTAB also showed no helicity (Figure S7). The exciton-coupled spectra correspond to the π* ← polaron transition,60 strongly suggesting that the chiral CSA counterion is complexed to the TANI and inducing local chirality in this chromophore, which is translated into helicity in the nanowire as a whole.
Figure 7.
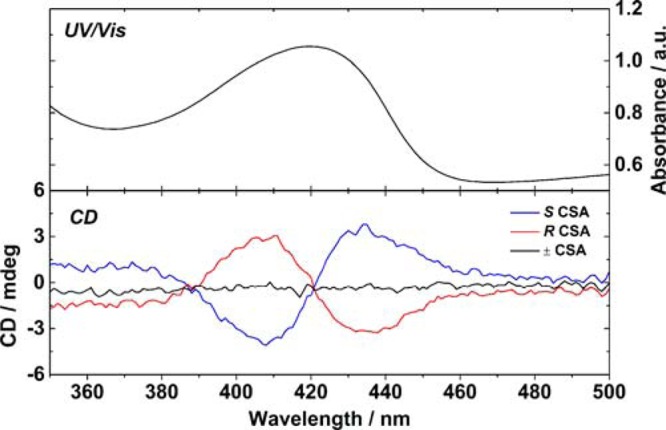
Exciton-coupled CD spectra of TANI-PTAB doped with both oppositely handed CSA enantiomers and racemic CSA to form TANI(CSA)2-PTAB, with UV–vis of the same spectral region.
Thin films of TANI(CSA)2-PTAB could be easily prepared directly from aqueous solution by spin coating onto glass substrates, without the addition of any plasticizing additives or further processing. We investigated the conductivity of these films after drying in vacuum to remove residual water, with prepatterned bottom-contact gold electrodes in a collinear 4-point probe configuration, with electrode separation of 250 μm. Conductivity of 2.7 ± 0.3 mS cm–1 was calculated based on 4-point probe resistance measurements (Figure 8a).61 This is comparable for previous bulk film measurements of TANI,45,62 although lower than the highest observed values for crystalline TANI microstructures.63 However, when compared to the processing steps for producing previously reported films or microcrystals, the facile processing by which TANI-PTAB nanowire thin films were obtained may make TANI-PTAB a more attractive candidate for device applications in future.
Figure 8.
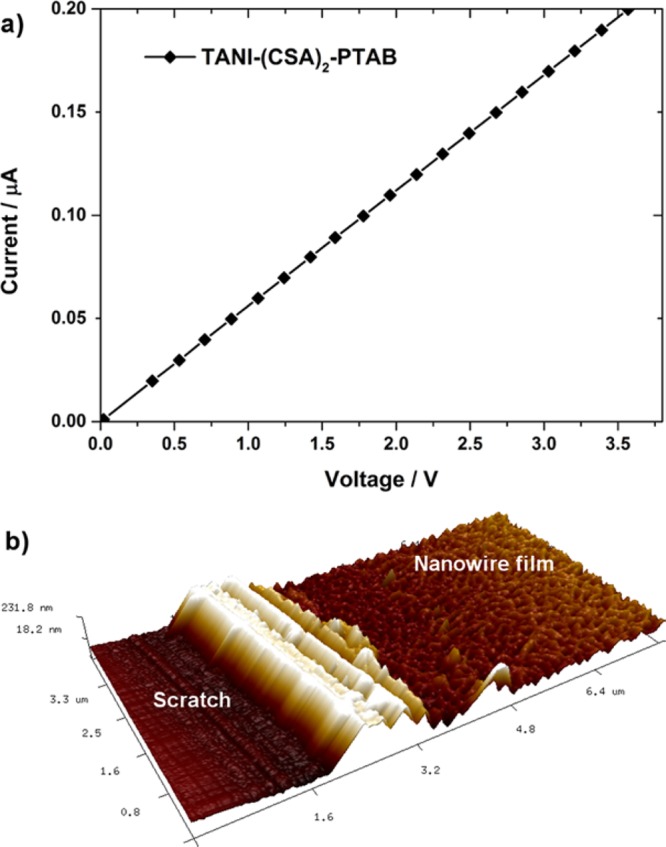
(a) Representative I–V curve for TANI(CSA)2-PTAB spin-cast on glass substrates with prepatterned gold electrodes in bottom-contact collinear 4-point probe configuration (separation 250 μm). (b) AFM image of spin-cast TANI(CSA)2-PTAB film (a scratch has been made in the film to expose the substrate and allow film thickness calculation).
