

INTRODUCTION
Coeliac disease (CD) is an enteropathy, characterised by villous atrophy, which occurs in genetically susceptible individuals. It affects mainly the proximal small intestine, and is caused by an intolerance to cereal storage proteins found in wheat, barley and rye. Due to earlier diagnosis, and the recognition of ‘silent’ or ‘latent’ forms of the disease, the very severe symptoms that were seen previously are not very common now[1]. Malabsorption, with steatorrhoea and weight loss, occur less frequently. Anaemia, vitamin deficiencies, complications of pregnancy and associated autoimmune diseases, such as insulin dependent diabetes mellitus or thyroid disease are often the clues which lead to the diagnosis of coeliac disease. Coeliac disease affects people from all ethnic groups, though it is most common in people originating in Europe, including people in North America and Australia. It is rarely seen in people from an Afro-Caribbean background[2].
In the past, the prevalence of coeliac disease has been thought to be 1 in 1500 of the population in Western countries, based on the number of identified cases. However, recent screening studies of blood donors has shown a far higher prevalence of 1 in 250 in Sweden[3] and the United States [4]. In Italy, a population screening study of 17000 school children between the ages of 6 and 15 revealed a prevalence rate of 1 in 184[5]. This appears to be uniform throughout most of Europe, with some areas of higher incidence, such as the west coast of Ireland. It affects males and females equally.
The reason for this discrepancy between clinically apparent cases, and the number of individuals with positive screening results, lies in the concept of the ‘coeliac iceberg’[2]. The majority of people with coeliac disease are symptom-free, or have only mild symptoms, and do not approach a health care professional for a diagnosis. These individuals have ‘silent’ coeliac disease, if they have abnormal screening antibodies and an abnormal small bowel biopsy, but no symptoms. Individuals with abnormal screening antibody tests, but a normal small bowel biopsy and no symptoms, have ‘latent’ coeliac disease[6]. Thus, there are large numbers of people who are undiagnosed, in the ‘coeliac iceberg’ analogy this would be the vast portion of the iceberg which is not visible, whereas the diagnosed individuals with symptoms form the tip of the iceberg (Figure 1).
Figure 1.
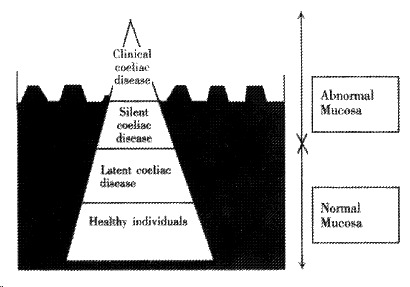
The coeliac iceberg. After A Ferguson[2]
HISTOPATHOLOGY
In the small intestine, the abnormalities are most marked proximally and decrease in severity with distal progression through the small intestine. In severe cases, the lesion may affect the ileum and even the stomach and rectum[7].
Flattening of the mucosa can vary from mild, through partial villous atrophy, to a total absence of villi. Classically, in untreated celiac disease, there is a flat mucosa with no villi (total villous atrophy), but more usually there is a reduction in the normal villous height, resulting in the villous height: crypt depth ratio being reduced from its normal value of between 3-5∶1. The thickness of the mucosa is usually increased because of crypt hyperplasia.
The surface epithelial cells become pseudostratified compared to their normal tall columnar shape with resultant fall in enterocyte height. Crypt mitotic activity is no longer confined to the base and, although the histological appearance is usually normal, crypt abscesses have been described. Cell migration from the crypt base to the villous tip is reduced in untreated coeliac disease to 12-24 h compared with the normal 3-5 d.
There is a chronic inflammatory cell infiltrate in the mucosa of the small intestine in untreated coeliac disease with a rise in the number of plasma cells in the lamina propria. There is an increase in the ratio of intra-epithelial lymphocytes to surface enterocytes in active disease. Most of the intra-epithelial lymphocytes express the common leucocyte antigen CD3, 70% express the suppressor/cytotoxic leucocyte antigen CD8, 5% express the helper/inducer CD4 phenocyte, whereas 20% of the cells are CD3+ve, CD4-ve and CD8-ve. Most of these cells express the more primitive γ/δ rather than the more usual α/β T-cell receptor. This results in a significant increase in the number of γ/δ T-cell receptor+ve lymphocytes in the surface epithelium of the small intestine, both in treated and untreated coeliac disease[7].
