

Supplemental Digital Content is available in the text.
Keywords: cell proliferation, endothelial cells, microRNAs, transcription activation, vascular endothelial growth factor A
Rationale:
Several lines of evidence indicate that the regulation of microRNA (miRNA) levels by different stimuli may contribute to the modulation of stimulus-induced responses. The miR-17–92 cluster has been linked to tumor development and angiogenesis, but its role in vascular endothelial growth factor–induced endothelial cell (EC) functions is unclear and its regulation is unknown.
Objective:
The purpose of this study was to elucidate the mechanism by which VEGF regulates the expression of miR-17–92 cluster in ECs and determine its contribution to the regulation of endothelial angiogenic functions, both in vitro and in vivo. This was done by analyzing the effect of postnatal inactivation of miR-17–92 cluster in the endothelium (miR-17–92 iEC-KO mice) on developmental retinal angiogenesis, VEGF-induced ear angiogenesis, and tumor angiogenesis.
Methods and Results:
Here, we show that Erk/Elk1 activation on VEGF stimulation of ECs is responsible for Elk-1-mediated transcription activation (chromatin immunoprecipitation analysis) of the miR-17–92 cluster. Furthermore, we demonstrate that VEGF-mediated upregulation of the miR-17–92 cluster in vitro is necessary for EC proliferation and angiogenic sprouting. Finally, we provide genetic evidence that miR-17–92 iEC-KO mice have blunted physiological retinal angiogenesis during development and diminished VEGF-induced ear angiogenesis and tumor angiogenesis. Computational analysis and rescue experiments show that PTEN (phosphatase and tensin homolog) is a target of the miR-17–92 cluster and is a crucial mediator of miR-17-92–induced EC proliferation. However, the angiogenic transcriptional program is reduced when miR-17–92 is inhibited.
Conclusions:
Taken together, our results indicate that VEGF-induced miR-17–92 cluster expression contributes to the angiogenic switch of ECs and participates in the regulation of angiogenesis.
Vascular endothelial growth factor-A (VEGF) is a major regulator of blood vessel formation and pathological processes.1 As a key proangiogenic factor for endothelial cells (ECs), VEGF regulates their proliferation, migration, and survival.1 During adulthood, the majority of ECs within vasculature remain quiescent2 and only proliferate after angiogenic activation, mostly via VEGF stimulation.1
In This Issue, see p 2
Editorial, see p 9
MicroRNAs (miRNAs), important regulators of gene expression,3 modulate a wide range of biological processes, including angiogenesis.4 Although a large fraction of miRNAs is constitutively expressed, others can be dynamically regulated in ECs in response to a variety of stimuli.5–10 However, the mechanisms by which different factors regulate the expression of specific miRNAs in ECs, in comparison with other cell systems, have not yet been studied and remain an area of considerable interest.
The miR-17–92 cluster is a polycistronic miRNA gene encoding 7 miRNAs.11 The cluster was initially described as an oncogene and was later demonstrated to drive key physiological responses during development and disease.11,12 Several laboratories, including ours, have shown that human ECs exhibit low baseline levels of miR-17–92 cluster members in quiescent condition that are induced on VEGF stimulation.5,9,10 However, the mechanism by which VEGF stimulates the expression of the miR-17–92 cluster has not yet been examined. Members of the miR-17–92 cluster have been implicated in controlling different EC functions.6,9,13–15 However, the role of miR-17–92 cluster in regulating angiogenic EC functions is unclear and there is no genetic evidence for the role of endothelial miR-17–92 cluster in angiogenesis. Here, we report the mechanism by which VEGF stimulates the expression of the miR-17–92 cluster in ECs and the effect of endothelial postnatal genetic inactivation of this cluster on angiogenesis.
Methods
Cell culture (extended), inhibition of miR-17–92 cluster in vitro, silencing RNA knockdown of Elk-1 or PTEN (phosphatase and tensin homolog), vector construction, transient transfection and reporter gene assay, RNA isolation, quantitative reverse transcription polymerase chain reaction, Western blot analysis, bioinformatics analysis (promoter analysis, transcription factor prediction, and miRNA target prediction analysis), chromatin immunoprecipitation assays, gene expression polymerase chain reaction array, flow cytometry analyses of apoptosis and bromodeoxyuridine incorporation, cell number assessment, fibrin gel bead assay, mice (extended), mouse lung and retina EC isolation, mouse retina vascular system analysis, tumor-induced neovascularization, ear angiogenesis, and statistical analysis are described in the online Data Supplement.
Cell Culture
Human umbilical vein ECs (HUVECs) were obtained from the tissue culture core laboratory of the Vascular Biology and Therapeutics program (Yale University, New Haven, CT). Human aortic ECs were from Lonza, Allendale, NJ.
Mice
To generate inducible vascular EC–specific miR-17–92 iEC-KO mice, we crossed miR-17–92flox/flox mice16 with a tamoxifen-inducible expressed Cre-recombinase (Cre-ERT2) under the regulation of vascular endothelial cadherin 5 promoter17 to achieve specific inactivation of miR-17–92 cluster in ECs.
Results
Regulation of MiR-17–92 Cluster Expression by VEGF
To analyze the effect of VEGF on miR-17–92 expression in ECs, we first measured the levels of miR-17–92 cluster primary transcript (pri-miR-17–92) in quiescent freshly isolated HUVECs and on stimulation with VEGF. Primary transcript levels were barely detectable in quiescent cells (t=0 hours), but strongly stimulated when ECs lose their quiescent state after culture in the presence of VEGF-A (t=2, 4, and 6 hours; Figure 1A). Concomitantly, the levels of all 7 components of the cluster were increased on VEGF stimulation (Figure 1B). As the miR-17–92 cluster is conserved in the mouse genome, we also established that the loss of quiescence in freshly isolated mouse ECs via stimulation with VEGF was accompanied by an induction of pri-miR-17–92 as well as its 7 mature miRNAs (Online Figure VA). Similarly, the expression of pri-miR-17–92 was also stimulated by VEGF in HUVECs that were previously serum starved (Online Figure IA). Furthermore, when HUVECs were pretreated with actinomycin D, before VEGF stimulation, the induction of pri-miR-17–92 expression was not observed (Figure 1C). These data indicate that VEGF transcriptionally induces the expression of the miR-17–92 cluster in ECs.
Figure 1.
