

Abstract
An ongoing challenge in modern catalysis is to identify and understand new modes of reactivity promoted by earth-abundant and inexpensive first-row transition metals. Herein, we report a mechanistic study of an unusual copper(I)-catalyzed 1,3-migration of 2-bromostyrenes that reincorporates the bromine activating group into the final product with concomitant borylation of the aryl halide bond. A combination of experimental and computational studies indicated this reaction does not involve any oxidation state changes at copper; rather, migration occurs through a series of formal sigmatropic shifts. Insight provided from these studies will be used to expand the utility of aryl copper species in synthesis and develop new ligands for enantioselective copper-catalyzed halogenation.
Graphical abstract
INTRODUCTION
The field of base metal catalysis is a vibrant area of research and offers many potential advantages over more widely utilized precious metal catalysts. Not only are earth-abundant, first-row transition metals significantly less expensive and better from an environmental perspective, but their differing electronic structures compared to second- and third-row metals provide promise for uncovering new reactivities that proceed through novel mechanistic pathways. While the mechanisms of precious metal-catalyzed reactions have been studied extensively, the emphasis on better mechanistic understanding of reactions catalyzed by base metals is more recent.1 In this vein, our group has recently described a new mode of reactivity for Cu(I) that exhibits some unusual features compared to traditional cross-couplings.2–4 Key to the further development of this chemistry into synthetically useful, Cu-catalyzed carbon–carbon and carbon–heteroatom bond-forming methodologies is an understanding of the mechanistic details of this new reactivity.
The functionalization of arenes via cross-coupling is arguably one of the most important and versatile reactions in organic synthesis.5 The majority of these reactions use aryl or vinyl halides with an organometallic coupling partner, such as a boronic acid or ester (Suzuki), organozinc (Negishi), organostannane (Stille), organosilane (Hiyama), or Grignard reagent (Kumada).5,6 While the majority of cross-coupling reactions are catalyzed by palladium, there has been much interest recently in promoting these types of transformations using first-row transition metals, including nickel, iron, and copper.5 However, there are challenges associated with promoting the typical mechanistic pathway invoked for cross-coupling, which involves the oxidative addition of the metal into the aryl halide or pseudohalide bond, transmetalation of the coupling partner to the metal, and a final reductive elimination to yield the product.5 Typically, first-row transition metals prefer one-electron oxidation state changes, as in the oft-invoked Cu(I)/Cu(II) and Fe(II)/Fe(III) mechanistic cycles.7 This renders oxidative addition more challenging with earth-abundant metals, often requiring the use of less convenient pseudohalides, high temperatures, long reaction times, or a combination of forcing conditions.8,9 Furthermore, one-electron chemistry can complicate the reaction pathways, making both spectroscopic and mechanistic analyses difficult.7 While great strides have been made by several groups to promote Pd-like mechanistic pathways with first-row metals,8,9 our group is taking an alternative approach by developing Cu-catalyzed reactions that invoke unusual mechanistic pathways involving no oxidation state changes at the metal center.2,3
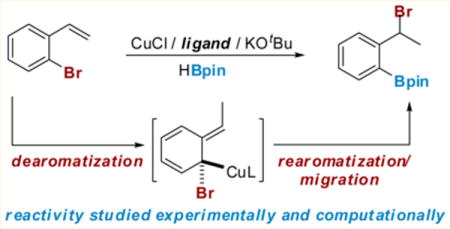
Our work in this area was stimulated by a serendipitous observation made during attempts to achieve a copper-catalyzed carboxylation of styrenes.4 In the course of these studies, we attempted to synthesize a benzyl boronic ester via a Cu-catalyzed hydroboration of a 2-bromostyrene using a method recently reported by the Yun group (Scheme 1, top).10 In Yun’s proposed mechanism, a phosphine-supported copper-hydride, 1.1, which is generated in situ, adds to the styrene in a Markovnikov fashion. A subsequent σ-bond metathesis of the benzyl copper species 1.2 with pinacol borane (HBpin) regenerates the catalyst and forms the product. However, when 2-bromostyrene 1.3 was utilized with this protocol, very little of the desired product was formed; instead we observed small amounts (<10%) of what was eventually identified as 1.4. Based on this initial observation, it appeared that the benzyl copper intermediate preferred to undergo an unexpected rearrangement, causing the bromine to migrate from the aryl ring to the benzyl position with concomitant borylation of the aryl carbon–bromine bond.2,3 The use of a bis(1,2-dicyclohexylphosphino)ethane (dCype) ligand further improved the yields of the 1,3-halogen migration/borylation reaction. We were intrigued by this unusual transformation and wanted to undertake a thorough mechanistic study to gain insight into the features of both the catalyst and the substrate that promote this pathway.11 With this mechanistic understanding in hand, we expected to be able to expand the scope and utility of copper-catalyzed arene and benzyl functionalization chemistries. The efforts described in this paper have allowed us to (1) establish a reasonable energy profile for the 1,3-halogen migration via density functional theory (DFT) studies, (2) identify the enantio-determining step in the asymmetric version of this reaction, and (3) gain insight into design principles for the development of new catalysts exhibiting increased substrate scope.
Scheme 1.
Cu-Catalyzed Hydroboration vs Halogen Migration
RESULTS AND DISCUSSION
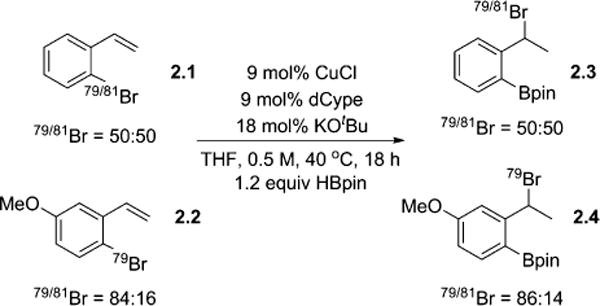
In our previous report, crossover experiments using an isotopically enriched styrene, 2.2 (Scheme 2), indicated that the halogen migration was most likely either an intramolecular or a rapid dissociation and recombination pathway.2 To rule out the possibility that the reaction proceeds via direct borylation of the aryl halide, followed by bromination of the styrene, 1,4-dibromobenzene was subjected to the reaction conditions (eq 1). No reaction was observed, indicating that the olefin is necessary for reactivity.2 When 3-bromostyrene was subjected to the reaction conditions, only hydroboration occurred, showing that the olefin’s location relative to the bromine is also important (eq 2).2 This result supports the conclusion from our crossover experiments suggesting that the 1,3-halogen migration is an intramolecular reaction. Finally, subjecting the hydroboration product to the reaction conditions resulted in no further reaction (eq 3); thus, the benzyl boronic ester can be ruled out as a potential intermediate in this transformation.
 |
(1) |
 |
(2) |
 |
(3) |
Scheme 2.
Crossover Experiments via 79/81Br Isotopic Labeling
Initial studies to better understand the nature of the copper catalyst were undertaken. Although we suspected that the phosphine-supported copper(I) hydride was the active catalyst, these species are known to form dimers and other higher-order oligomers and aggregates, which could complicate the mechanistic picture.12 Fortunately, we have recently developed an asymmetric version of the 1,3-halogen migration reaction, which permitted a nonlinear effects study to gain insight into the nuclearity of the catalyst (Figure 1).3 The absence of nonlinear effects support the assumption that the active catalyst in solution is monomeric in nature and argues against the presence of off-cycle organocopper dimers or higher order aggregates.13,14
Figure 1.