CEREAL CHEMISTRY
Cereal storage proteins fall into four groups, the minor albumins, the globulins, the ethanol-insoluble glutenins, and the ethanol-soluble fraction termed prolamins.
Initial separation of wheat proteins depends on their relative solubility characteristics. Gliadins are soluble in 40%-90% ethanol and high molecular weight glutenins are insoluble in neutral aqueous solution, saline or ethanol, although low molecular weight glutenins are ethanol soluble. The gliadins may be subdivided according to their relative electrophoretic mobility into α, β, γ and ω subfractions or according to their N-terminal amino acid sequence into α, γ or ω subfractions, the previously described β-subfraction being reclassified as a type of α gliadin[8]. The molecular weight of these proteins rises from 32 K to 58 K daltons through α to ω gliadins.
Prolamins, the alcohol-soluble fraction of storage proteins are responsible for triggering the disease[9]. Wheat, barley and rye, being closely related, all contain prolamins (known respectively as gliadins, hordeins and secalins) with a high composition of glutamine and proline, whereas the prolamins of oats and more distantly related cereals, contain less glutamine and proline and more alanine and leucine[10]. The glutamine-rich peptide sequences appear to be responsible for the toxicity of wheat, barley and rye in coeliac disease.
Immune activation occurs after ingestion of these cereals, when these peptides are presented, in conjunction with MHC class II molecules, to activate CD4+ T helper lymphocytes, causing release of Th1 and Th2 cytokines, which encourage expansion of autoreactive B cell clones, and mucosal destruction. Much recent research has provided an insight into the mechanisms of pathogenesis by which this occurs.
Once the diagnosis of coeliac disease has been made, the patient needs to be established on a gluten free diet. All foods which contain wheat or wheat flour, as well as barley and rye must be avoided. There are a number of foods which contain hidden gluten, including soy sauce, mustard, mayonnaise and beer, which contains the barley prolamin, hordein. Gluten-free flour, bread, biscuits, pasta and snacks are available from a variety of companies.
An average Western diet contains 13 g of gluten, a “Codex-Alimentarius” gluten free diet contains < 0.3% gluten from grains, and is sufficiently gluten free for the majority of coeliac patients, however, some patients continue to have symptoms, and improve only when gluten is completely removed from the diet. This was shown in a study by Faulkner-Hogg[11], where 15 out of 22 patients with symptoms on a Codex-Alimentarius gluten free diet improved after removal of all detectable gluten.
In the past, it was assumed that the small bowel mucosa returns to normal after introduction of a gluten-free diet, however, despite improvement of symptoms, persistent small bowel mucosal abnormalities may occur, and are not necessarily an indication of poor dietary compliance[12].
Oats are a member of the avena tribe of the gramineae, or grass family, of plant s, whereas wheat, barley and rye belong to the triticeae tribe, both tribes belonging to the pooideae sub-family. Thus avenin, the prolamin of oats, is genetically less like gliadin than secalin and hordein (Figure 2).
Figure 2.
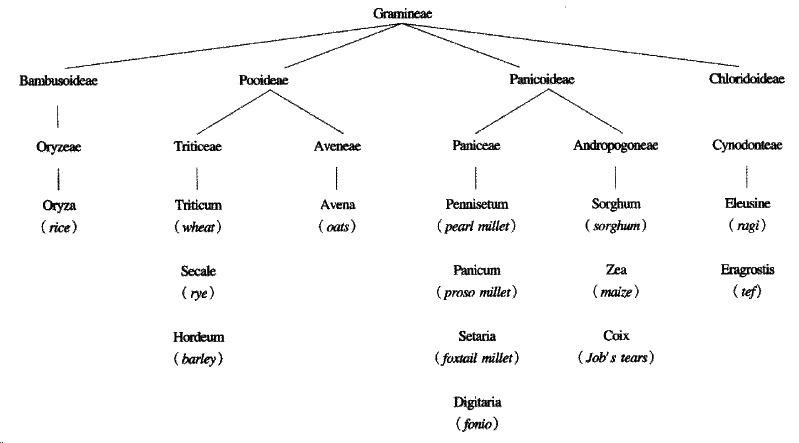
The taxonomic relationships of cereals. After P Shewry, A Tatham and D Kasarda[10]
The toxicity of oats to patients with coeliac disease has been a controversial issue, as early studies have shown conflicting results. Harmful effects were observed by some workers[9,13], but not by others[14,15], and some investigators found variable results[16,17]. However, a recent Finnish study[18] on newly diagnosed patients, as well as coeliac patients in remission on a gluten-free diet, have shown that moderate amounts (up to 60 g/day) of oats are not detrimental, as witnessed by no significant differences in gliadin and reticulin antibodies, as well as numbers of intra-epithelial lymphocytes before and after introduction of oats into the diet.