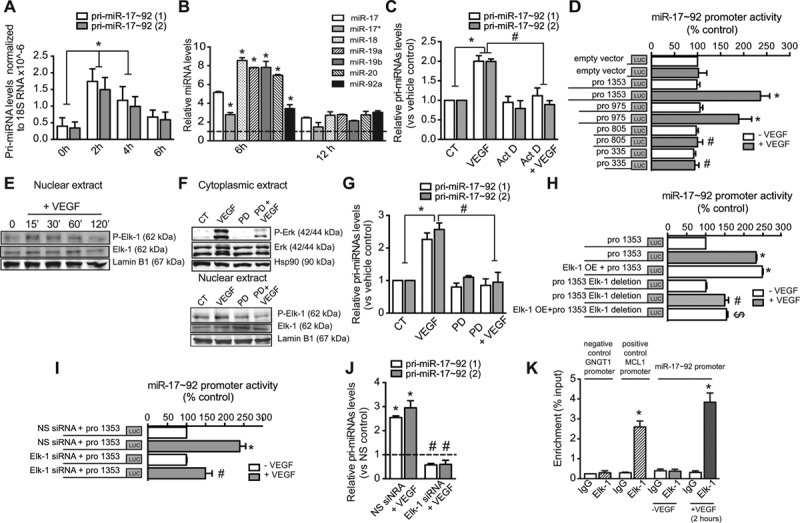
Vascular endothelial growth factor (VEGF) regulates the expression of the microRNA-17–92 cluster through Elk-1 transcription factor. Quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis of (A) pri-miRNA-17–92 levels or (B) different members of miR-17–92 cluster in quiescent freshly isolated human umbilical vein endothelial cells (HUVECs; t=0 hours) or after their stimulation with VEGF (50 ng/mL) at the indicated times. A, Data are expressed as pri-miRNA levels detected using 2 different primers, pri-miR-17–92 (1) and (2) normalized to 18S rRNA and correspond to the mean±SEM of 3 independent experiments. B, Data are expressed as miRNA levels normalized to U6 and correspond to 1 representative experiment out of 3e. C, qRT-PCR analysis of pri-miRNA-17–92 levels in HUVECs pretreated with actinomycin D (Act D; 4 μg/mL) for 2 hours before VEGF stimulation. D, Luciferase activity of human miR-17–92 promoter (pro 1353) and deletion constructs (pro 975, pro 805, and pro 335). E, Western blot analysis of P-Elk-1, Elk-1, and lamin B1 from nuclear extracts. Lamin B1 was used as nuclear-loading control. F, Western blot analysis of P-Erk, Erk, and Hsp90 from cytoplasmic extracts of HUVECs or P-Elk-1, Elk-1, and lamin B from nuclear extracts of HUVECs pretreated for 30 minutes with PD098059 (PD; 18 nmol/L) before VEGF stimulation. Hsp90 was used as loading control. E and F, Representative blots of 1 experiment out of 3 with similar results. G, qRT-PCR analysis of pri-miRNA-17–92 levels in HUVECs pretreated PD (18 nmol/L) before VEGF stimulation. H, miR-17–92 promoter activity in HUVECs cotransfected with Elk-1 overexpression construct. Effect of Elk-1-binding site deletion on miR-17–92 promoter activity in HUVECs treated with VEGF for 2 hours or cotransfected with Elk-1 overexpression construct. I, Luciferase activity of human miR-17–92 promoter in HUVECs transfected with Elk-1 silencing RNA (siRNA) or nonsilencing (NS) control, starved for 12 hours and then treated with VEGF for 2 hours. J, qRT-PCR analysis of pri-miRNA-17–92 levels in HUVECs transfected with Elk-1 siRNA or NS control treated with VEGF. C and G, Data correspond to the mean±SEM of 3 independent experiments. Data correspond to (D) 6 or (H and I) 3 independent experiments. K, Quantification of promoter-specific Elk-1 bound antibody was carried by PCR with primers proximal to the predicted binding site of miR-17–92 promoter in HUVECs. GNGT1 and MCL1 promoters are used as negative and positive controls for promoter genes nontarget and target by Elk-1, respectively.18 Data were normalized to input chromatin and expressed as enrichment compared with untreated cells. *P≤0.05 compared with vehicle control; #P≤0.05 compared with VEGF alone; $P≤0.05 compared with cotrantrasfection with Elk-1 overexpression+pro 1353.
To ascertain the mechanism by which VEGF regulates miR-17–92 cluster expression in ECs, we transfected HUVECs with a luciferase reporter vector harboring 1353-bp upstream of the miR-17–92 gene.19 We observed increased promoter activity in ECs treated with VEGF (Figure 1D). In VEGF-stimulated conditions, 5′ deletions of the full-length promoter fragment revealed a reduction in promoter activity with the −805 bp fragment, suggesting the presence of cis-acting elements between −975 and −805 bp (Figure 1D). Sequence promoter analysis for cis-acting elements within this region identified Elk-1 as a potential transacting factor of miR-17–92 promoter activation by VEGF. Elk-1 belongs to the ETS (E-twenty-six) domain family proteins and plays a critical role in gene expression on mitogen stimulation and activation of the mitogen-activated protein kinase cascade.20 Interestingly, we observed an increase of Elk-1 phosphorylation on VEGF stimulation of ECs (Figure 1E). PD098059-mediated inhibition of MEK1/2, a direct upstream modulator of extracelular signal regulated kinase (ERK), blocked VEGF-induced activation of both ERK and Elk-1 (Figure 1F), and abrogated the VEGF-induced upregulation of pri-miR-17–92 expression (Figure 1G). Overexpression of Elk-1 also elicited a stimulatory effect on promoter activity in non-VEGF stimulated conditions (Figure 1H). In contrast, stimulation with neither VEGF nor Elk-1 overexpression increased miR-17–92 promoter activity when the Elk-1-binding site was deleted from the promoter reporter construct (Figure 1H). Furthermore, silencing of Elk-1 (Online Figure II) significantly reduced VEGF-mediated stimulation of miR-17–92 promoter activity (Figure 1I) and pri-miR-17–92 expression (Figure 1J). In line with these findings, using chromatin immunoprecipitation analysis, we demonstrated that on VEGF stimulation of ECs, Elk-1 binds to the miR-17–92 promoter region (Figure 1K). Taken together, these results indicate that the VEGF/ERK/ELK-1 pathway is involved in the transcriptional regulation of the miR-17–92 cluster in human ECs. In addition to VEGF, fibroblast growth factor 2, a well-documented angiogenic growth factor that stimulates the mitogen-activated protein kinase pathway,21 slightly induced expression of the miR-17–92 cluster in HUVECs (Online Figure IIIA). Altogether, these data indicate that growth factor activation of the mitogen-activated protein kinase pathway is one mechanism to induce expression of miR-17–92 cluster in ECs.
Our results indicate that VEGF stimulates the expression of miR-17–92 cluster in human macrovascular venous ECs (Figure 1A and 1B; Online Figure I) as well as in mouse lung ECs (Online Figure VA), which are mostly microvascular. We also tested if the same regulatory mechanisms observed mostly in the venous context were present in arterial ECs. Interestingly, human aortic ECs stimulated with VEGF did not exhibit a significant upregulation of pri-miR-17–92 expression (Online Figure IIIB).
Regulation of EC Angiogenic Responses In Vitro by VEGF-Induced MiR-17–92 Expression
We next assessed if VEGF-mediated upregulation of the miR-17–92 cluster participates in regulating EC angiogenic responses in vitro. Inhibition of the miR-17–92 cluster (Figure 2A) resulted in reduced EC proliferation, evaluated by cell number (Figure 2B) and bromodeoxyuridine incorporation (Online Figure IVA). To test if induction of cell death mediates the reduction in cell number on miR-17–92 cluster inhibition, apoptosis was blocked by preincubation with the caspase inhibitor z-VAD-FMK (carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone). Because zVAD-FMK addition did not affect cell number (Online Figure IVB and IVC), we concluded that the effect on cell number was not because of cell death. By using the fibrin gel bead assay that recapitulates several crucial steps of angiogenesis22 (Figure 2C–G), we found that inhibition of the miR-17–92 cluster in VEGF-stimulated conditions decreased cumulative sprout length (Figure 2D), number of sprouts per spheroid (Figure 2E), and the early formation of branch points (Figure 2F). However, inhibition of the miR-17–92 cluster increased the number of detached cells (Figure 2G), indicative of vessel regression.22 Taken together, our results indicate that in VEGF-stimulated conditions, miR-17–92 cluster is required for proliferation and angiogenic sprouting, and therefore participates in the angiogenic switch of the quiescent endothelium.