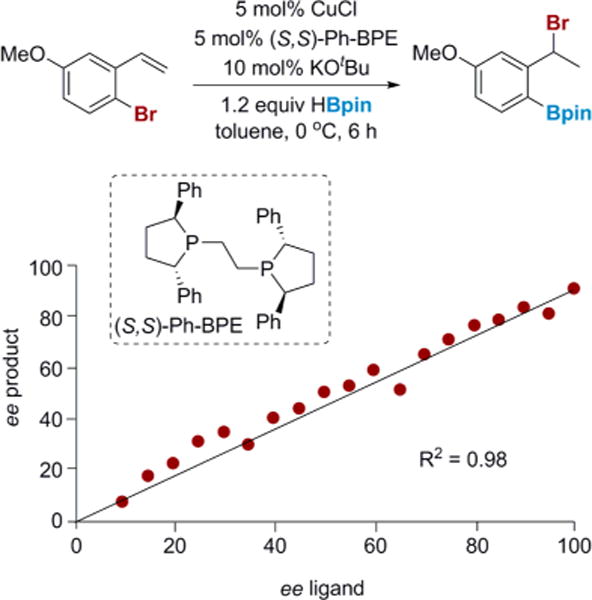
Dependence of product enantiomeric excess (ee) on ligand ee.
An investigation of the kinetics of the 1,3-halogen migration reaction using dCype as the ligand showed that there is no rate dependence on either 2-bromostyrene or HBpin and that the reaction is first order in catalyst at low catalyst concentrations (Figure 2). At higher catalyst loadings, the reaction displayed saturation behavior, presumably due to the decreased solubility of the metal complex at higher concentrations.
Figure 2.
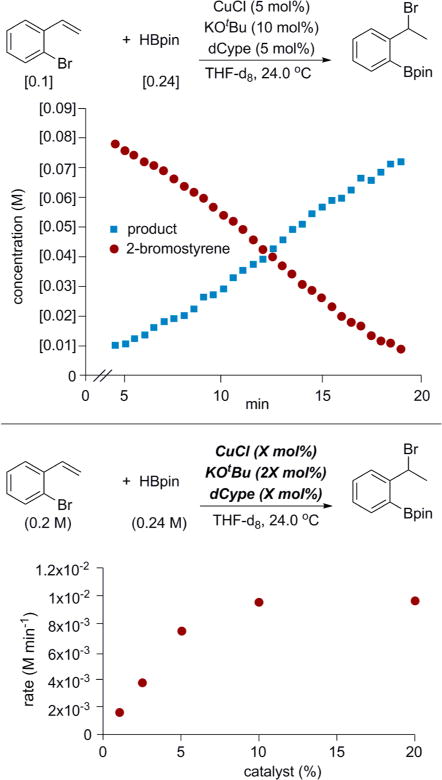
Time course of 1,3-halogen migration and rate dependence on catalyst loading.
Deuteration of the terminal position of the olefin resulted in a secondary kinetic isotope effect of 1.19 (eq 4).15 This
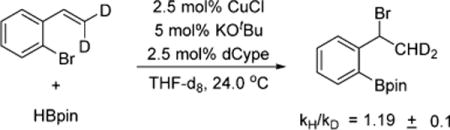 |
(4) |
indicates that the terminal carbon of the styrene is undergoing a hybridization change in the rate-determining step, despite the fact that the overall reaction is zero-order in styrene. To explain this result, we hypothesize that the active catalyst is the ligand-supported CuH (Scheme 3); however, interaction of the Cu–H with the styrene forms a π-olefin complex.16 Since the styrene is in high concentration relative to the CuH, the concentration of this intermediate is dictated by the concentration of the CuH. The CuH then adds across the olefin in the rate-determining step. This proposed pre-equilibrium accounts for the zero-order rate dependence on the styrene, but still allows it to be involved in the rate-determining step.17
Scheme 3.
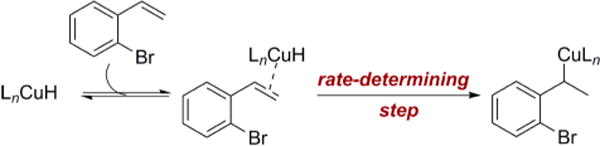
Pre-equilibrium Step Forming π-Olefin Complex and Rate-Determining Step
In an effort to elucidate possible intermediates spectroscopically, stoichiometric studies were undertaken. The phosphine-supported CuH was generated in situ by treating Cu(OAc)2 and dCype with poly(methylhydrosiloxane) (PMHS), and then 1 equiv of 2-bromostyrene was added. Upon warming, approximately 1% of what appeared to be an aryl copper was rapidly formed (eq 5). At lower temperatures, the intermediate
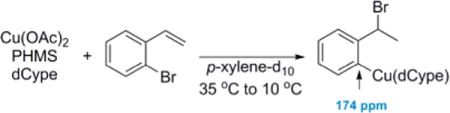 |
(5) |
was stable long enough to acquire a variety of spectra. In spite of the fact that only a small amount of the ArCu was formed, nearly all of the carbons and protons from the aryl skeleton could be assigned. Notably, the aryl carbon bearing copper has a chemical shift of 174 ppm. While this is an unusual chemical shift for aryl carbons, it is typical for aryl carbons bearing copper(I).14h,18 For more details and all spectra, see the Supporting Information.
Based on these preliminary studies, a partial mechanism was proposed (Scheme 4). The CuH 4.1 adds to the olefin in a regioselective manner to give the benzyl copper intermediate 4.3. This species then undergoes a formal 1,3-rearrangement to generate an aryl copper, 4.4, followed by σ-bond metathesis with HBpin to form the benzyl bromide product and regenerate the catalyst.19 The competing pathway involves direct metathesis of the benzyl copper 4.3 with HBpin to afford the unobserved benzyl boronic ester 4.6 originally reported by Yun.10 This proposed mechanistic pathway raises two important questions that need to be answered. First, it is unclear what parameters in both the substrate and the catalyst contribute to the tendency of the benzyl copper 4.3 to undergo rearrangement, as opposed to immediate borylation. Second, the nature of the migration of the benzyl copper 4.3 to the aryl copper species 4.4 needs to be understood, especially as it relates to transfer of chiral information in the enantioselective version of the 1,3-halogen migration.
Scheme 4.