Sequence homologies, and weak immunological cross reactivity, have been found between avenin and the prolamins of wheat, barley and rye[10,19,20]. Additionally, only 5% to 15% of the total protein in oats is avenin, whereas 40% to 50% of the total protein in wheat, barley and rye are made up of their respective prolamins[21]. Thus, there is a smaller amount of avenin per gram if oat seed, and there are fewer toxic epitopes per gram of avenin. This suggests that a small amount of oats can be consumed by patients with coeliac disease, as long as the oats are not contaminated by wheat flour. In many mills however, the same equipment is used to grind wheat, as that used to grind oats, causing enough contamination to have a detrimental effect on the health of sensitive coeliac patients.
THE IMMUNODOMINANT PEPTIDE
The precise structure of the gluten proteins that exacerbate coeliac disease remains unknown, although there have been considerable recent advances in this area. Peptides from the fully sequenced α-gliadin, A-gliadin, have been used in a number of studies to try to identify the toxic epitope, which induces intestinal inflammation. A peptide corresponding to amino acids 31 to 49 of A gliadin was found to cause significant histological changes in small bowel biopsy specimens after in vivo challenge by intraduodenal infusion [22]. Anderson et al[23] used 51 overlapping synthetic 15-amino acid peptides, spanning the complete sequence of the A-gliadin. They assessed optimal peripheral blood mononuclear cell (PBMC) secretion of the Th1 cytokine gamma interferon (IFN-γ) in response to incubation with each of the peptides and demonstrated a transient, disease-specific, DQ2 restricted, CD4 T-cell response to a single dominant epitope. This peptide corresponded to amino acids 57-73 of A-gliadin, which had been partially deamidated by tissue transglutaminse at position 65 (Q65E).
Arentz-Hansen et al[24] produced eleven different recombinant antigens from α-gliadin, to demonstrate that the intestinal T cell response to α-gliadin in adult coeliac disease patients is focused on two immunodominant, DQ2 restricted peptides that overlap by a seven residue fragment. Gluten-specific T cell lines from small intestinal biopsies of 16 different patients all responded to one or both of the deamidated peptides, indicating that these epitopes are highly relevant to disease pathology. The peptides correspond to amino acids 62-75 (α2) and 57-68 (α9) with Q65E.
The identification of these peptide sequences, which act as potent T cell epitopes, may lead to the development of antigen specific therapy for coeliac disease. Once a target has been defined for immunomodulation, it may be possible to create non-toxic cereal based wheat, by removal or modification of the antigenic sequence in gliadin proteins.
TISSUE TRANSGLUTAMINASE
Tissue transglutaminase (tTG) is a ubiquitous cytoplasmic enzyme, which is found mainly in respiratory and gut epithelial cells. It is important in prevention of tissue damage, by catalysing protein cross-linkage, causing formation of isopeptide bonds between glutamine and lysine residues. tTG also deaminates glutamine residues to glutamic acid.
Native gluten proteins have very few negatively charged residues, as they contain approximately 40% glutamine and 20% proline, however, several of these glutamines are converted to glutamic acid in the presence of tTG. Deamidation of glutamine residues to glutamic acid was found to strongly enhance T cell reactivity, due to the formation of negatively charged amino acids needed for efficient binding to DQ molecules, thus inducing maximal T cell proliferation[25].
Virtually all patients with CD have been found to express either HLA-DQ2 or -DQ8 class II molecules. HLA class II molecules are responsible for binding exogenous protein antigens and presenting them to CD4+ T cells. These molecules have a characteristic binding groove, which differ in size, shape and position between class II alleles, and which can be used to predict the sequence of peptides needed to fit into it. Both DQ2 and DQ8 require negatively charged amino acids at certain positions for effective binding. Gluten specific HLA-DQ2 and -DQ8 restricted T cell clones can be isolated from small intestinal biopsy samples of patients with CD, and have been used to characterise gluten derived peptides capable of stimulating T cells[26].