Figure 2.
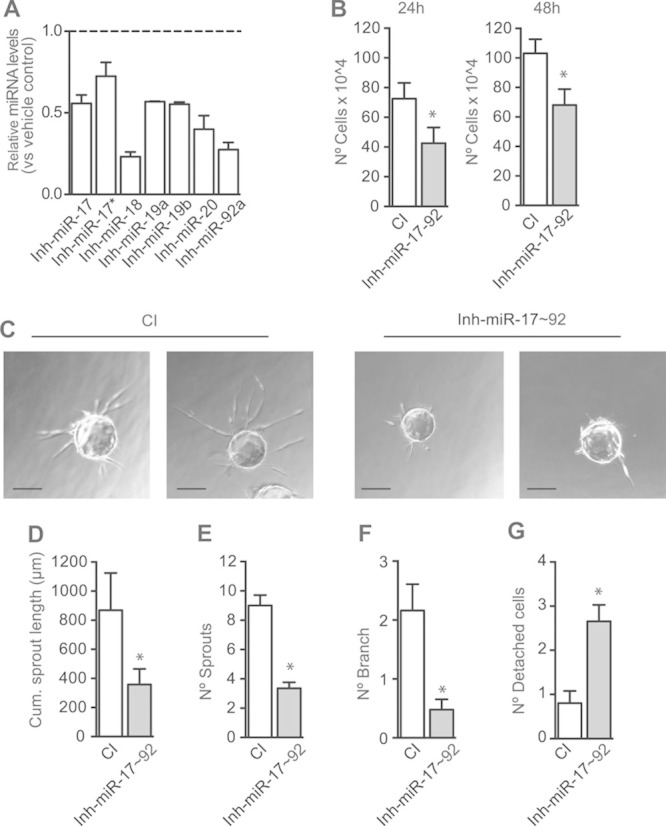
Inhibition of microRNA-17–92 cluster decreases proliferation and angiogenic sprouting in endothelial cells. Human umbilical vein endothelial cells (HUVECs) were transfected with 70 nmol/L mix of miR-17–92 inhibitors (Inh-miR-17–92) or control inhibitor (CI). A, Quantitative reverse transcription polymerase chain reaction analysis of individual miR-17–92 cluster members analyzed 36 hours post-transfection. Data are expressed as relative miRNA levels vs CI. B, Total number of cells, 24 or 48 hours post-transfection. C–G, Fibrin gel bead assay was performed 36 hours post-transfection. HUVECs were allowed to undergo morphogenesis for 2 days in the presence of EGM-2 (endothelial cell growth factor medium 2) media containing VEGF (vascular endothelial growth factor). C, Representative images of HUVECs bead spheres. D–G, Quantification of angiogenic sprouting: (D) cumulative sprout length, (E) number of sprouts, (F) number of branch, and (G) number of detached cells. Data are expressed as mean±SEM of 3 independent experiments. *P≤0.05, compared with cells transfected with CI (Scale, 84 μm).
Role of MiR-17–92 Cluster in Angiogenesis
Even though the miR-17–92 cluster was among the first miRNAs linked to tumor14 and corpus luteum angiogenesis,23 there is no genetic evidence for the role of endothelial miR-17–92 cluster in angiogenesis. We tested the effect of postnatal inactivation of miR-17–92 cluster in the endothelium of developing mouse retinal vasculature because this is a model highly dependent on VEGF24,25 and useful for analyzing the molecular and cellular mechanisms regulating angiogenesis.17 Interestingly, we found that miR-17–92 cluster is highly expressed in mouse retina ECs at early stages and declines as development proceeds (Online Figure VB). Consistently, several studies have shown that miR-17–92 cluster is highly expressed in mouse embryonic stem cells, with expression levels decreasing during embryonic development.26,27 Furthermore, in a human embryonic stem cell vasculogenesis model, the expression of all members of the miR-17–92 cluster is reduced after 21 days of differentiation.10 These reports support the idea that high levels of miR-17–92 cluster may promote cell proliferation and, therefore, could participate in promoting endothelial angiogenic response.
Endothelial postnatal inactivation of miR-17–92 cluster resulted in significant reduction of miR-17–92 cluster members (Online Figure VC). As such, retinal angiogenesis was impaired, as shown by reduced endothelial coverage and delayed radial outgrowth (Figure 3A and 3B, 3D–3G; Online Figure VI), without affecting growth (Figure 3C). Overall, miR-17–92 iEC-KO retinas exhibited significantly less branching, fewer segment numbers, fewer hole numbers, and shorter vessel length (Figure 3D–3G; Online Figure VI). Endothelial tip cells, localized at the retinal front during development, are characterized by specialized apical filopodia and are vital for the development of new capillaries.25 miR-17–92 deletion significantly reduced the number of filipodia projections at the leading edge of the growing vascular front (Figure 3H and 3I). Importantly, the endothelial loss of miR-17–92 decreased proliferation, quantified by nuclear phosphohistone 3 staining (Figure 3J and 3K) and induced vessel regression, as denoted by an increased number of collagen IV–positive, isolectin B4 (Iso b4)–negative empty sleeves (Figure 3 L and 3M). Interestingly, the number of apoptotic ECs was similar in control and miR-17–92i EC-KO mice (Online Figure VII). Therefore, premature vessel regression observed in miR-17–92 iEC-KO is likely a consequence of reduced cell proliferation rather than induced apoptosis.28,29 These data indicate that loss of miR-17–92 cluster reduces retinal vascular coverage. These results show, for the first time, that endothelial expression of miR-17–92 cluster regulates normal retinal angiogenesis. Thus, although miR-17–92 is an important proangiogenic signal during embryonic and retinal development, it may also play a role in the mature circulatory system. To ascertain this, we examined the importance of miR-17–92 cluster in VEGF-induced ear angiogenesis. miR-17–92 iEC-KO and littermate controls were injected intradermally into the ear with an adenovirus expressing murine VEGF 164 (Ad-VEGF) and angiogenesis was assessed after 3 days. As expected, VEGF increased the number of angiogenic (platelet/endothelial cell adhesion molecule 1)-positive vascular structures, whereas injection of a control virus expressing enhanced green fluorescent protein (Ad-eGFP) did not. However, the angiogenic response to VEGF was reduced in miR-17–92 iEC-KO mice (Figure 4A and 4B). We additionally tested the effects miR-17–92 cluster loss in the Lewis lung carcinoma tumor-induced angiogenesis model (Figure 4C–4E). Interestingly, and in agreement with the previously described effects of miR-17–92 cluster in the angiogenic responses of ECs, in physiological retinal angiogenesis, and in VEGF-induced ear angiogenesis, a reduction of platelet/endothelial cell adhesion molecule 1–positive structures (in cross-sections; Figure 4C and 4D) in the tumor-associated vasculature was observed in miR-17–92 iEC-KO, as well as an overall reduction of tumor growth (weight; Figure 4E). Here, we show for the first time that miR-17–92 cluster participates in tumor-induced angiogenesis by regulating VEGF-induced angiogenic EC functions. Altogether, these data suggest that miR-17–92 cluster participates in the regulation of both developmental angiogenesis, as well as angiogenesis during adulthood.
Figure 3.