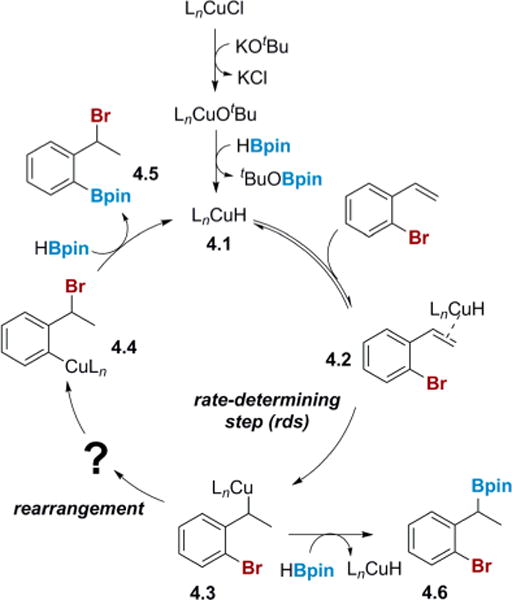
Partial Proposed Mechanism for Cu(I)-Catalyzed 1,3-Halogen Migration Reaction
To answer the question as to why migration occurs only with certain ligands, while other ligands give the benzyl boronic ester, a blend of experimental and computational studies were carried out. All structures throughout our studies were optimized with Gaussian 0920 using the M06 functional21 with a 6-311G* basis set22 for H, B, C, O, P, and Br and a LANL2TZ+ basis set23 for copper (results from calculations using other basis sets and functionals are given in the Supporting Information). A Solvent Model Density (SMD) continuum model was used with tetrahydrofuran (THF) as the solvent (except where noted).24 All minima were checked for absence of imaginary vibrational modes, and all transition-state structures were checked for one imaginary vibrational mode and confirmed with intrinsic reaction coordinate calculations.25 The barriers of the σ-bond metathesis leading to the benzyl boronic esters were calculated for the dCype, dMepe (1,2-bis(dimethylphosphino)ethane), and dtBupe (1,2-bis(di-tert-butylphosphino)ethane) ligands. The barrier for the dCype ligand (TS-5.2) was higher than that of the dMepe ligand (TS-5.2b) (Scheme 5). Since the dCype ligand prefers migration, the barrier for hydroboration can provide an upper limit for the energy required for migration, whereas the hydroboration using the dMepe ligand may provide the lower limit. Thus, the barrier for migration is most likely between 22.7 and 19.1 kcal/mol. Presumably, this difference in barriers can be attributed to steric bulk. Indeed, the barrier for hydroboration using dtBupe (TS-5.2a) was the highest of the three, predicting that a more sterically bulky ligand should favor migration to a greater extent for substrates that currently give mixtures of products. This design principle will be tested in future investigations of new ligands for 1,3-halogen migration.
Scheme 5. Impact of Steric Bulk on Hydroborationa.
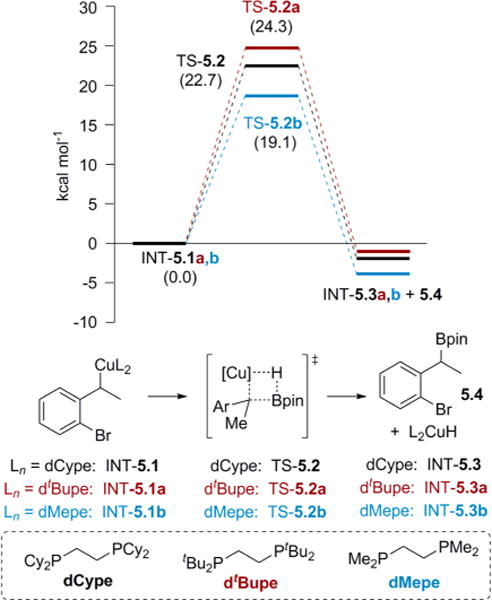
aFree energies shown are in kcal/mol relative to 5.1, 5.1a, and 5.1b computed at SMD(THF)-M06/6-311G(d):LANL2TZ+ (Cu).
To examine the role the steric and electronic features of both the substrate and the ligand play in controlling the distribution of products between migration and hydroboration, a series of ligands were investigated with a variety of substrates (Table 1). In the case of 2-bromostyrene itself (entries 1–4), as predicted by our calculations (Scheme 5), the smaller dMepe and dEtpe (1,2-bis(diethylphosphino)ethane) ligands switched the reactivity from exclusively migration to exclusively hydroboration (entries 2 and 3). The larger (S,S)-Ph-BPE (1,2-bis[(2S,5S)-2,5-diphenylphospholano]ethane) ligand (entry 4), used as a surrogate for dtBupe, still gave exclusively the migration product, albeit in much lower yield than dCype (entry 1). As the substrate becomes more electron-poor, the hydroboration product is increasingly favored with dCype, indicating that the rate of halogen migration slows with electron-poor aromatics (entries 5 and 7). However, the bulkier (S,S)-Ph-BPE ligand still favored the migration product with these bromostyrenes, showing that greater steric bulk does promote the migration (entries 6 and 8). We were curious if increasing the electron density of the 2-bromostyrene might enable less bulky ligands to catalyze the 1,3-halogen migration (entries 9–14); however, this did not appear to be the case when only one electron-donating group was present on the arene (entries 9–11). Nonetheless, the combination of a bulky, electron-rich catalyst and an electron-rich aromatic gave near-quantitative yields of the migration product. Increasing the electron density of the substrate even further resulted in some migration product, even with dMepe and dEtpe ligands (entries 13 and 14). From these results, we can conclude that increasing the electron density of the aromatic ring favors migration to a greater extent, while decreasing electron density disfavors migration. Addtionally, increasing the steric bulk of the ligand disfavors hydroboration, while small ligands favor hydroboration.
Table 1.
Factors Impacting the Product Distribution in the 1,3-Halogen Migration
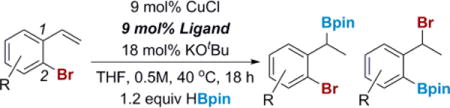
| ||||
|---|---|---|---|---|
| entry | R | ligandb | yield (%)a
|
|
| hydroboration | migration | |||
| 1 | H | dCype | 0 | 94 |
| 2 | H | dMepe | 91 | 0 |
| 3 | H | dEtpe | 98 | 0 |
| 4 | H | Ph-BPE | 0 | 37 |
| 5 | 5-F | dCype | 28 | 49 |
| 6 | 5-F | Ph-BPE | 0 | 21 |
| 7 | 5-CF3 | dCype | 31 | 28 |
| 8 | 5-CF3 | Ph-BPE | 6 | 14 |
| 9 | 5-OMe | dCype | 0 | 87 |
| 10 | 5-OMe | dMepe | 52 | 0 |
| 11 | 5-OMe | dEtpe | 100 | 0 |
| 12 | 3,5-OMe | dCype | 0 | 98 |
| 13 | 3,5-OMe | dMepe | 40 | 16 |
| 14 | 3,5-OMe | dEtpe | 32 | 17 |
Ligands:
NMR yields using 1,1,1,2-tetrachloroethane as an internal standard.
The second question concerning the mechanism involves the nature of the migration event that transforms the benzyl copper 4.3 to an aryl copper species, 4.4. To better understand the nature of the 1,3-halogen migration, an experiment was designed to explore how stereochemical information in the reaction is transferred from the benzyl copper to the aryl copper species, and eventually to the final product. Such insight would be valuable in determining which migration pathways are feasible. A deuterated dihydronaphthalene substrate, 6.1, was employed (Scheme 6) to determine if the stereochemistry set during the initial hydrocupration step was retained, inverted or ablated during the 1,3-halogen migration event. Since the hydrocupration of a π-bond is precedented to occur in a syn fashion to give 6.3,26 the stereochemistry of the bromine relative to the deuterium in the product 6.2 should indicate whether migration occurs with retention or inversion of stereochemistry. In the case of retention, the product 6.7 should display an anti relationship between the two protons on adjacent carbons. If inversion is taking place, then product 6.6 should display a syn relationship between these same two protons. Interestingly, the product obtained from this experiment was a mixture of 1:1 of diastereomers, indicating the presence of a stereoablative step in the migration process.