Arentz-Hansen proposes that conditions may exist in the gut where T cell epitopes are both created and trapped locally by tTG, prohibiting their presentation by tolerogenic APCs in the gut. Alternatively, tTG may prevent these epitopes from spreading systemically as soluble antigen, a factor thought to be important in oral tolerance. Thus, it may be possible to administer soluble deamidated gliadin peptide to patients with coeliac disease, to induce tolerance to gliadin[24].
IMMUNOGENETICS
The precise mode of inheritance of coeliac disease is unknown, although l0%-15% of first degree relatives of probands are similarly affected[27,28]. There is 70%-100% concordance in affected monozygotic twins and 30%-50% concordance in human leukocyte antigen[29] (HLA)-identical siblings. Efforts to understand the mechanisms and genetics of polygenic human diseases have focused on the identification of DNA or protein products and protein molecules that segregate in both populations and families.
The most significant observation was the increased frequency of specific serologically defined lymphoid cell surface proteins, termed HLA class II molecules, in people with coeliac disease. These are glycosylated transmembrane heterodimers comprising both α and β-chains, the genes for which are organised into three related subregions DR, DP and DQ. The genes are encoded within the HLA-class II region of the major histocompatibility complex on the short arm of chromosome 6. The association of particular HLA-DR and DQ types with coeliac disease is well established[30]. Associations with the HLA-DP region and the TNF-α genes have been reported but are thought to be secondary to linkage disequilibrium with HLA-DR and DQ haplotypes[31,32]. The genes most strongly associated with coeliac disease are DQAl *0501, DQB1 *0201[33,34]. 98% of northern Europeans with coeliac disease have these alleles in cis (DQ2) whereas in Southern Europe a third of the population with the condition express the same class II molecule from these alleles in trans (DR5, 7)[30]. Italian and Argentinian coeliac disease populations are also reported to have a increased frequency of the DR5, 7 haplotype[35]. In Israel there is also an association with the haplotype HLA-DR4, DQ8[36]. This genotype encodes a class II molecule with considerable similarity in the peptide binding groove configuration to that produced by the DQ2 genes, supporting a central role for the class IImolecule in an immune mediated model of coeliac disease.
Twenty-five percent of people in the general population of Northern Europe have HLA-DQ2. It has been shown in epidemiological studies that only 30% of the genetic susceptibility to coeliac disease lies in the HLA region[37], the remaining 70% being elsewhere in the human genome. Two genome-wide linkage studies to identify these remaining susceptibility alleles have been undertaken using sibling pair analysis and have produced conflicting results. The first group from Ireland performed an autosomal screen using 40 affected sibling pairs, and identified five areas of interest at the following locations: 6p23, 7q31.3, 11p11, 15q26 and 22cen[38]. The second group from Italy using 110 sibling pairs, failed to confirm linkage in the areas identified in the previous study, but proposed to further areas of interest, at 5qter, and in a subgroup of patients at 11qter[39].
Our group, employing analysis of multi-generation families, identified two new potential susceptibility loci at 10q23.1 and 16q23.3, and found evidence of linkage on chromosome 7, close to the γ T-cell receptor gene and on chromosome 2, near the CTLA4 gene[40].
The CTLA4/CD28 region, on chromosome 2q33, has been independently implicated in an association study from France[41] and a linkage study from Finland[42]. These genes control T cell proliferation, and play a part in other autoimmune dieases, such as type I insulin dependent diabetes mellitus and Graves’ disease.
IMMUNE RESPONSE
It has been suggested that in coeliac disease there is a primary abnormal immune response of the small intestine to gluten proteins that produces an allergic phenomenon. There is considerable evidence to support this hypothesis, although the observed aberrant immune response may be secondary to an unrecognised alternative aetiopathology. There is a dense infiltration of the small intestinal lamina propria with lympho cytes and plasma cells in patients with untreated coeliac disease. There is also an increased ratio of intra-epithelial lymphocytes to surface enterocytes with the majority of these cells expressing the suppresser/cytotoxic phenotype and having the appearance of immunoblasts[43], and there is a strong association with the histocompatibility antigen HLA-DQ2 which is also associated with other auto-immune disorders.