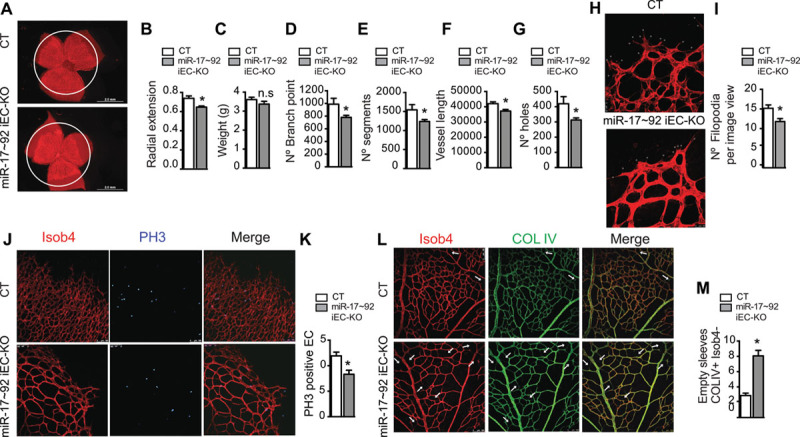
Postnatal deletion of microRNA-17–92 cluster in the endothelium reduces developmental retinal angiogenesis. Effect of postnatal endothelial-specific miR-17–92 cluster deletion on retinal vasculature (mice injected with tamoxifen P1–P3, assessed at P6; control n=10; miR-17–92 iEC-KO n=17). A, Retinal whole mounts, isolectin B4 (Isob4 immunostaining representative images. B, Radial vessel extension quantification. C, Effect of endothelial-specific miR-17–92 cluster deletion on overall growth (weight). D–G, Quantification of the effects of postnatal endothelial-specific miR-17–92 cluster deletion on retinal vasculature: (D) number of branch point, (E) number of segments, (F) vessel length, and (G) number of holes. H, Retinal vasculature front, IsoB4 immunostaining representative images (magnification ×63). I, Filipodia was quantified as number of filipodia per view. J, Retinal vasculature front, IsoB4 and phosphohistone 3 (PH3) immunostaining representative images. K, Proliferation was quantified as a ratio of PH3-positive endothelial cells (ECs) to IsoB4-positive vessels. L, Retinal vascular plexus, IsoB4 and collagen IV (COL IV) immunostaining representative images. M, Vessel regression was quantified as number of empty sleeves COL IV–positive and IsoB4-negative. J and L, Magnification: ×20. For each retina, 4 images were quantified. Data are expressed mean±SEM.*P≤0.05, compared with control littermates.
Figure 4.
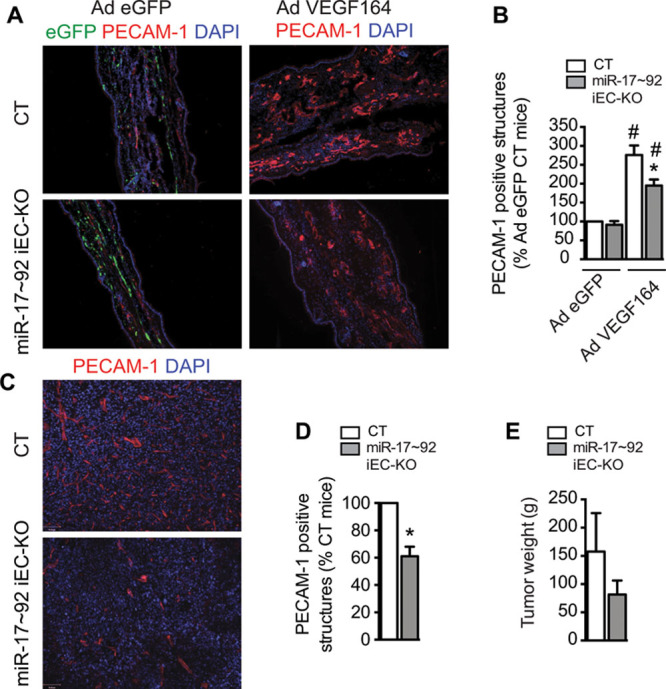
Postnatal deletion of microRNA-17–92 cluster in the endothelium reduces vascular endothelial growth factor (VEGF)–induced ear angiogenesis and tumor-induced angiogenesis. A and B, Effect of postnatal endothelial-specific miR-17–92 cluster deletion on ear angiogenesis model. Ad-VEGF or Ad-eGFP were injected intradermally into the right and left ears, respectively of 6-week-old mice (n=6 per group). A, Representative images of cross-sections from ears immunostained stained for platelet/endothelial cell adhesion molecule 1 (PECAM-1) and counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Enhanced green fluorescent protein (eGFP) for basal nonstimulated control of infection. B, VEGF-induced angiogenesis was quantified as PECAM-1-positive structures. For each animal, 2 images from 3 sections were quantified. C–E, Postnatal endothelial-specific miR-17–92 cluster deletion on tumor angiogenesis model. Lewis lung carcinoma cells (106) were injected subcutaneously in the dorsal flank of 6-week-old mice (control n=9; miR-17–92 iEC-KO n=6). After 12 days, animals were euthanized and tumor tissues were collected. C, Cross-sections from tumors, PECAM-1 and DAPI immunostaining representative images. D, Tumor-induced angiogenesis was quantified as PECAM-1-positive structures. For each tumor, 2 images from 3 sections from 2 different tumor pieces were quantified. E, Tumor weight. Data are expressed mean±SEM.*P≤0.05, compared with control littermates. #P≤0.05, compared with Ad-eGFP control.