AFM measurements show that films were networks of conductive nanowires and had consistent thickness over large areas (Figure 8b). No conductivity was observed for films of EB TANI-PTAB prepared in an identical manner, as expected for the typically insulating EB-state TANI materials. The conductivity of our TANI(CSA)2-PTAB nanowires is over 300 times greater than reported for a similar self-assembling fiber system (from organic solvents) based on oligo(thiophene)s, further demonstrating the advantage of our material.64
Conclusion
A novel tetra(aniline)-based amphiphile has been prepared and found to self-assemble into nanofibers. This process was investigated by a variety of complementary methods. Self-assembly fitted an isodesmic model, with SAXS and cryo-TEM confirming the presence of nanowires of 3 nm width in solution. A model of the structure within the self-assembled nanowires is suggested on the basis of LD and TD-DFT. The morphology of the self-assembled nanowires could be controlled by addition of a chiral acid dopant, which induced helicity in the nanowires, with handedness dependent on the dopant enantiomer used. These nanowires could be spin-coated from aqueous solution into thin films of conducting nanowires on glass substrates, exhibiting conductivity of 2.7 ± 0.3 mS cm–1. We have demonstrated a new and promising amphiphilic oligo(aniline) material that, with facile aqueous processing, tunable conductivity and supramolecular morphology, has great potential for use in devices and sensors.
Acknowledgments
This work was supported by the EPSRC (EP/J500379/1), ERC (20110209), and the Academy of Finland. This work made use of the Aalto University Nanomicroscopy Centre (Aalto-NMC) instrumentation. The Ganesha X-ray scattering apparatus used for this research and the PeakForce atomic force microscopy carried out in the Chemical Imaging Facility, University of Bristol,was purchased under EPSRC Grant “Atoms to Applications” Grant ref. EP/K035746/1. MS analysis was performed on instrumentation bought through the Core Capability for Chemistry Research - Strategic Investment in Mass Spectrometry EPSRC grant (EP/K03927X/1). F. Brömmel thanks the Deutsche Forschungsgemeinschaft for funding under Grant no. BR 4346/1-1. The facilities and staff of the Electron Microscopy Unit, Mass Spectrometry service and NMR service, as well as computing resources from the Centre for Computational Chemistry at the University of Bristol are kindly acknowledged.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b06892.
Instrumentation, experimental details, synthetic methods and characterization, additional UV–vis, Van’t Hoff thermodynamic analysis, SAXS of TANI(CSA)2-PTAB, AFM showing EB TANI-PTAB and R-CSA-doped TANI-PTAB, and computational chemistry details. (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Cölfen H.; Mann S. Angew. Chem., Int. Ed. 2003, 42, 2350–2365 10.1002/anie.200200562. [DOI] [PubMed] [Google Scholar]
- Jatsch A.; Schillinger E.-K.; Schmid S.; Bäuerle P. J. Mater. Chem. 2010, 20, 3563–3578 10.1039/b926594e. [DOI] [Google Scholar]
- Würthner F. Chem. Commun. 2004, 1564–1579 10.1039/b401630k. [DOI] [PubMed] [Google Scholar]