High titres of antibodies to gliadin occur in coeliac disease as well as antibodies to reticulin and endomysium, which also implies an immune mediated mechanism [44]. The role of these antibodies in the disease pathogenesis is unknown. However, it has recently been shown that the antigen for anti-endomysial antibodies is the enzyme tissue transglutaminase, which is present in the small intestine and causes selective partial deamidation of gluten molecules.
There are an increased number of cells that express the cytokines IL-2, interferon-γ (IFN-γ) tumour necrosis factor-α (TNF-α) and tumour necrosis factor-β (TNF-β), IL-10, IL-1β and transforming growth factor-β (TGF-β). The expression of both Th1 (IFN-γ and IL-2) and Th2 (IL-4 and IL-10) associated cytokine transcripts in the same biopsies indicate the activation of Th0 cells. The expression of IL-2 and IL-4 mRNA was not observed in the peripheral blood samples of patients with inactive coeliac disease, implying that they are associated with disease activity[45]. This also supports the immune-mediated hypothesis. The isolation of gluten-sensitive T-cells both from both the peripheral blood and small intestinal biopsies from coeliac patients[34,46], the HLA-DQ2 restriction of the majority of these cells and their production of pro-inflammatory cytokines when stimulated with wheat gliadin[47], provide further evidence that these represent an hyperimmune sensitivity to certain cereal peptides.
“Gliadin (or gluten) shock”, is a rare condition which affects treated coeliac patients. In this condition a gluten challenge is followed by collapse with vomiting and tachycardia. The condition responds to treatment with corticosteroids.
In patients who do not respond to a strict gluten free diet, or who relapse on a gluten free diet, steroids can be used, such as prednisolone 20 mg/day, with a reducing course over the next 6 wk. Azathioprine can also be used as a steroid-sparing agent[48]. Cyclosporine does not appear to be useful[49], indeed, some patients’ symptoms increase, and budesonide, despite reducing the risk of osteoporosis, is not useful, as it is formulated to be slowly released in the terminal ileum, usually beyond the coeliac small bowel lesion, and is absorbed only in very small amounts.
Footnotes
Edited by Rampton DS and Ma JY
References
- 1.Visakorpi JK. Changing features of coeliac disease. In: Maki M, Collin P, Visakorpi JK, editors. Coeliac Disease. Tampere: Tampere University; 1997. pp. 1–7. [Google Scholar]
- 2.Ferguson A. The Coeliac Iceberg. CME J Gastroenterol Hepatol Nutr. 1999;2:52–56. [Google Scholar]
- 3.Grodzinsky E, Franzen L, Hed J, Ström M. High prevalence of celiac disease in healthy adults revealed by antigliadin antibodies. Ann Allergy. 1992;69:66–70. [PubMed] [Google Scholar]
- 4.Fasano A. Where have all the American celiacs gone. Acta Paediatr Suppl. 1996;412:20–24. doi: 10.1111/j.1651-2227.1996.tb14242.x. [DOI] [PubMed] [Google Scholar]
- 5.Catassi C, Fabiani E, Rätsch IM, Coppa GV, Giorgi PL, Pierdomenico R, Alessandrini S, Iwanejko G, Domenici R, Mei E, et al. The coeliac iceberg in Italy. A multicentre antigliadin antibodies screening for coeliac disease in school-age subjects. Acta Paediatr Suppl. 1996;412:29–35. doi: 10.1111/j.1651-2227.1996.tb14244.x. [DOI] [PubMed] [Google Scholar]
- 6.Ferguson A, Arranz E, O'Mahony S. Clinical and pathological spectrum of coeliac disease--active, silent, latent, potential. Gut. 1993;34:150–151. doi: 10.1136/gut.34.2.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marsh MN. Mucosal pathology in gluten sensitivity. In: Michael N Marsh., editor. Coeliac Disease. Oxford: Blackwell Scientific Publications; 1992. pp. 136–191. [Google Scholar]