Targeting Activity of MiR-17–92 Cluster
To date, several targets have been described, in ECs, for individual members of the miR-17–92 cluster.13,15 Our data indicate that, as a result of angiogenic activation of ECs, miR-17–92 is transcriptionally stimulated; therefore, the expression of individual member of the entire cluster is increased. As indicated, miR-17–92 cluster consists of 7 highly conserved miRNAs and this complicates the identification of a bona fide unique target that may explain the role of miR-17–92 as a unit. The 7 miRNAs in the cluster can be grouped in 4 different seed families and, therefore, 4 different target specificities.3 To investigate the targets of miR-17–92 cluster that may explain the observed antiangiogenic phenotype after its inhibition in vitro or conditional deletion in vivo, we performed a bioinformatics analysis to identify targets common to the 4 different seeds (Figure 5A), conserved between human and mouse, and relevant to ECs (Figure 5A). From this unbiased bioinformatics analysis, we identified 4 targets expressed in ECs that are common to the 4 seed families and conserved in human and mouse. On the basis of a bibliographic search of the role of the identified predicted targets, PTEN stands out as a strong candidate to explain the antiangiogenic phenotype observed in vitro on inhibition of the cluster in ECs and in vivo after deletion of the miR-17–92 cluster. Indeed, PTEN has been reported to regulate angiogenesis.30,31 Importantly, the targeting activity of different members toward PTEN 3′ untranslated region is well documented.11,12 Thus, we decided to investigate if the inhibition or the conditional deletion of the cluster on ECs results in an upregulation of PTEN because of the lack of miR-17–92 targeting activity. Our results indicate that PTEN expression (both mRNA and protein) was increased in HUVECs where the miR-17–92 cluster is inhibited (Figure 5B). Similarly, protein PTEN levels (major direct effect of miRNA action32) were also increased in mouse lung ECs isolated from tamoxifen-treated miR-17–92 iEC-KO mice (Figure 5C). These data suggest that the antiangiogenic phenotype observed both in vitro and in vivo may, in part, be explained through the stimulation of PTEN expression in ECs when miR-17–92 cluster is inhibited. To test if the effects of miR-17–92 inhibition could be completely recapitulated by PTEN targeting, we have overexpressed or silenced PTEN together with inhibition of miR-17–92 cluster. As expected, overexpression of PTEN drastically reduced proliferation of ECs both in control inhibitor condition and on inhibition of miR-17–92 cluster. Concomitant inhibition of miR-17–92 cluster and PTEN overexpression does not further affect proliferation compared with its respective control inhibitor (Figure 5D). These findings are in agreement with a previous work that shows that overexpression of PTEN inhibited the proliferative and angiogenic effects of VEGF30 and suggest that overexpression of PTEN overcomes the effects of miR-17–92 inhibition. Conversely, silencing of PTEN significantly increases proliferation (Figure 5E). However, although the difference in proliferation between control inhibitor and miR-17–92 inhibitor is ≈20% in nonsilencing control conditions, the defect in proliferation in miR-17–92 inhibition is partially rescued when PTEN is silenced (−11% versus −20%; Figure 5E). Because the silencing RNA considerably reduces the levels of PTEN to a similar extent than in control inhibitor condition or after miR-17–92 cluster inhibition, the differences we observe in proliferative responses are likely because of additional targeting activity of the miR-17–92 cluster. To gain a global insight on the effects of miR-17–92 cluster inhibition on angiogenic programs, we performed an expression analysis of genes implicated in the regulation of angiogenesis in HUVECs cultured in the presence of VEGF with or without miR-17–92 cluster inhibitors. As expected, the expression of the negative regulator of angiogenesis thrombospondin 1 was increased on miR-17–92 cluster inhibition9 (Online Figure VIIIA). Overall, by analyzing gene expression with VEGF stimulation, we found that HUVECs with inhibited expression of the miR-17–92 cluster were predicted to have a less angiogenic phenotype than their noninhibited controls. In fact, the expression pattern of most genes analyzed indicates that the pathways of angiogenesis and proliferation of ECs were reduced in EC where miR-17–92 was inhibited (Online Figure VIIIB). Altogether, our present data indicate that VEGF stimulates miR-17–92 to promote angiogenic functions of ECs (Figure 5F).
Figure 5.
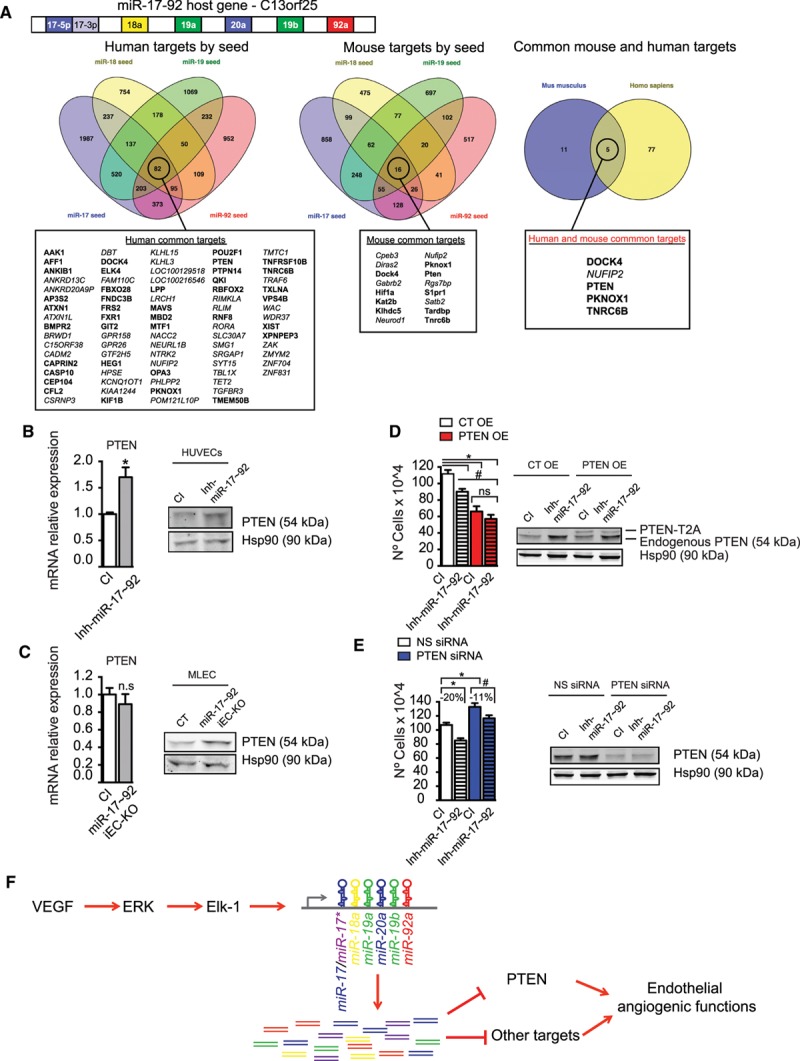
MicroRNA-17≈92 (miR-17–92) cluster targeting unit regulates phosphatase and tensin homolog (PTEN) expression and promote and angiogenic phenotype in endothelial cells (ECs). A, Schematic representation of the polycistronic miR-17–92 cluster and its host gene. The 4 seed families are indicated in different colors.11 Common targets for each seed sequence of the miRNA-17–92 cluster were obtained from miRNA target prediction algorithms. Targets common for all the seed sequences of the miRNA-17–92 cluster were found for mouse (16 genes) and human (82 genes). Among the targets common for all seeds, 5 were conserved among human and mouse. The targets in bold are genes that are expressed in ECs.33 B, Quantitative reverse transcription polymerase chain reaction (qRT-PCR) and Western blot analysis of PTEN in human umbilical vein ECs (HUVECs) transfected with 70 nmol/L mix of miR-17–92 inhibitors (Inh-miR-17–92) or control inhibitor (CI) for 36 hours. C, qRT-PCR or Western blot analysis of PTEN in mouse lung ECs (MLECs) from miR-17–92 iEC-KO or CT mice. Data are expressed as mRNA relative expression. D, Total number of cells and Western blot analysis of PTEN in HUVECs cotransfected with PTEN-T2A-EGFP overexpression vector (PTEN overexpression [OE]) or empty control (CT OE) and 70 nmol/L mix of miR-17–92 inhibitors (Inh-miR-17–92) or CI. E, Total number of cells and Western blot analysis of PTEN in HUVECs cotransfected with PTEN silencing RNA (siRNA) or nonsilencing (NS) control and 70 nmol/L mix of miR-17–92 inhibitors (Inh-miR-17–92) or CI. D and E, Analysis is 36 hours post-transfection. B–E, Endogenous PTEN is shown and Hsp90 was used as loading control. D, PTEN overexpression fused to the cleaved T2A peptide (PTEN-T2A). Representative blots of 1 experiment out of 3 with similar results. Data are expressed as total number of cells and correspond to the mean±SEM of 3 independent experiments. *P≤0.05, compared with CI (B and C) and * or #P≤0.05 comparisons are as indicated (D and E). F, Graphic summary. VEGF stimulates the transcription of the miR-17–92 cluster in ECs through the activation of ERK/Elk-1 pathway and regulate VEGF-induced angiogenic functions by targeting PTEN among other targets.
