
Figure 1.
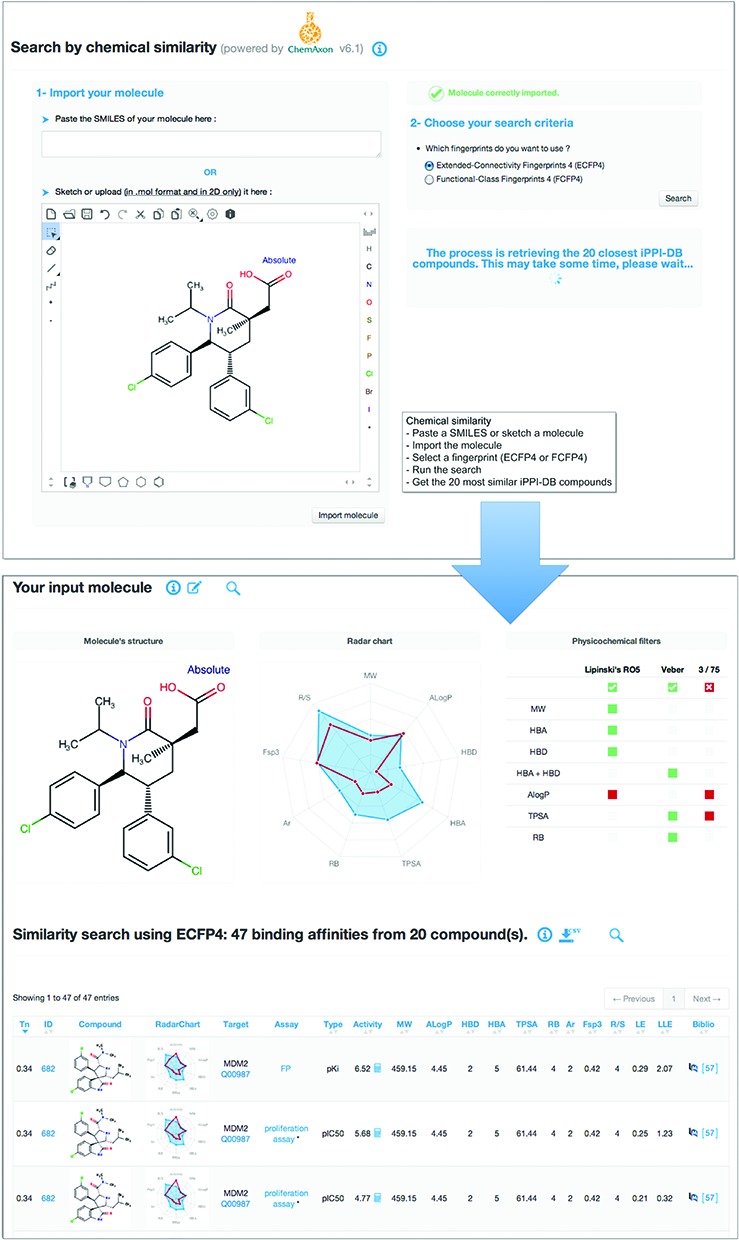
Example of a query on iPPI-DB using the mode ‘Chemical Similarity’. As the query compound, the user can either copy and paste a SMILES string into the interface, or directly sketch the compound within a Marvin JS editor. Here, we show an example with the structure with a recently identified piperidinone-based inhibitor of the MDM2/p53 interaction (25). Then, the compound can properly be imported, and the desired type of fingerprints for the chemical similarity search has to be chosen between ECFP4 and FCFP4. The search provides the user with all the binding data (here 47) of the 20 most similar iPPI-DB compounds in a table that can be sorted according to various criteria: molecular descriptors, activity, efficiencies or bibliographic IDs. The interface also provides a summary of the properties of the input compound using a radar chart based on nine physicochemical properties (molecular weight–MW, hydrophobicity–AlogP, the number of H-bond donors–HBD and acceptors–HBA, Topological Polar Surface Area–TPSA, the number of rotatable bonds RB, the number of aromatic rings–Ar, the proportion of sp3 carbon atoms–Fsp3 and the number of chiral centers–R/S) and its compliance to the three chemistry rules, Lipinski's RO5, Veber's and Pfizer's 3/75.