Scheme 6. Transfer of Stereochemical Information during the 1,3-Halogen Migrationa.
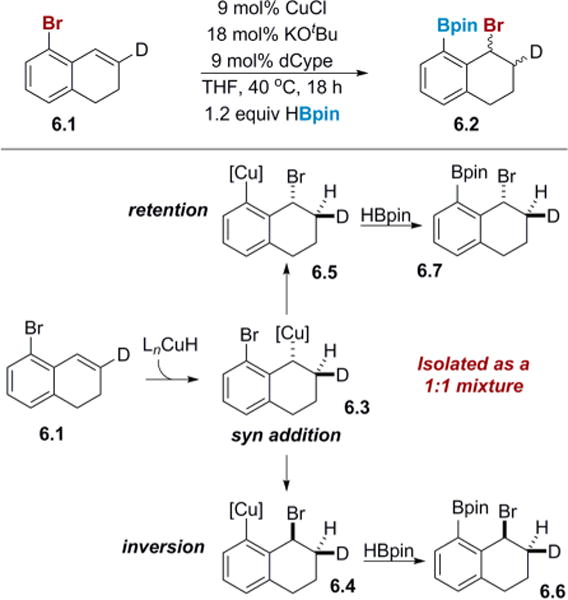
aDiastereomeric ratio was determined by displacing the benzyl bromide with LiSePh and then performing a selenoxide elimination.
In our initial report, we proposed that the migration might occur through an oxidative addition/reductive elimination pathway via INT-3.4a (Figure 3, bottom).27 However, when we investigated this pathway computationally, INT-3.4a could not be optimized as a minimum on the potential energy surface. To accommodate the formation of two new bonds in the oxidative addition step, one of the phosphine ligands was dissociated to yield INT-3.4. The energy required to form the strained cupracycle INT-3.4, containing a high oxidation state Cu(III), was very high at 50 kcal/mol; in addition, this pathway did not account for the stereoablative step. Moreover, oxidative addition generally proceeds more rapidly with electron deficient Ar–X bonds; however, electron-poor substrates in our system disfavor migration, further suggesting this chemistry does not proceed through an oxidative addition/reductive elimination pathway (for other pathways involving Cu(III), see Supporting Information).28
Figure 3.
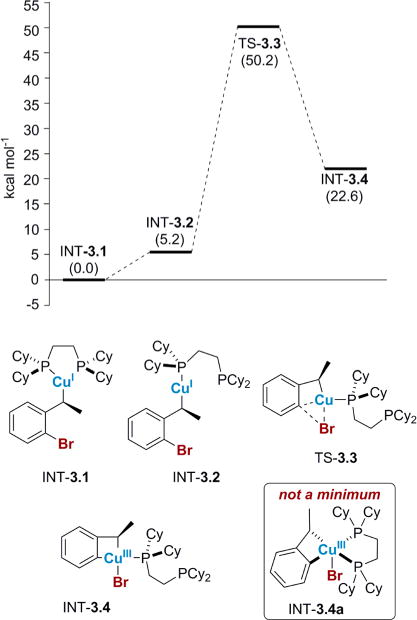
Proposed oxidative addition pathway proceeding through a Cu(I)/Cu(III) cycle. Free energies shown are in kcal/mol relative to 3.1 computed at SMD(THF)-M06/6-311G(d):LANL2TZ+ (Cu).
Since a Cu(I)/Cu(III) pathway seemed unlikely, we considered a pathway that would take advantage of a facile Cu(I)/Cu(II) oxidation state change (Scheme 7).7 In this pathway, bromine abstraction by copper would form an aryl radical/Cu(II) species, 7.2. Homolytic cleavage of the benzyl carbon–copper bond, followed by rapid recombination with the aryl radical, would produce a relatively stable benzyl radical intermediate, 7.4. From this intermediate, the bromine could potentially undergo a 1,4-shift to form an aryl Cu(I) species, 7.5. Unfortunately, the aryl radical 7.2 was high in energy, making the formation of this intermediate unlikely. Another possible pathway that could lead to the benzyl radical/aryl copper species 7.4 involves homolytic cleavage of the benzyl carbon–copper bond of 7.1 to produce a benzyl radical and a phosphine-supported Cu(0) intermediate, 7.3, followed by oxidative addition of the metal into the aryl carbon–bromine bond. This pathway proved to be even higher in energy than the Cu(I)/Cu(II) pathway and was not considered further.
Scheme 7. Pathways Invoking Potential Cu(II) Intermediatesa.
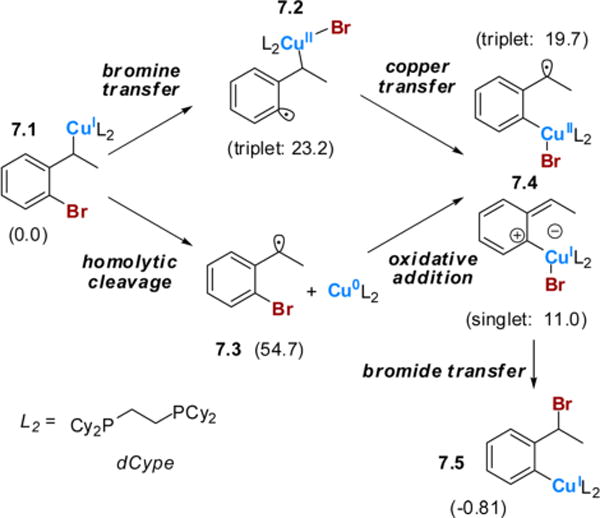
aFree energies shown are in kcal/mol relative to 7.1, computed at SMD(THF)-M06/6-311G(d):LANL2TZ+ (Cu).
We next investigated a dearomatization pathway that did not involve changes in the oxidation state at copper (Figure 4).11b,29 The proposed mechanism consists of a three-step process. While M06 results are shown here, calculations with various theoretical methods (functionals and basis sets) on systems with both bidentate and monodentate bisphosphine coordination were performed (see Supporting Information); the mechanistic pathways found for these systems were qualitatively the same. Initially, INT-4.1 proceeds through a dearomative η3 Cu(I) transition state, leading to non-aromatic INT-4.3, where both bromine and Cu(I) are bound to the same carbon. The carbon bearing the bromine, C2, can be thought of as the “nucleophile” in the dearomatizing step, whereas the copper can be thought of as the electrophile. According to this model, increasing the electron density at this carbon should favor migration; this has been borne out in studies where the electronics of the substrate have been varied (see Table 1). In INT-4.3, the Cu(I) occupies the pseudoaxial position of the tetrahedral carbon while the bromine is in the equatorial position, making bromine and the benzyl carbon, C7, nearly coplanar. Since the bromine needs to add to the π-system at C7, it must come from either above or below C7, not in the C7–C2–Br plane. Thus, this geometry does not allow for the correct trajectory for bromine migration to the benzyl carbon without a conformational change. However, calculations revealed a 1,2-Br shift onto the Cu(I) atom that proceeds through a low-barrier transition-state structure to restore aromaticity in INT-4.5. In this intermediate, the bromine and the benzyl carbon are no longer coplanar, allowing the bromine to be oriented in a manner that would permit transfer to the benzylic position. A final 1,4-Br shift from Cu(I) to the benzylic position results in the aryl copper INT-4.7. It should be noted that INT-4.5 allows for free rotation to take place around the C2–Cu(I) bond; thus, Br could be delivered to either face of the benzylic carbon. The barrier for rotation (see Supporting Information) was found to be comparable to the barrier from INT-4.5 to TS-4.6 and the INT-4.5 → INT-4.7 reaction is reversible, allowing stereochemistry to be scrambled at this waypoint. The barrier for rotation was smaller than the barrier from INT-4.5 to TS-4.6, confirming that stereochemistry can be scrambled at this waypoint. This energetically viable dearomative pathway accounts for the stereoablative step; furthermore, this pathway is consistent with the electronic effects of the substrate that impact selectivity between migration and hydroboration (see Table 1). Additionally, the same pathway computed with the dMepe ligand showed that the migration pathway was higher in energy than hydroboration (see Supporting Information).