- Weingarten A. S.; Kazantsev R. V.; Palmer L. C.; McClendon M.; Koltonow A. R.; Samuel A. P. S.; Kiebala D. J.; Wasielewski M. R.; Stupp S. I. Nat. Chem. 2014, 6, 964–970 10.1038/nchem.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biedermann F.; Nau W. M.; Schneider H.-J. Angew. Chem., Int. Ed. 2014, 53, 11158–11171 10.1002/anie.201310958. [DOI] [PubMed] [Google Scholar]
- Kauzmann W. In Advances in Protein Chemistry; Elsevier: London, 1959; Vol. 14, pp 1–63. [DOI] [PubMed] [Google Scholar]
- Chandler D. Nature 2005, 437, 640–647 10.1038/nature04162. [DOI] [PubMed] [Google Scholar]
- Hoeben F. J. M.; Jonkheijm P.; Meijer E. W.; Schenning A. P. H. J. Chem. Rev. 2005, 105, 1491–1546 10.1021/cr030070z. [DOI] [PubMed] [Google Scholar]
- Babu S. S.; Prasanthkumar S.; Ajayaghosh A. Angew. Chem., Int. Ed. 2012, 51, 1766–1776 10.1002/anie.201106767. [DOI] [PubMed] [Google Scholar]
- Schenning A. P. H. J.; Meijer E. W. Chem. Commun. 2005, 3245–3258 10.1039/b501804h. [DOI] [PubMed] [Google Scholar]
- Tovar J. D. Acc. Chem. Res. 2013, 46, 1527–1537 10.1021/ar3002969. [DOI] [PubMed] [Google Scholar]
- Diegelmann S. R.; Gorham J. M.; Tovar J. D. J. Am. Chem. Soc. 2008, 130, 13840–13841 10.1021/ja805491d. [DOI] [PubMed] [Google Scholar]
- Morris K. L.; Chen L.; Raeburn J.; Sellick O. R.; Cotanda P.; Paul A.; Griffiths P. C.; King S. M.; O’Reilly R. K.; Serpell L. C.; Adams D. J. Nat. Commun. 2013, 4, 1480. 10.1038/ncomms2499. [DOI] [PubMed] [Google Scholar]
- Wei Z.; Faul C. F. J. Macromol. Rapid Commun. 2008, 29, 280–292 10.1002/marc.200700741. [DOI] [Google Scholar]
- Kang E. Prog. Polym. Sci. 1998, 23, 277–324 10.1016/S0079-6700(97)00030-0. [DOI] [Google Scholar]
- Geniès E. M.; Boyle A.; Lapkowski M.; Tsintavis C. Synth. Met. 1990, 36, 139–182 10.1016/0379-6779(90)90050-U. [DOI] [Google Scholar]
- Dane T. G.; Cresswell P. T.; Bikondoa O.; Newby G. E.; Arnold T.; Faul C. F. J.; Briscoe W. H. Soft Matter 2012, 8, 2824–2832 10.1039/C2SM06492H. [DOI] [Google Scholar]
- Ford W. E.; Gao D.; Scholz F.; Nelles G.; Von Wrochem F. Chem. Mater. 2013, 25, 3603–3613 10.1021/cm401330s. [DOI] [Google Scholar]
- Wang Y.; Tran H. D.; Liao L.; Duan X.; Kaner R. B. J. Am. Chem. Soc. 2010, 132, 10365–10373 10.1021/ja1014184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Liu J.; Tran H. D.; Mecklenburg M.; Guan X. N.; Stieg A. Z.; Regan B. C.; Martin D. C.; Kaner R. B. J. Am. Chem. Soc. 2012, 134, 9251–9262 10.1021/ja301061a. [DOI] [PubMed] [Google Scholar]
- Shao Z.; Yu Z.; Hu J.; Chandrasekaran S.; Lindsay D. M.; Wei Z.; Faul C. F. J. J. Mater. Chem. 2012, 22, 16230–16234 10.1039/c2jm32278a. [DOI] [Google Scholar]
- Udeh C. U.; Fey N.; Faul C. F. J. J. Mater. Chem. 2011, 21, 18137–18153 10.1039/c1jm12557e. [DOI] [Google Scholar]
- Lee K.; Cho S.; Heum Park S.; Heeger A. J.; Lee C.-W.; Lee S.-H. Nature 2006, 441, 65–68 10.1038/nature04705. [DOI] [PubMed] [Google Scholar]
- Mishra A.; Ma C.-Q.; Bäuerle P. Chem. Rev. 2009, 109, 1141–1276 10.1021/cr8004229. [DOI] [PubMed] [Google Scholar]
- Xia C.; Locklin J.; Youk J. H.; Fulghum T.; Advincula R. C. Langmuir 2002, 18, 955–957 10.1021/la015587p. [DOI] [Google Scholar]
- Brustolin F.; Surin M.; Lemaur V.; Romanazzi G.; Sun Q.; Cornil J.; Lazzaroni R.; Sommerdijk N. A. J. M.; Leclère P.; Meijer E. W. Bull. Chem. Soc. Jpn. 2007, 80, 1703–1715 10.1246/bcsj.80.1703. [DOI] [Google Scholar]