Discussion
A growing body of evidence indicates that precise regulation of miRNA expression is critical for diverse physiological and pathophysiological processes. In this study, we have investigated the mechanism by which VEGF regulates the expression of miR-17–92 cluster in ECs to determine its contribution to the regulation of angiogenic functions of ECs. Our present results indicate that the VEGF/ERK/ELK-1 pathway is involved in the transcriptional regulation of the miR-17–92 cluster in ECs. Previous studies in other cell types have shown that the miR-17–92 cluster is transcriptionally regulated by different transcription factors, including c-Myc.19,34,35 Under physiological conditions, c-Myc expression is dependent on mitogens. This control is lost in cancer cells, resulting in elevated levels of c-Myc oncoprotein, which in turn induces the expression of the miR-17–92 cluster.35 c-Myc regulation of the miR-17–92 cluster is not likely to be operative in primary ECs because in normal cells, c-Myc activation triggers the apoptotic program.36 In the context of ECs, it has been reported that histone deacetylase 9 participates in the repression of miR-17–92 cluster,37 therefore, it is possible that histone deacetylase 9 dissociates from the miR-17–92 cluster promoter in VEGF-stimulated conditions. Interestingly, fibroblast growth factor 2 also induced the expression of the miR-17–92 cluster ECs although in a less efficient manner. Given that fibroblast growth factor also stimulates the mitogen-activated protein kinase pathway21 in ECs, it is likely that Elk-1 participate in its transcriptional regulation. However, further investigation is needed to reveal if additional mechanisms are involved in fibroblast growth factor–mediated regulation of miR-17–92 cluster in ECs.
Each vascular bed has unique structural and functional properties. Indeed, ECs lining distinct blood vessels behave differently.38 We have shown that VEGF stimulates the expression of miR-17–92 cluster human macrovascular venous ECs as well as in mouse lung ECs, which are mostly microvascular, but did not affect its expression in arterial ECs. Several lines of evidence suggest a role for hemodynamics in mediating arterial/venous identity;39 however, studies in zebrafish embryos suggest that many artery- and vein-specific properties are epigenetically programmed before the onset of blood flow.40 An epigenetically repressed promoter may explain the lack of upregulation of pri-miR-17–92 on VEGF stimulation in arterial ECs, therefore, suggesting different roles of miR-17–92 depending on the vascular bed. Future investigation is warranted to ascertain the nature of these differences and their physiological relevance.
Previous reports have shown that single overexpression of pre-miR-17, pre-miR-18a, pre-miR-19a, or pre-miR-20a significantly inhibits 3-dimensional spheroid sprouting in vitro, whereas their individual inhibition has an opposite outcome.15 This is in contrast to our present and previous findings9 that indicate that in VEGF-stimulated conditions, miR-17–92 cluster is required for proliferation and angiogenic sprouting, and therefore participates in the angiogenic switch of quiescent endothelium. A possible explanation for these discrepancies is that miR-17–92 levels vary significantly in response to VEGF, suggesting that growth and culture conditions influence basal miRNA expression, and therefore, cluster function in cultured ECs. Given the potential for opposing effects of the miR-17–92 miRNAs depending on cellular compartment,9,14,15,23 we postulated that deciphering the role of this cluster in angiogenic functions in vivo might required a genetic cell type–specific deletion. We generated a mouse model of inducible deletion of the miR-17–92 cluster in the vascular endothelium and we analyzed its effects on models of angiogenesis that are highly dependent on VEGF (eg, retinal angiogenesis, VEGF-induced ear angiogenesis, and tumor-induced neovascularization). Postnatal inducible deletion of the miR-17–92 cluster reduced normal retinal angiogenesis. Because VEGF levels are increased in patients with active proliferative diabetic retinopathy,24 one might speculate that miR-17–92 levels in ECs would increase and lead to pathological angiogenesis. However, in the murine retinopathy of prematurity model, miR-17/20 downregulation in whole retina has been linked to increased angiogenesis and increased VEGF secretion by retinoblastoma cells.41 The proposed targeting effect of VEGF is likely to occur on glial and Muller cells, which are responsible for the majority of VEGF secretion24,25 and not directly on ECs. In fact, one could hypothesize that miR-17/20 downregulation in retinal non-ECs promotes VEGF secretion to induce angiogenic responses in ECs, in part, via miR-17–92 upregulation, as indicated by our present findings. This corroborates the notion that modulation of miR-17–92 expression is specific in different cell types and physiological situations, and governs context-specific and developmental changes in endothelial behavior. However, the miR-17–92 cluster, also known as oncomir-1, is among the most potent oncogenic miRNAs.11,12 Our present data show, for the first time, that miR-17–92 cluster also participates in tumor-induced angiogenesis by regulating VEGF-induced angiogenic EC functions. Altogether, our present work shows that endothelial miR-17–92 cluster regulates both developmental angiogenesis, as well as angiogenesis during adulthood.
The miR-17–92 cluster consists of 7 highly conserved miRNAs, therefore, complicating the identification of a bona fide target that may explain the role of the miR-17–92 as a unit. It is recognized that the coordinated expression of miRNAs belonging to different seed families from a single transcriptional unit suggests functional cooperation.42 Our bioinformatics analysis to ascertain for targets common to the 4 different seeds, conserved between mouse and human and relevant to ECs, identified PTEN as an important candidate to explain the antiangiogenic phenotype observed in vitro on inhibition of the cluster in ECs and in vivo after deletion of the miR-17–92 cluster. Indeed, PTEN inhibits proliferation, vascular sprouting, and endothelial tube formation induced by VEGF.30 The Tie2CrePtenflox/+ mice, with allelic reduction of PTEN, display enhanced tumorigenesis because of an increase in angiogenesis driven by vascular growth factors.31 Our experimental approach to determine if targeting of PTEN is mechanistically involved in promoting the angiogenic phenotype suggests that, in addition to PTEN, the various members of the miR-17–92 cluster cooperatively regulate, either directly or indirectly, a large number of genes affecting EC functions. Their effects are likely to be temporal and context dependent.42 Previous studies on individual members of the miR-17–92 cluster and their effect on angiogenic properties of ECs have characterized integrin α5 or Janus kinase as relevant targets for their reported antiangiogenic function.13,15 In our study, we report a proangiogenic action for the entire miR-17–92 cluster when studied as a single targeting unit. These discrepancies on the role of the miR-17–92 cluster on the regulation of EC functions are not contradictory, if one takes into consideration their context, their expression level, and their individual-dependent actions.42 Altogether, our data and previous studies suggest coexistence of functional cooperation and specialization among members of this cluster that can account for the complex biological functions of this important miRNA cluster in EC biology.
Acknowledgments
We thank Nicole E. Calabro for the editing work of the article; Gwendolyn Davis-Arrington for helping with human umbilical vein endothelial cell isolation and culture; Dr Huanjiao Zhou for suggestions on the fibrin bead gel assay, and Dr Anne Eichmann for critical discussions on the retinal angiogenesis model during the preparation of this article.
Sources of Funding
This work was supported by National Institutes of Health Grants R01 HL105945 (to Y. Suárez) R01 HL64793, R01 HL61371, and P01 HL1070295 (to W.C. Sessa), R01 HL107953 R01 and HL 106063 (to C. Fernández-Hernando) and R01 HL126933 (to J. Yu). W.C. Sessa and C. Fernández-Hernando were supported by the Leducq Fondation (MIRVAD network). J. Yu was supported by American Heart Association grant 14GRNT20450093. M.Y. Lee and S. Landskroner-Eiger were supported by a National Institutes of Health Training Program F32HL119147-01 and F32HL107078-02, respectively. E. Araldi was supported by a Howard Hughes Medical Institute International Student Research Fellowship.