Figure 4.
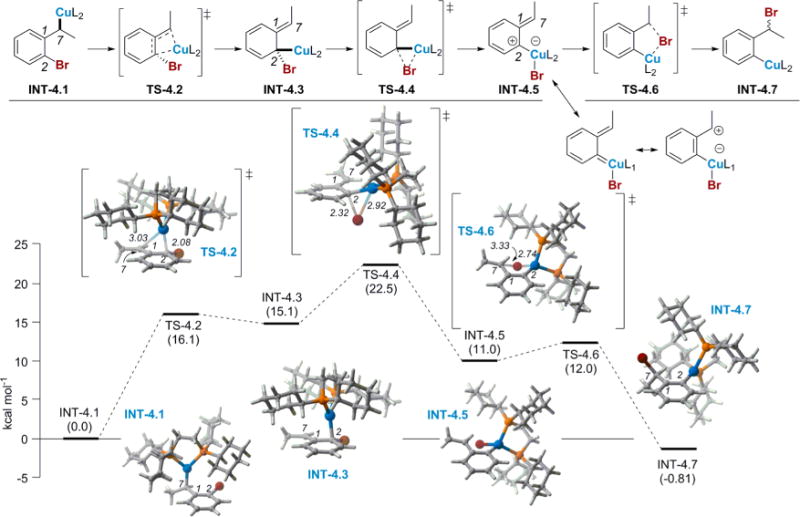
Proposed Cu-catalyzed 1,3-halogen migration pathway invoking a dearomatized intermediate. Free energies shown are in kcal/mol relative to INT-4.1 computed at SMD(THF)-M06/6-311G(d):LANL2TZ+ (Cu). Select bond lengths shown are in angstroms.
With our current understanding of the mechanism in hand, we wanted to return to the asymmetric 1,3-halogen migration to investigate the nature of the enantio-determining step.10a,30 If we assume the dearomatization pathway described in Figure 4 is the operative mechanism, the step that sets the stereochemistry of the final benzyl bromide product is the 1,4-bromide shift from the copper of INT-4.5 to the benzyl carbon of INT-4.7. When the transition-state structures that lead to both the (R) and the (S) products were optimized with the (S,S)-Ph-BPE ligand, the transition-state structure that leads to the (S) enantiomer (5.2) was slightly higher in energy, which corresponds to our observed stereochemistry (Figure 5). While the energy difference is relatively small, the expected energy difference given the enantiomeric ratio for this substrate (92:8) and the reaction conditions is ~1.4 kcal/mol.31 In transition-state structure 5.1 that leads to the observed stereochemistry, the geometry is reinforced by a π-stacking interaction between the substrate and one of the phenyl rings on the ligand. In transition state 5.2, which leads to the minor enantiomer, the phenyl ring of the ligand is directed away from the substrate and, consequently, does not have any favorable π-stacking interactions. Replacing the phenyl groups on the ligand with π-extended aromatics, such as naphthyl groups, would likely result in higher enantioselectivity; these design principles will be incorporated into future studies.
Figure 5.
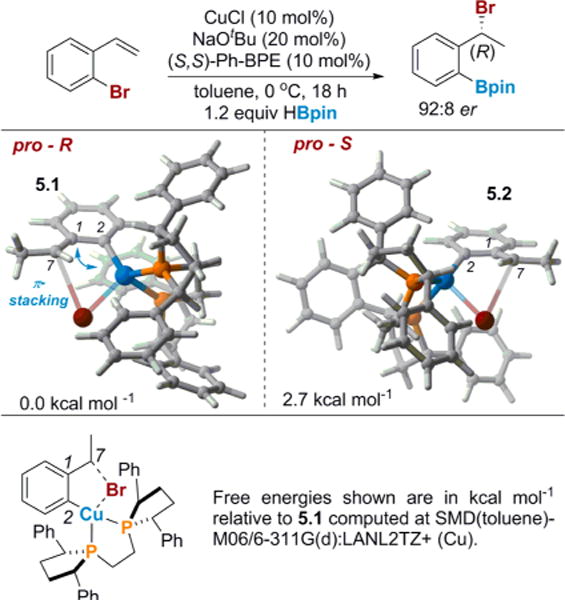
Transition-state structures for the enantio-determining step.
The full proposed mechanism is illustrated in Scheme 8. Phosphine-supported CuH 8.1 is the active catalyst that forms a π-olefin complex, 8.2, with 2-bromostyrene. This complex undergoes hydrometalation to form a benzyl copper, 8.3. Notably, the hydrometalation transition state is the highest in energy, making this step the rate-determining step, which fits well with our kinetics studies (see Figure 2). The benzyl copper 8.3 then either reacts with HBpin to form the undesired hydroboration product or undergoes a 1,3-copper shift that dearomatizes the ring. As the hydroboration pathway is higher in energy than the dearomatization pathway, or any subsequent transition state, the benzyl copper 8.3 prefers to undergo migration in the presence of the appropriate ligand. While the difference in energy between the hydroboration and 1,3-migration pathway is small,32 this energy difference may not be the only factor influencing selectivity. It is worth noting that the hydroboration of 8.3 to 8.8 is a bimolecular process, while conversion of 8.3 to 8.4 is unimolecular; thus, concentration might be expected to impact selectivity. Indeed, monitoring selectivity in the reaction of 2-bromo-5-fluorostyrene (which produces a mixture of migration and hydroboration, see Table 1) shows an initial migration:hydroboration ratio of 1:1, which increases to a final selectivity of 2:1 as the reaction progresses. The dearomatized intermediate 8.4 rearomatizes in a 1,2-bromide shift to form 8.5. This intermediate then transfers bromine from copper to the benzyl carbon, which in the presence of chiral ligand constitutes the enantio-determining step. The aryl copper 8.6 then reacts with HBpin to form the product and regenerate the CuH catalyst.
Scheme 8. Complete Catalytic Cycle for the Cu-Catalyzed 1,3-Halogen Migration/Borylationa.
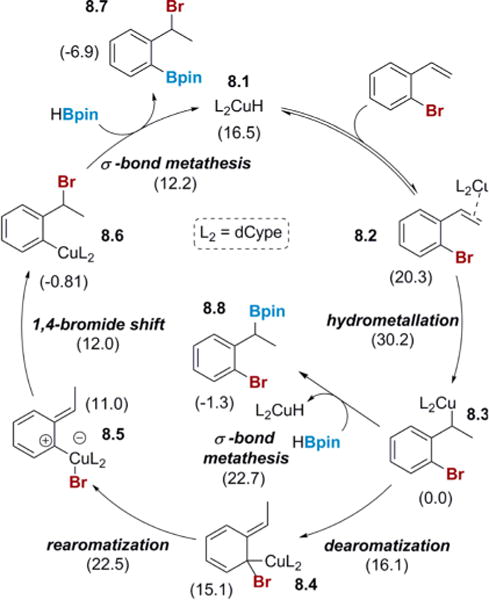
aFree energies shown are in kcal/mol relative to 8.3 computed at SMD(THF)-M06/6-311G(d):LANL2TZ+ (Cu).