- Stone D. A.; Hsu L.; Stupp S. I. Soft Matter 2009, 5, 1990–1993 10.1039/b904326h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaytan A. K.; Schillinger E.-K. K.; Khalatur P. G.; Mena-Osteritz E.; Hentschel J.; Börner H. G.; Bäuerle P.; Khokhlov A. R. ACS Nano 2011, 5, 6894–6909 10.1021/nn2011943. [DOI] [PubMed] [Google Scholar]
- Jiang L.; Hughes R. C.; Sasaki D. Y. Chem. Commun. 2004, 1028–1029 10.1039/b316657k. [DOI] [PubMed] [Google Scholar]
- Van Rijn P.; Janeliunas D.; Brizard A. M.; Stuart M. C. a.; Koper G. J. M.; Eelkema R.; van Esch J. H. New J. Chem. 2011, 35, 558–567 10.1039/C0NJ00760A. [DOI] [Google Scholar]
- Janeliunas D.; Eelkema R.; Nieto-Ortega B.; Ramírez Aguilar F. J.; López Navarrete J. T.; van der Mee L.; Stuart M. C. A.; Casado J.; van Esch J. H. Org. Biomol. Chem. 2013, 11, 8435–8442 10.1039/c3ob41645c. [DOI] [PubMed] [Google Scholar]
- Vadehra G. S.; Wall B. D.; Diegelmann S. R.; Tovar J. D. Chem. Commun. 2010, 46, 3947–3949 10.1039/c0cc00301h. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Bai H.; Chen Q.; Li C.; Shi G. Sens. Actuators, B 2009, 138, 563–571 10.1016/j.snb.2009.02.047. [DOI] [Google Scholar]
- Ma T.; Li C.; Shi G. Langmuir 2008, 24, 43–48 10.1021/la702559m. [DOI] [PubMed] [Google Scholar]
- Yang R.; Xu Y.; Dang X. D.; Nguyen T. Q.; Cao Y.; Bazan G. C. J. Am. Chem. Soc. 2008, 130, 3282–3283 10.1021/ja711068d. [DOI] [PubMed] [Google Scholar]
- Duarte A.; Pu K. Y.; Liu B.; Bazan G. C. Chem. Mater. 2011, 23, 501–515 10.1021/cm102196t. [DOI] [Google Scholar]
- Huang F.; Niu Y. H.; Zhang Y.; Ka J. W.; Liu M. S.; Jen A. K. Y. Adv. Mater. 2007, 19, 2010–2014 10.1002/adma.200602967. [DOI] [Google Scholar]
- Ortony J. H.; Chatterjee T.; Garner L. E.; Chworos A.; Mikhailovsky A.; Kramer E. J.; Bazan G. C. J. Am. Chem. Soc. 2011, 133, 8380–8387 10.1021/ja202776b. [DOI] [PubMed] [Google Scholar]
- Du J.; Thomas A. W.; Chen X.; Garner L. E.; Vandenberg C. A.; Bazan G. C. Chem. Commun. 2013, 49, 9624–9626 10.1039/c3cc45094e. [DOI] [PubMed] [Google Scholar]
- Costa T.; Garner L. E.; Knaapila M.; Thomas A. W.; Rogers S. E.; Bazan G. C.; Burrows H. D. Langmuir 2013, 29, 10047–10058 10.1021/la401871x. [DOI] [PubMed] [Google Scholar]
- Yang R.; Chao D.; Liu H.; Berda E. B.; Wang S.; Jia X.; Wang C. Electrochim. Acta 2013, 93, 107–113 10.1016/j.electacta.2013.01.109. [DOI] [Google Scholar]
- Kim H.; Jeong S.-M.; Park J.-W. J. Am. Chem. Soc. 2011, 133, 5206–5209 10.1021/ja200297j. [DOI] [PubMed] [Google Scholar]
- Kim H.; Kim T.-G.; Park J.-W. Macromol. Res. 2013, 21, 815–820 10.1007/s13233-013-1093-7. [DOI] [Google Scholar]
- Jin W.; Fukushima T.; Niki M.; Kosaka A.; Ishii N.; Aida T. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 10801–10806 10.1073/pnas.0500852102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z.; Rannou P.; Sadki S.; Fey N.; Lindsay D. M.; Faul C. F. J. Chem. - Eur. J. 2011, 17, 12512–12521 10.1002/chem.201101697. [DOI] [PubMed] [Google Scholar]
- Eelkema R.; Anderson H. L. Macromolecules 2008, 41, 9930–9933 10.1021/ma801697w. [DOI] [Google Scholar]
- Chen R.; Benicewicz B. C. Macromolecules 2003, 36, 6333–6339 10.1021/ma030033+. [DOI] [Google Scholar]
- CAC was determined by comparison of the ratio of intensity of the I1 (373 nm) and I3 (383 nm) fluorescence emission bands of a pyrene fluorescence probe, which has been shown to be sensitive to the polarity of the pyrene probe’s environment. See refs (49, 50).