Disclosures
None.
Supplementary Material
Nonstandard Abbreviations and Acronyms
- miRNA
- microRNA
- EC
- endothelial cell
- VEGF
- vascular endothelial growth factor
- HUVECs
- human umbilical vein endothelial cells
- PTEN
- phosphatase and tensin homolog
In September 2015, the average time from submission to first decision for all original research papers submitted to Circulation Research was 12.75 days.
This manuscript was sent to Ingrid Fleming, Consulting Editor, for review by expert referees, editorial decision, and final disposition.
The online-only Data Supplement is available with this article at http://circres.ahajournals.org/lookup/suppl/doi:10.1161/CIRCRESAHA.115.307408/-/DC1.
Novelty and Significance
What Is Known?
Vascular endothelial growth factor (VEGF) controls angiogenesis mainly by targeting the vascular endothelium.
The microRNA-17-92 (miR-17–92) cluster consists of 7 highly conserved miRNAs, previously shown to regulate cell proliferation and tumorigenesis.
Individual miRNAs of the miR-17–92 cluster can exhibit antiangiogenic activity.
What New Information Does This Article Contribute?
VEGF stimulates the expression of the miR-17–92 cluster in endothelial cells (ECs) by activating the Erk/Elk1 pathway.
In vitro loss-of-function studies (miR-17–92 cluster inhibition in human ECs) show that miR-17–92 cluster is required for endothelial proliferation and angiogenic sprouting.
Endothelial postnatal genetic inactivation of miR-17–92 reduces physiological retinal angiogenesis during development, and diminishes VEGF-induced ear angiogenesis and tumor angiogenesis.
On angiogenic VEGF stimulation, the miR-17–92 cluster targets PTEN to promote endothelial proliferation.
The angiogenic process involves a switch from normal quiescent vasculature to an activated state, by which ECs acquire a proliferative, migratory, and morphogenic phenotype. The miR-17–92 cluster has been linked to tumorigenesis and angiogenesis, but its role in VEGF-induced EC functions is unclear, and its regulation by this key angiogenic factor remains unknown. In this report, we elucidate the mechanism by which VEGF stimulates the expression of the miR-17–92 cluster in ECs. Furthermore, we provide evidence that this stimulation is important for promoting angiogenesis. We found that the endothelial miR-17–92 cluster participates in the regulation of both developmental angiogenesis and angiogenesis during adulthood. These data and previous studies suggest functional cooperation among the members of this cluster that can account for the complex biological functions of miR-17–92 in regulating angiogenesis.
References
- 1.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–936. doi: 10.1038/nature04478. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 2.Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7:803–815. doi: 10.1038/nri2171. doi: 10.1038/nri2171. [DOI] [PubMed] [Google Scholar]
- 3.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suárez Y, Sessa WC. MicroRNAs as novel regulators of angiogenesis. Circ Res. 2009;104:442–454. doi: 10.1161/CIRCRESAHA.108.191270. doi: 10.1161/CIRCRESAHA.108.191270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brock M, Trenkmann M, Gay RE, Michel BA, Gay S, Fischler M, Ulrich S, Speich R, Huber LC. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ Res. 2009;104:1184–1191. doi: 10.1161/CIRCRESAHA.109.197491. doi: 10.1161/CIRCRESAHA.109.197491. [DOI] [PubMed] [Google Scholar]
- 6.Suárez Y, Wang C, Manes TD, Pober JS. Cutting edge: TNF-induced microRNAs regulate TNF-induced expression of E-selectin and intercellular adhesion molecule-1 on human endothelial cells: feedback control of inflammation. J Immunol. 2010;184:21–25. doi: 10.4049/jimmunol.0902369. doi: 10.4049/jimmunol.0902369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chamorro-Jorganes A, Araldi E, Penalva LO, Sandhu D, Fernández-Hernando C, Suárez Y. MicroRNA-16 and microRNA-424 regulate cell-autonomous angiogenic functions in endothelial cells via targeting vascular endothelial growth factor receptor-2 and fibroblast growth factor receptor-1. Arterioscler Thromb Vasc Biol. 2011;31:2595–2606. doi: 10.1161/ATVBAHA.111.236521. doi: 10.1161/ATVBAHA.111.236521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chamorro-Jorganes A, Araldi E, Rotllan N, Cirera-Salinas D, Suárez Y. Autoregulation of glypican-1 by intronic microRNA-149 fine tunes the angiogenic response to FGF2 in human endothelial cells. J Cell Sci. 2014;127:1169–1178. doi: 10.1242/jcs.130518. doi: 10.1242/jcs.130518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suárez Y, Fernández-Hernando C, Yu J, Gerber SA, Harrison KD, Pober JS, Iruela-Arispe ML, Merkenschlager M, Sessa WC. Dicer-dependent endothelial microRNAs are necessary for postnatal angiogenesis. Proc Natl Acad Sci U S A. 2008;105:14082–14087. doi: 10.1073/pnas.0804597105. doi: 10.1073/pnas.0804597105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anand S, Majeti BK, Acevedo LM, Murphy EA, Mukthavaram R, Scheppke L, Huang M, Shields DJ, Lindquist JN, Lapinski PE, King PD, Weis SM, Cheresh DA. MicroRNA-132-mediated loss of p120RasGAP activates the endothelium to facilitate pathological angiogenesis. Nat Med. 2010;16:909–914. doi: 10.1038/nm.2186. doi: 10.1038/nm.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mendell JT. miRiad roles for the miR-17-92 cluster in development and disease. Cell. 2008;133:217–222. doi: 10.1016/j.cell.2008.04.001. doi: 10.1016/j.cell.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mogilyansky E, Rigoutsos I. The miR-17/92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013;20:1603–1614. doi: 10.1038/cdd.2013.125. doi: 10.1038/cdd.2013.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonauer A, Carmona G, Iwasaki M, et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009;324:1710–1713. doi: 10.1126/science.1174381. doi: 10.1126/science.1174381. [DOI] [PubMed] [Google Scholar]
- 14.Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT, Thomas-Tikhonenko A. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet. 2006;38:1060–1065. doi: 10.1038/ng1855. doi: 10.1038/ng1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doebele C, Bonauer A, Fischer A, Scholz A, Reiss Y, Urbich C, Hofmann WK, Zeiher AM, Dimmeler S. Members of the microRNA-17-92 cluster exhibit a cell-intrinsic antiangiogenic function in endothelial cells. Blood. 2010;115:4944–4950. doi: 10.1182/blood-2010-01-264812. doi: 10.1182/blood-2010-01-264812. [DOI] [PubMed] [Google Scholar]
- 16.Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, Newman J, Bronson RT, Crowley D, Stone JR, Jaenisch R, Sharp PA, Jacks T. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008;132:875–886. doi: 10.1016/j.cell.2008.02.019. doi: 10.1016/j.cell.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pitulescu ME, Schmidt I, Benedito R, Adams RH. Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nat Protoc. 2010;5:1518–1534. doi: 10.1038/nprot.2010.113. doi: 10.1038/nprot.2010.113. [DOI] [PubMed] [Google Scholar]