The mechanistic proposal described herein is somewhat unusual. It does not represent the most direct pathway to products, and it involves uncommon steps. For example, the conversion of INT-4.1 to INT-4.7 can be considered to be a formal dyotropic rearrangement. Dyotropic rearrangements—migrations of two groups, often past each other so that they exchange positions—have a long history.33 The dyotropic rearrangement described herein is only formal in nature; i.e., it is stepwise rather than concerted. While other examples of stepwise dyotropic processes are known,33,34 ours is unusual in terms of the different pathways traveled by the two migrating groups: while the copper migrates directly to its new position, the bromine follows a more convoluted route. In addition, the final step of our mechanism has a very small associated barrier, comparable to that for stereochemical scrambling of INT-4.5. This opens the door for nonstatistical dynamic control35 of product stereochemistry.
CONCLUSION
We have reported computational and experimental studies to elucidate the mechanism of an unusual transformation in which an aryl bromide is reincorporated into the product via a dearomative 1,3-shift. These in-depth studies have revealed that the catalyst is probably monomeric in nature and that the rate-determining step is hydrometalation. Additionally, we have shown that this transformation is unlikely to involve oxidation state changes at the metal center, but rather favors a dearomative 1,3-shift that does not require copper to change its oxidation state. In the enantioselective 1,3-halogen migration, the enantio-determining step is a 1,4-bromide shift from copper to the benzyl carbon. Current studies are focused on using 1,3-halogen migration as a strategy to enable a broad range of aromatic functionalizations. Additionally, insights gained from this study will be employed in the design of improved catalysts for asymmetric 1,3-halogen migration.
Supplementary Material
Acknowledgments
The authors thank Dr. David Grigg for many helpful conversations. Sigma-Aldrich and Strem are thanked for a kind donation of phosphine ligands used in this study. Dr. Charlie Fry, Dr. Heike Hofstetter, and Dr. Monika Ivancic of the University of Wisconsin are thanked for their assistance with NMR spectroscopy and Dr. Martha Vestling for her assistance with MS data. This research was supported by start-up funds provided by the University of Wisconsin–Madison and the ACS-PRF No. 53146-ND1. The NMR facilities at UW-Madison are funded by the National Science Foundation (NSF; CHE-9208463, CHE-9629688) and National Institutes of Health (NIH; RR08389-01). The National Magnetic Resonance Facility at Madison is supported by the NIH (P41GM103399, S10RR08438, S10RR029220) and the NSF (BIR-0214394). The computational cluster at UW-Madison is funded by NSF Grant CHE-0840494. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by NSF (ACI-1053575 and CHE030089). B.M.H. was supported by a U.S. Department of Education GAANN Fellowship and H.B.W. was supported by a NSF Graduate Research Fellowship.
Footnotes
Supporting Information
Experimental procedures, computational details, and characterization data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.For selected references, see:; (a) Ritleng V, Sirlin C, Pfeffer M. Chem Rev. 2002;102:1731. doi: 10.1021/cr0104330. [DOI] [PubMed] [Google Scholar]; (b) Lyons TW, Sanford MS. Chem Rev. 2010;110:1147. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Diederich F, Stang PJ, editors. Metal-catalyzed Cross-coupling Reactions. Wiley-VCH; Weinheim: 1998. [Google Scholar]; (d) Phapale VB, Cardenas D. Chem Soc Rev. 2009;38:1598. doi: 10.1039/b805648j. [DOI] [PubMed] [Google Scholar]; (e) McDonald RI, Liu G, Stahl SS. Chem Rev. 2011;111:2981. doi: 10.1021/cr100371y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Farina V. Adv Synth Catal. 2004;346:1553. [Google Scholar]; (g) Jana R, Pathak TP, Sigman MS. Chem Rev. 2011;111:1417. doi: 10.1021/cr100327p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Xu LM, Li BJ, Yang Z, Shi ZJ. Chem Soc Rev. 2010;39:712. doi: 10.1039/b809912j. [DOI] [PubMed] [Google Scholar]
- 2.Grigg RD, Van Hoveln R, Schomaker JM. J Am Chem Soc. 2012;134:16131–16134. doi: 10.1021/ja306446m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Hoveln RJ, Schmid SC, Tretbar M, Buttke CT, Schomaker JM. Chem Sci. 2014;5:4763. doi: 10.1039/C4SC02040E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grigg RD, Rigoli JW, Van Hoveln R, Neale S, Schomaker JM. Chem—Eur J. 2012;30:9391–9396. doi: 10.1002/chem.201200642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Negishi E-I, de Meijere A, editors. Handbook of Organo-palladium Chemistry for Organic Synthesis. Wiley; New York: 2002. [Google Scholar]; (b) de Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. 2nd. Wiley-VCH; Weinheim: 2004. [Google Scholar]
- 6.(a) Tamaru Y, editor. Modern Organonickel Chemistry. Wiley; Weinheim: 2005. [Google Scholar]; (b) Krause N, editor. Modern Organocopper Chemistry. Wiley; Weinheim: 2002. [Google Scholar]; (c) Rosen BM, Quasdorf KW, Wilson DA, Zhang N, Resmerita A, Garg NK, Percec V. Chem Rev. 2011;111:1346. doi: 10.1021/cr100259t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Alonso DA, Najera C. In: Science of Synthesis. de Meijere A, editor. 47a. Thieme; New York/Stuttgart: 2009. pp. 439–479. [Google Scholar]; (e) Jana R, Pathak TP, Sigman MS. Chem Rev. 2011;111:1417. doi: 10.1021/cr100327p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Li B, Xu L, Wu Z, Guan B, Sun C, Wang B, Shi Z. J Am Chem Soc. 2009;131:14656. doi: 10.1021/ja907281f. [DOI] [PubMed] [Google Scholar]; (g) Han FS. Chem Soc Rev. 2013;42:5270. doi: 10.1039/c3cs35521g. [DOI] [PubMed] [Google Scholar]