- 8.Ejllnckd AJC. N-terminal amino acid sequencing of prolamins of wheat and related species. Nature. 1979;282:527–529. [Google Scholar]
- 9.VAN DE KAMER JH, WEIJERS HA, DICKE WK. Coeliac disease. IV. An investigation into the injurious constituents of wheat in connection with their action on patients with coeliac disease. Acta Paediatr. 1953;42:223–231. doi: 10.1111/j.1651-2227.1953.tb05586.x. [DOI] [PubMed] [Google Scholar]
- 10.Peter R Shewry ASTDDK. Cereal proteins and coeliac disease. In: Michael N Marsh., editor. Coeliac Disease. Oxford: Blackwell Scientific Publications; 1992. pp. 305–348. [Google Scholar]
- 11.Faulkner-Hogg KB, Selby WS, Loblay RH. Dietary analysis in symptomatic patients with coeliac disease on a gluten-free diet: the role of trace amounts of gluten and non-gluten food intolerances. Scand J Gastroenterol. 1999;34:784–789. doi: 10.1080/003655299750025714. [DOI] [PubMed] [Google Scholar]
- 12.Selby WS, Painter D, Collins A, Faulkner-Hogg KB, Loblay RH. Persistent mucosal abnormalities in coeliac disease are not related to the ingestion of trace amounts of gluten. Scand J Gastroenterol. 1999;34:909–914. doi: 10.1080/003655299750025390. [DOI] [PubMed] [Google Scholar]
- 13.DICKE WK, WEIJERS HA, VAN DE KAMER JH. Coeliac disease. II. The presence in wheat of a factor having a deleterious effect in cases of coeliac disease. Acta Paediatr. 1953;42:34–42. doi: 10.1111/j.1651-2227.1953.tb05563.x. [DOI] [PubMed] [Google Scholar]
- 14.SHELDON W. Coeliac disease. Lancet. 1955;269:1097–1103. doi: 10.1016/s0140-6736(55)92946-9. [DOI] [PubMed] [Google Scholar]
- 15.Dissanayake AS, Truelove SC, Whitehead R. Lack of harmful effect of oats on small-intestinal mucosa in coeliac disease. Br Med J. 1974;4:189–191. doi: 10.1136/bmj.4.5938.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baker PG, Read AE. Oats and barley toxicity in coeliac patients. Postgrad Med J. 1976;52:264–268. doi: 10.1136/pgmj.52.607.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MOULTON AL. The place of oats in the coeliac diet. Arch Dis Child. 1959;34:51–55. doi: 10.1136/adc.34.173.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janatuinen EK, Pikkarainen PH, Kemppainen TA, Kosma VM, Järvinen RM, Uusitupa MI, Julkunen RJ. A comparison of diets with and without oats in adults with celiac disease. N Engl J Med. 1995;333:1033–1037. doi: 10.1056/NEJM199510193331602. [DOI] [PubMed] [Google Scholar]
- 19.Troncone R, Auricchio S, De Vincenzi M, Donatiello A, Farris E, Silano V. An analysis of cereals that react with serum antibodies in patients with coeliac disease. J Pediatr Gastroenterol Nutr. 1987;6:346–350. doi: 10.1097/00005176-198705000-00007. [DOI] [PubMed] [Google Scholar]
- 20.Freedman AR, Galfre G, Gal E, Ellis HJ, Ciclitira PJ. Western immunoblotting of cereal proteins with monoclonal antibodies to wheat gliadin to investigate coeliac disease. Int Arch Allergy Appl Immunol. 1988;85:346–350. doi: 10.1159/000234530. [DOI] [PubMed] [Google Scholar]
- 21.Shewry PR, Napier JA, Tatham AS. Seed storage proteins: structures and biosynthesis. Plant Cell. 1995;7:945–956. doi: 10.1105/tpc.7.7.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sturgess R, Day P, Ellis HJ, Lundin KE, Gjertsen HA, Kontakou M, Ciclitira PJ. Wheat peptide challenge in coeliac disease. Lancet. 1994;343:758–761. doi: 10.1016/s0140-6736(94)91837-6. [DOI] [PubMed] [Google Scholar]