- 18.Boros J, Donaldson IJ, O’Donnell A, Odrowaz ZA, Zeef L, Lupien M, Meyer CA, Liu XS, Brown M, Sharrocks AD. Elucidation of the ELK1 target gene network reveals a role in the coordinate regulation of core components of the gene regulation machinery. Genome Res. 2009;19:1963–1973. doi: 10.1101/gr.093047.109. doi: 10.1101/gr.093047.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woods K, Thomson JM, Hammond SM. Direct regulation of an oncogenic micro-RNA cluster by E2F transcription factors. J Biol Chem. 2007;282:2130–2134. doi: 10.1074/jbc.C600252200. doi: 10.1074/jbc.C600252200. [DOI] [PubMed] [Google Scholar]
- 20.Sharrocks AD. The ETS-domain transcription factor family. Nat Rev Mol Cell Biol. 2001;2:827–837. doi: 10.1038/35099076. doi: 10.1038/35099076. [DOI] [PubMed] [Google Scholar]
- 21.Presta M, Dell’Era P, Mitola S, Moroni E, Ronca R, Rusnati M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005;16:159–178. doi: 10.1016/j.cytogfr.2005.01.004. doi: 10.1016/j.cytogfr.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 22.Welch-Reardon KM, Ehsan SM, Wang K, Wu N, Newman AC, Romero-Lopez M, Fong AH, George SC, Edwards RA, Hughes CC. Angiogenic sprouting is regulated by endothelial cell expression of Slug. J Cell Sci. 2014;127:2017–2028. doi: 10.1242/jcs.143420. doi: 10.1242/jcs.143420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Otsuka M, Zheng M, Hayashi M, Lee JD, Yoshino O, Lin S, Han J. Impaired microRNA processing causes corpus luteum insufficiency and infertility in mice. J Clin Invest. 2008;118:1944–1954. doi: 10.1172/JCI33680. doi: 10.1172/JCI33680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gariano RF, Gardner TW. Retinal angiogenesis in development and disease. Nature. 2005;438:960–966. doi: 10.1038/nature04482. doi: 10.1038/nature04482. [DOI] [PubMed] [Google Scholar]
- 25.Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, Jeltsch M, Mitchell C, Alitalo K, Shima D, Betsholtz C. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol. 2003;161:1163–1177. doi: 10.1083/jcb.200302047. doi: 10.1083/jcb.200302047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu Y, Thomson JM, Wong HY, Hammond SM, Hogan BL. Transgenic over-expression of the microRNA miR-17-92 cluster promotes proliferation and inhibits differentiation of lung epithelial progenitor cells. Dev Biol. 2007;310:442–453. doi: 10.1016/j.ydbio.2007.08.007. doi: 10.1016/j.ydbio.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomson JM, Parker J, Perou CM, Hammond SM. A custom microarray platform for analysis of microRNA gene expression. Nat Methods. 2004;1:47–53. doi: 10.1038/nmeth704. doi: 10.1038/nmeth704. [DOI] [PubMed] [Google Scholar]
- 28.Franco CA, Jones ML, Bernabeu MO, Geudens I, Mathivet T, Rosa A, Lopes FM, Lima AP, Ragab A, Collins RT, Phng LK, Coveney PV, Gerhardt H. Dynamic endothelial cell rearrangements drive developmental vessel regression. PLoS Biol. 2015;13:e1002125. doi: 10.1371/journal.pbio.1002125. doi: 10.1371/journal.pbio.1002125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Phng LK, Potente M, Leslie JD, Babbage J, Nyqvist D, Lobov I, Ondr JK, Rao S, Lang RA, Thurston G, Gerhardt H. Nrarp coordinates endothelial Notch and Wnt signaling to control vessel density in angiogenesis. Dev Cell. 2009;16:70–82. doi: 10.1016/j.devcel.2008.12.009. doi: 10.1016/j.devcel.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang J, Kontos CD. PTEN modulates vascular endothelial growth factor-mediated signaling and angiogenic effects. J Biol Chem. 2002;277:10760–10766. doi: 10.1074/jbc.M110219200. doi: 10.1074/jbc.M110219200. [DOI] [PubMed] [Google Scholar]
- 31.Hamada K, Sasaki T, Koni PA, et al. The PTEN/PI3K pathway governs normal vascular development and tumor angiogenesis. Genes Dev. 2005;19:2054–2065. doi: 10.1101/gad.1308805. doi: 10.1101/gad.1308805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cloonan N. Re-thinking miRNA-mRNA interactions: intertwining issues confound target discovery. Bioessays. 2015;37:379–388. doi: 10.1002/bies.201400191. doi: 10.1002/bies.201400191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang B, Day DS, Ho JW, Song L, Cao J, Christodoulou D, Seidman JG, Crawford GE, Park PJ, Pu WT. A dynamic H3K27ac signature identifies VEGFA-stimulated endothelial enhancers and requires EP300 activity. Genome Res. 2013;23:917–927. doi: 10.1101/gr.149674.112. doi: 10.1101/gr.149674.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kayali S, Giraud G, Morlé F, Guyot B. Spi-1, Fli-1 and Fli-3 (miR-17-92) oncogenes contribute to a single oncogenic network controlling cell proliferation in friend erythroleukemia. PLoS One. 2012;7:e46799. doi: 10.1371/journal.pone.0046799. doi: 10.1371/journal.pone.0046799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 36.Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Waters CM, Penn LZ, Hancock DC. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- 37.Kaluza D, Kroll J, Gesierich S, Manavski Y, Boeckel JN, Doebele C, Zelent A, Rössig L, Zeiher AM, Augustin HG, Urbich C, Dimmeler S. Histone deacetylase 9 promotes angiogenesis by targeting the antiangiogenic microRNA-17-92 cluster in endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33:533–543. doi: 10.1161/ATVBAHA.112.300415. doi: 10.1161/ATVBAHA.112.300415. [DOI] [PubMed] [Google Scholar]
- 38.Aird WC, Edelberg JM, Weiler-Guettler H, Simmons WW, Smith TW, Rosenberg RD. Vascular bed-specific expression of an endothelial cell gene is programmed by the tissue microenvironment. J Cell Biol. 1997;138:1117–1124. doi: 10.1083/jcb.138.5.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moyon D, Pardanaud L, Yuan L, Bréant C, Eichmann A. Plasticity of endothelial cells during arterial-venous differentiation in the avian embryo. Development. 2001;128:3359–3370. doi: 10.1242/dev.128.17.3359. [DOI] [PubMed] [Google Scholar]
- 40.Torres-Vázquez J, Kamei M, Weinstein BM. Molecular distinction between arteries and veins. Cell Tissue Res. 2003;314:43–59. doi: 10.1007/s00441-003-0771-8. doi: 10.1007/s00441-003-0771-8. [DOI] [PubMed] [Google Scholar]
- 41.Nunes DN, Dias-Neto E, Cardó-Vila M, et al. Synchronous down-modulation of miR-17 family members is an early causative event in the retinal angiogenic switch. Proc Natl Acad Sci U S A. 2015;112:3770–3775. doi: 10.1073/pnas.1500008112. doi: 10.1073/pnas.1500008112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Han YC, Vidigal JA, Mu P, Yao E, Singh I, González AJ, Concepcion CP, Bonetti C, Ogrodowski P, Carver B, Selleri L, Betel D, Leslie C, Ventura A. An allelic series of miR-17 ~ 92-mutant mice uncovers functional specialization and cooperation among members of a microRNA polycistron. Nat Genet. 2015;47:766–775. doi: 10.1038/ng.3321. doi: 10.1038/ng.3321. [DOI] [PMC free article] [PubMed] [Google Scholar]