- Aguiar J.; Carpena P.; Molina-Bolívar J. A.; Carnero Ruiz C. J. Colloid Interface Sci. 2003, 258, 116–122 10.1016/S0021-9797(02)00082-6. [DOI] [Google Scholar]
- Basu Ray G.; Chakraborty I.; Moulik S. P. J. Colloid Interface Sci. 2006, 294, 248–254 10.1016/j.jcis.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Smulders M. M. J.; Nieuwenhuizen M. M. L.; De Greef T. F. A.; Van Der Schoot P.; Schenning A. P. H. J.; Meijer E. W. Chem. - Eur. J. 2010, 16, 362–367 10.1002/chem.200902415. [DOI] [PubMed] [Google Scholar]
- Lydon J. J. Mater. Chem. 2010, 20, 10071–10099 10.1039/b926374h. [DOI] [Google Scholar]
- Tam-Chang S.-W.; Huang L. Chem. Commun. 2008, 1957–1967 10.1039/b714319b. [DOI] [PubMed] [Google Scholar]
- Echue G.; Lloyd-Jones G. C.; Faul C. F. J. Chem. - Eur. J. 2015, 21, 5118–5128 10.1002/chem.201406094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shacklette L. W.; Wolf J. F.; Gould S.; Baughman R. H. J. Chem. Phys. 1988, 88, 3955–3961 10.1063/1.453844. [DOI] [Google Scholar]
- Pouget J. P.; Jozefowicz M. E.; Epstein A. J.; Tang X.; MacDiarmid A. G. Macromolecules 1991, 24, 779–789 10.1021/ma00003a022. [DOI] [Google Scholar]
- Wei Z.; Laitinen T.; Smarsly B.; Ikkala O.; Faul C. F. J. Angew. Chem., Int. Ed. 2005, 44, 751–756 10.1002/anie.200460928. [DOI] [PubMed] [Google Scholar]
- Kohlman R. S.; Zibold A.; Tanner D. B.; Ihas G. G.; Ishiguro T.; Min Y. G.; MacDiarmid A. G.; Epstein A. J. Phys. Rev. Lett. 1997, 78, 3915–3918 10.1103/PhysRevLett.78.3915. [DOI] [Google Scholar]
- Xia Y.; Wiesinger J. M.; MacDiarmid A. G.; Epstein A. J. Chem. Mater. 1995, 7, 443–445 10.1021/cm00051a002. [DOI] [Google Scholar]
- Xia Y.; MacDiarmid A. G.; Epstein A. J. Macromolecules 1994, 27, 7212–7214 10.1021/ma00102a033. [DOI] [Google Scholar]
- Films were dried in a vacuum before measurement to remove water, ensuring that measured conductivity was electronic rather than ionic.
- Wei Z.; Laitinen T.; Smarsly B.; Ikkala O.; Faul C. F. J. Angew. Chem. 2005, 117, 761–766 10.1002/ange.200460928. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Tran H. D.; Liao L.; Duan X.; Kaner R. B. J. Am. Chem. Soc. 2010, 132, 10365–10373 10.1021/ja1014184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone D. A.; Tayi A. S.; Goldberger J. E.; Palmer L. C.; Stupp S. I. Chem. Commun. 2011, 47, 5702–5704 10.1039/c1cc10809c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.