- 23.Anderson RP, Degano P, Godkin AJ, Jewell DP, Hill AV. In vivo antigen challenge in celiac disease identifies a single transglutaminase-modified peptide as the dominant A-gliadin T-cell epitope. Nat Med. 2000;6:337–342. doi: 10.1038/73200. [DOI] [PubMed] [Google Scholar]
- 24.Arentz-Hansen H, Körner R, Molberg O, Quarsten H, Vader W, Kooy YM, Lundin KE, Koning F, Roepstorff P, Sollid LM, et al. The intestinal T cell response to alpha-gliadin in adult celiac disease is focused on a single deamidated glutamine targeted by tissue transglutaminase. J Exp Med. 2000;191:603–612. doi: 10.1084/jem.191.4.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Molberg O, Mcadam SN, Körner R, Quarsten H, Kristiansen C, Madsen L, Fugger L, Scott H, Norén O, Roepstorff P, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med. 1998;4:713–717. doi: 10.1038/nm0698-713. [DOI] [PubMed] [Google Scholar]
- 26.van de Wal Y, Kooy YM, van Veelen PA, Peña SA, Mearin LM, Molberg O, Lundin KE, Sollid LM, Mutis T, Benckhuijsen WE, et al. Small intestinal T cells of celiac disease patients recognize a natural pepsin fragment of gliadin. Proc Natl Acad Sci USA. 1998;95:10050–10054. doi: 10.1073/pnas.95.17.10050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ellis A. Coeliac Disease: previous family studies. In: McConnell RB, editor. The Genetics of Coeliac Disease. Lancaster, England: MTP; 1981. pp. 197–200. [Google Scholar]
- 28.Marsh MN. Gluten, major histocompatibility complex, and the small intestine. A molecular and immunobiologic approach to the spectrum of gluten sensitivity ('celiac sprue') Gastroenterology. 1992;102:330–354. [PubMed] [Google Scholar]
- 29.Polanco I BIVLAea. Gluten sensitive enteropathy in Spain: genetic and environmental factors. In: R B McConnell, ed , editors. The genetics of Coeliac disease. Lancaster: MTP Press Limited; 1981. pp. 211–231. [Google Scholar]
- 30.Sollid LM, Markussen G, Ek J, Gjerde H, Vartdal F, Thorsby E. Evidence for a primary association of celiac disease to a particular HLA-DQ alpha/beta heterodimer. J Exp Med. 1989;169:345–350. doi: 10.1084/jem.169.1.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hall MA, Lanchbury JS, Bolsover WJ, Welsh KI, Ciclitira PJ. Celiac disease is associated with an extended HLA-DR3 haplotype which includes HLA-DPw1. Hum Immunol. 1990;27:220–228. doi: 10.1016/0198-8859(90)90052-q. [DOI] [PubMed] [Google Scholar]
- 32.Tighe MR, Hall MA, Barbado M, Cardi E, Welsh KI, Ciclitira PJ. HLA class II alleles associated with celiac disease susceptibility in a southern European population. Tissue Antigens. 1992;40:90–97. doi: 10.1111/j.1399-0039.1992.tb01965.x. [DOI] [PubMed] [Google Scholar]
- 33.Sollid LM, Thorsby E. HLA susceptibility genes in celiac disease: genetic mapping and role in pathogenesis. Gastroenterology. 1993;105:910–922. doi: 10.1016/0016-5085(93)90912-v. [DOI] [PubMed] [Google Scholar]
- 34.Lundin KE, Scott H, Hansen T, Paulsen G, Halstensen TS, Fausa O, Thorsby E, Sollid LM. Gliadin-specific, HLA-DQ (alpha 1*0501, beta 1*0201) restricted T cells isolated from the small intestinal mucosa of celiac disease patients. J Exp Med. 1993;178:187–196. doi: 10.1084/jem.178.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herrera M, Chertkoff L, Palavecino E, Mota A, Guala MC, Fainboim L, Satz ML. Restriction fragment length polymorphism in HLA class II genes of Latin-American Caucasian celiac disease patients. Hum Immunol. 1989;26:272–280. doi: 10.1016/0198-8859(89)90005-0. [DOI] [PubMed] [Google Scholar]