- 7.(a) Wendlandt AE, Suess AM, Stahl SS. Angew Chem, Int Ed. 2011;50:11063–11087. doi: 10.1002/anie.201103945. [DOI] [PubMed] [Google Scholar]; (b) Beletskaya IP, Cheprakov AV. Coord Chem Rev. 2004;248:2337–2364. [Google Scholar]; (c) Sperotto E, van Klink GPM, van Koten G, de Vries JG. Dalton Trans. 2010;39:10338–10351. doi: 10.1039/c0dt00674b. [DOI] [PubMed] [Google Scholar]; (d) Sherry BD, Fürstner A. Acc Chem Res. 2008;41:1500–1511. doi: 10.1021/ar800039x. [DOI] [PubMed] [Google Scholar]; (e) Czaplik WM, Mayer M, Cvengros J, Jacobi A. Chem Sus Chem. 2009;2:396–417. doi: 10.1002/cssc.200900055. [DOI] [PubMed] [Google Scholar]
- 8.(a) Watson MP, Jacobsen EN. J Am Chem Soc. 2008;130:12504–12505. doi: 10.1021/ja805094j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mao J, Guo J, Fang F, Ji SJ. Tetrahedron. 2008;64:3905–3911. [Google Scholar]; (c) Li JH, Li JL, Wang DP, Pi SF, Xie YX, Zhang MB, Hu XC. J Org Chem. 2007;72:2053–2057. doi: 10.1021/jo0623742. [DOI] [PubMed] [Google Scholar]; (d) Evinder G, Batey RA. J Org Chem. 2006;71:1802–1808. doi: 10.1021/jo051927q. [DOI] [PubMed] [Google Scholar]
- 9.(a) Ehle AR, Zhou Q, Watson MP. Org Lett. 2012;14:1202–1205. doi: 10.1021/ol203322v. [DOI] [PubMed] [Google Scholar]; (b) Guan BT, Wang Y, Li BJ, Yu DG, Shi ZJ. J Am Chem Soc. 2008;130:14468–14470. doi: 10.1021/ja8056503. [DOI] [PubMed] [Google Scholar]; (c) Quasdorf KW, Antoft-Finch A, Liu P, Silberstein AL, Komaromi A, Blackburn T, Ramgren SD, Houk KN, Snieckus V, Garg NK. J Am Chem Soc. 2011;133:6352–6363. doi: 10.1021/ja200398c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Noh D, Chea H, Ju J, Yun J. Angew Chem, Int Ed. 2009;48:6062. doi: 10.1002/anie.200902015. [DOI] [PubMed] [Google Scholar]; (b) Won J, Noh D, Yun J, Lee JY. J Phys Chem A. 2010;114:12112. doi: 10.1021/jp1081966. [DOI] [PubMed] [Google Scholar]
- 11.For other examples of copper-catalyzed 1,3-migration, see:; (a) Alcaide B, Almendros P, Alonso JM, Cembellin S, Fernandez I, Martinez del Campo T, Torres MRSL. Chem Commun. 2013;49:7779–7781. doi: 10.1039/c3cc44073g. [DOI] [PubMed] [Google Scholar]; (b) Yang Y, Buchwald S. Angew Chem, Int Ed. 2014;53:8677–8681. doi: 10.1002/anie.201402449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Bezman SA, Churchill MR, Osborn JA, Wormald J. J Am Chem Soc. 1971;93:2063–2065. [Google Scholar]; (b) Mankad NP, Laitar DS, Sadighi JP. Organometallics. 2004;23:3369–3371. [Google Scholar]; (c) Lemmen TH, Folting K, Huffman JC, Caulton KG. J Am Chem Soc. 1985;107:7774–7775. [Google Scholar]
- 13.(a) Girard C, Kagan HB. Angew Chem, Int Ed. 1998;37:2922–2959. doi: 10.1002/(SICI)1521-3773(19981116)37:21<2922::AID-ANIE2922>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]; (b) Blackmond DG. Acc Chem Res. 2000;33:402–411. doi: 10.1021/ar990083s. [DOI] [PubMed] [Google Scholar]
- 14.For examples of monomeric organocopper complexes, see:; (a) Russo V, Herron JR, Ball ZT. Org Lett. 2010;12:220–223. doi: 10.1021/ol902458v. [DOI] [PubMed] [Google Scholar]; (b) Laitar DS, Tsui EY, Sadighi JP. Organometallics. 2006;25:2405–2408. [Google Scholar]; (c) Mankad NP, Laitar DS, Sadighi JP. Organometallics. 2004;23:3369–3371. [Google Scholar]; (d) Mankad NP, Gray TG, Laitar DS, Sadighi JP. Organometallics. 2004;23:1191–1193. [Google Scholar]; (e) Hope H, Olmstead MM, Power PP, Sandell J, Xu X. J Am Chem Soc. 1985;107:4337–4338. [Google Scholar]; (f) Rucker RP, Whittaker AM, Dang H, Lalic G. J Am Chem Soc. 2012;134:6571–6574. doi: 10.1021/ja3023829. [DOI] [PubMed] [Google Scholar]; (g) Laitar DS, Tsui EY, Sadighi JP. J Am Chem Soc. 2006;128:11036–11037. doi: 10.1021/ja064019z. [DOI] [PubMed] [Google Scholar]; (h) Gurung SK, Thapa S, Kafle A, Dickie DA, Giri R. Org Lett. 2014;16:1264–1267. doi: 10.1021/ol500310u. [DOI] [PubMed] [Google Scholar]
- 15.Anslyn EV, Dougherty DA. Modern Physical Organic Chemistry. University Science Books; Sausalito, CA: 2006. pp. 428–430. [Google Scholar]
- 16.Yamamoto Y. J Org Chem. 2007;72:7817–7831. doi: 10.1021/jo070579k. [DOI] [PubMed] [Google Scholar]
- 17.(a) Goering HL, Kantner SS, Seitz EP. J Org Chem. 1985;50:5495–5499. [Google Scholar]; (b) House HO, Wilkins JM. J Org Chem. 1978;43:2443–2454. [Google Scholar]
- 18.Herron JR, Ball ZT. J Am Chem Soc. 2008;130:16486–16487. doi: 10.1021/ja8070804. [DOI] [PubMed] [Google Scholar]
- 19.Van Hoveln RJ, Schmid SC, Schomaker JM. Org Biomol Chem. 2014;12:7655. doi: 10.1039/c4ob01294a. [DOI] [PubMed] [Google Scholar]
- 20.(a) Frisch MJ, et al. Gaussian 03, Revision D.01. Gaussian, Inc.; Wallingford, CT: 2004. [Google Scholar]; (b) Frisch MJ, et al. Gaussian 09, Revision B.01; Gaussian, Inc.; Wallingford, CT: 2009. See SI for full references. [Google Scholar]
- 21.(a) Zhao Y, Truhlar DG. Theor Chem Acc. 2008;120:215–241. [Google Scholar]; (b) Karton A, Tarnopolsky A, Lamere JF, Schatz GC, Martin JML. J Phys Chem A. 2008;112:12868–12886. doi: 10.1021/jp801805p. [DOI] [PubMed] [Google Scholar]; (c) Zhao Y, Truhlar DG. J Chem Phys. 2006;125:194101–194118. doi: 10.1063/1.2370993. [DOI] [PubMed] [Google Scholar]; (d) Zhao Y, Truhlar DG. J Phys Chem A. 2006;110:13126–13130. doi: 10.1021/jp066479k. [DOI] [PubMed] [Google Scholar]
- 22.(a) Hay PJ, Wadt WR. J Chem Phys. 1985;82:270–283. [Google Scholar]; (b) Yang Y, Weaver MN, Merz KM., Jr J Phys Chem A. 2009;113:9843–9851. doi: 10.1021/jp807643p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Su MD, Chu SY. J Am Chem Soc. 1997;119:10178–10185. [Google Scholar]; (d) Vigalok A, Uzan O, Shimon JW, Ben-David Y, Martin JML, Milstein D. J Am Chem Soc. 1998;120:12539–12544. [Google Scholar]; (e) Kadyrov R, Börner A, Selke R. Eur J Inorg Chem. 1999:705–711. [Google Scholar]; (f) Su MD, Chu SY. Chem—Eur J. 1999;5:198–207. [Google Scholar]; (g) Yu ZX, Wender PA, Houk KN. J Am Chem Soc. 2004;126:9154–9155. doi: 10.1021/ja048739m. [DOI] [PubMed] [Google Scholar]; (h) Luo X, Tang D, Li M. THEOCHEM. 2005;714:61–72. [Google Scholar]; (i) Luo X, Tang D, Li M. Int J Quantum Chem. 2005;105:108–123. [Google Scholar]; (j) Ardura D, López R, Sordo TL. J Org Chem. 2006;71:7315–7321. doi: 10.1021/jo0610356. [DOI] [PubMed] [Google Scholar]; (k) Alagona G, Chio C, Rocchiccioli S. J Mol Model. 2007;13:823–837. doi: 10.1007/s00894-007-0205-8. [DOI] [PubMed] [Google Scholar]; (l) Yu ZX, Cheong PHY, Liu P, Legault CY, Wender PA, Houk KN. J Am Chem Soc. 2008;130:2378–2379. doi: 10.1021/ja076444d. [DOI] [PubMed] [Google Scholar]; (m) Siebert MR, Yudin AK, Tantillo DJ. Eur J Org Chem. 2011;3:553–561. [Google Scholar]
- 23.(a) Hay PJ, Wadt WR. J Chem Phys. 1985;82:270. [Google Scholar]; (b) Hay PJ, Wadt WR. J Chem Phys. 1985;82:284. [Google Scholar]; (c) Hay PJ, Wadt WR. J Chem Phys. 1985;82:299. [Google Scholar]
- 24.Marenich AV, Cramer CJ, Truhlar DG. J Phys Chem B. 2009;113:6378–6396. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- 25.(a) Gonzalez CS, Schlegel HB. J Phys Chem. 1990;94:5523. [Google Scholar]; (b) Fukui K. Acc Chem Res. 1981;14:363–368. [Google Scholar]; (c) Maeda S, Harabuchi Y, Ono Y, Taketsugu T, Morokuma K. Int J Quantum Chem. 2015;115:258–269. [Google Scholar]
- 26.(a) Deutsch C, Krause N, Lipshutz BH. Chem Rev. 2008;108:2916–2927. doi: 10.1021/cr0684321. [DOI] [PubMed] [Google Scholar]; (b) Semba K, Fujihara T, Terao J, Tsuji Y. Chem—Eur J. 2012;18:4179–4184. doi: 10.1002/chem.201103612. [DOI] [PubMed] [Google Scholar]; (c) Reeker H, Norrby PO, Krause N. Organometallics. 2012;31:8024–8030. [Google Scholar]
- 27.Selected examples of Cu(III) species from C–X oxidative addition:; (a) Maiti D, Sarjeant AAN, Itoh S, Karlin KD. J Am Chem Soc. 2008;130:5644–5645. doi: 10.1021/ja800795b. [DOI] [PubMed] [Google Scholar]; (b) Casitas A, King AE, Parella T, Costas M, Stahl SS, Ribas X. Chem Sci. 2010;1:326–330. [Google Scholar]
- 28.Hartwig JF. Organotransition Metal Chemistry. University Science Books; Sausalito, CA: 2010. pp. 301–317. [Google Scholar]
- 29.(a) Zhu S, MacMillan DWC. J Am Chem Soc. 2012;134:10815–10818. doi: 10.1021/ja305100g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dattelbaum AM, Martin JD. Inorg Chem. 1999;38:6200–6205. doi: 10.1021/ic991064h. [DOI] [PubMed] [Google Scholar]
- 30.For other examples of CuH asymmetric transformations, see:; (a) Zhu S, Niljianskul N, Buchwald SL. J Am Chem Soc. 2013;135:15746–15749. doi: 10.1021/ja4092819. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Appella DH, Moritani Y, Shintani R, Ferreira EM, Buchwald SL. J Am Chem Soc. 1999;121:9473–9474. [Google Scholar]; (c) Jurkauskas V, Buchwald SL. J Am Chem Soc. 2002;124:2892–2893. doi: 10.1021/ja025603k. [DOI] [PubMed] [Google Scholar]
- 31.Walsh PJ, Kozlowski MC. Fundamentals of Asymmetric Catalysis. University Science Books; Sausalito, CA: 2009. [Google Scholar]
- 32.DFT calculations of the sort used herein are expected to predict relative transition-state energies for isomeric systems with kcal/mol, sometimes sub-kcal/mol, accuracy. Errors are expected to be larger for predictions of absolute barrier heights—often several kcal/mol—and when comparing competing unimolecular to bimolecular processes (since one depends on the concentration of one species and the other on the concentration of two species; accurately computing entropies for solution reactions is also a challenge:; Plata RE, Singleton DA. J Am Chem Soc. 2015;137:3811–3826. doi: 10.1021/ja5111392. Despite these caveats, we are encouraged that the predicted barrier for hydroboration/σ-bond metathesis with dMepe is 2 kcal/mol lower than that for migration (hydroboration observed experimentally), while the predicted barrier for hydroboration/σ-bond metathesis is slightly higher than that for migration with dCype (migration observed experimentally). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.(a) Reetz MT. Angew Chem, Int Ed. 1972;11:129–130. [Google Scholar]; (b) Reetz MT. Angew Chem, Int Ed. 1972;11:130–131. [Google Scholar]; (c) Reetz MT. Tetrahedron. 1973;29:2189–2194. [Google Scholar]; (d) Reetz MT. Adv Organomet Chem. 1977;16:33–65. [Google Scholar]; (e) Hoffmann R, Williams JE., Jr Helv Chim Acta. 1972;55:67–75. [Google Scholar]; (f) Buenker RJ, Peyerimhoff SD, Allen LC, Whitten JL. J Chem Phys. 1966;45:2835–2847. [Google Scholar]; (g) Gutierrez O, Tantillo DJ. J Org Chem. 2012;77:8845–8850. doi: 10.1021/jo301864h. [DOI] [PubMed] [Google Scholar]; (h) Braddock DC, Roy D, Lenoir D, Moore E, Rzepa HS, Wu JIC, Schleyer PVR. Chem Commun. 2012;48:8943–8945. doi: 10.1039/c2cc33676f. [DOI] [PubMed] [Google Scholar]; (i) Fernandez I, Cossío FP, Sierra MA. Chem Rev. 2009;109:6687–6711. doi: 10.1021/cr900209c. [DOI] [PubMed] [Google Scholar]
- 34.(a) Leverett CA, Purohit VC, Johnson AG, Davis RA, Tantillo DJ, Romo D. J Am Chem Soc. 2012;134:13348–13356. doi: 10.1021/ja303414a. [DOI] [PubMed] [Google Scholar]; (b) Davis RL, Leverett CA, Romo D, Tantillo DJ. J Org Chem. 2011;76:7167–7174. doi: 10.1021/jo2012175. [DOI] [PubMed] [Google Scholar]
- 35.(a) Carpenter BK. Annu Rev Phys Chem. 2005;56:57–89. doi: 10.1146/annurev.physchem.56.092503.141240. [DOI] [PubMed] [Google Scholar]; (b) Carpenter BK. Acc Chem Res. 1992;25:520–528. [Google Scholar]; (c) Carpenter BK. J Phys Org Chem. 2003;16:858–868. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.