- 36.Tighe MR, Hall MA, Ashkenazi A, Siegler E, Lanchbury JS, Ciclitira PJ. Celiac disease among Ashkenazi Jews from Israel. A study of the HLA class II alleles and their associations with disease susceptibility. Hum Immunol. 1993;38:270–276. doi: 10.1016/0198-8859(93)90554-e. [DOI] [PubMed] [Google Scholar]
- 37.Bevan S, Popat S, Braegger CP, Busch A, O'Donoghue D, Falth-Magnusson K, Ferguson A, Godkin A, Hogberg L, Holmes G, et al. Contribution of the MHC region to the familial risk of coeliac disease. J Med Genet. 1999;36:687–690. [PMC free article] [PubMed] [Google Scholar]
- 38.Zhong F, McCombs CC, Olson JM, Elston RC, Stevens FM, McCarthy CF, Michalski JP. An autosomal screen for genes that predispose to celiac disease in the western counties of Ireland. Nat Genet. 1996;14:329–333. doi: 10.1038/ng1196-329. [DOI] [PubMed] [Google Scholar]
- 39.Greco L, Corazza G, Babron MC, Clot F, Fulchignoni-Lataud MC, Percopo S, Zavattari P, Bouguerra F, Dib C, Tosi R, et al. Genome search in celiac disease. Am J Hum Genet. 1998;62:669–675. doi: 10.1086/301754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.King AL, Yiannakou JY, Brett PM, Curtis D, Morris MA, Dearlove AM, Rhodes M, Rosen-Bronson S, Mathew C, Ellis HJ, et al. A genome-wide family-based linkage study of coeliac disease. Ann Hum Genet. 2000;64:479–490. doi: 10.1046/j.1469-1809.2000.6460479.x. [DOI] [PubMed] [Google Scholar]
- 41.Djilali-Saiah I, Schmitz J, Harfouch-Hammoud E, Mougenot JF, Bach JF, Caillat-Zucman S. CTLA-4 gene polymorphism is associated with predisposition to coeliac disease. Gut. 1998;43:187–189. doi: 10.1136/gut.43.2.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holopainen P, Arvas M, Sistonen P, Mustalahti K, Collin P, Mäki M, Partanen J. CD28/CTLA4 gene region on chromosome 2q33 confers genetic susceptibility to celiac disease. A linkage and family-based association study. Tissue Antigens. 1999;53:470–475. doi: 10.1034/j.1399-0039.1999.530503.x. [DOI] [PubMed] [Google Scholar]
- 43.Marsh MN. Immunocytes, enterocytes and the lamina propria: an immunopathological framework of coeliac disease. J R Coll Physicians Lond. 1983;17:205–212. [PMC free article] [PubMed] [Google Scholar]
- 44.Yiannakou JY, Dell'Olio D, Saaka M, Ellis HJ, Rosen-Bronson S, Dumonde DC, Ciclitira PJ. Detection and characterisation of anti-endomysial antibody in coeliac disease using human umbilical cord. Int Arch Allergy Immunol. 1997;112:140–144. doi: 10.1159/000237445. [DOI] [PubMed] [Google Scholar]
- 45.Lahat N, Shapiro S, Karban A, Gerstein R, Kinarty A, Lerner A. Cytokine profile in coeliac disease. Scand J Immunol. 1999;49:441–446. doi: 10.1046/j.1365-3083.1999.00523.x. [DOI] [PubMed] [Google Scholar]
- 46.Gjertsen HA, Sollid LM, Ek J, Thorsby E, Lundin KE. T cells from the peripheral blood of coeliac disease patients recognize gluten antigens when presented by HLA-DR, -DQ, or -DP molecules. Scand J Immunol. 1994;39:567–574. doi: 10.1111/j.1365-3083.1994.tb03414.x. [DOI] [PubMed] [Google Scholar]
- 47.Przemioslo RT, Lundin KE, Sollid LM, Nelufer J, Ciclitira PJ. Histological changes in small bowel mucosa induced by gliadin sensitive T lymphocytes can be blocked by anti-interferon gamma antibody. Gut. 1995;36:874–879. doi: 10.1136/gut.36.6.874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hamilton JD, Chambers RA, Wynn-Williams A. Role of gluten, prednisone, and azathioprine in non-responsive coeliac disease. Lancet. 1976;1:1213–1216. doi: 10.1016/s0140-6736(76)92162-0. [DOI] [PubMed] [Google Scholar]
- 49.Duerksen DR, Ma MM, Jewell LD. Failure of immunosuppressive therapy to prevent relapse of celiac sprue. J Clin Gastroenterol. 1995;21:255–257. doi: 10.1097/00004836-199510000-00020. [DOI] [PubMed] [Google Scholar]