

Abstract
Objective
Myotonic dystrophy type 1 (DM1) is caused by the expansion of a CTG repeat in the 3′ untranslated region of DMPK. The transcripts containing an expanded CUG repeat (CUG exp) result in a toxic gain‐of‐function by forming ribonuclear foci that sequester the alternative splicing factor muscleblind‐like 1 (MBNL1). Although several small molecules reportedly ameliorate RNA toxicity, none are ready for clinical use because of the lack of safety data. Here, we undertook a drug‐repositioning screen to identify a safe and effective small molecule for upcoming clinical trials of DM1.
Methods
We examined the potency of small molecules in inhibiting the interaction between CUG exp and MBNL1 by in vitro sequestration and fluorescent titration assays. We studied the effect of lead compounds in DM1 model cells by evaluating foci reduction and splicing rescue. We also tested their effects on missplicing and myotonia in DM1 model mice.
Results
Of the 20 FDA‐approved small molecules tested, erythromycin showed the highest affinity to CUG exp and a capacity to inhibit its binding to MBNL1. Erythromycin decreased foci formation and rescued missplicing in DM1 cell models. Both systemic and oral administration of erythromycin in the DM1 model mice showed splicing reversal and improvement of myotonia with no toxicity. Long‐term oral administration of erythromycin at the dose used in humans also improved the splicing abnormality in the DM1 model mice.
Interpretation
Oral erythromycin treatment, which has been widely used in humans with excellent tolerability, may be a promising therapy for DM1.
Introduction
Myotonic dystrophy type 1 (DM1) is the most common type of muscular dystrophy in adults. This systemic disease presents with multiple symptoms, including myotonia, progressive muscle weakness, insulin resistance, cardiac conduction defects, and cognitive dysfunction.1 DM1 is caused by the expansion of a CTG repeat in a 3′ untranslated region, giving rise to toxic RNAs containing expanded CUG repeats.2 These mutant RNAs are retained in the nucleus, forming ribonuclear inclusions that sequester muscleblind‐like (MBNL) family‐member proteins.3 MBNL proteins are thought to play roles in the regulation of alternative splicing, mRNA localization, micro RNA processing, and alternative polyadenylation.4, 5, 6, 7 Accordingly, MBNL1 sequestration in DM1 leads to misregulation of alternative splicing of muscle‐specific chloride channel (CLCN1) and insulin receptor (INSR) RNAs, resulting in myotonia and glucose intolerance, respectively.8
Several therapeutic approaches have been recently developed to ameliorate RNA toxicity in DM1 models. One approach involves the use of antisense oligonucleotides to directly degrade toxic RNAs or inhibit MBNL1 sequestration.9, 10, 11, 12, 13, 14 The other approach is to modulate toxic RNAs with small molecules. Pentamidine and its derivatives reportedly decrease the production of toxic RNAs in DM1 mice.15, 16 Several compounds, including aminoglycoside derivatives, are believed to block the binding between toxic RNAs and MBNL1.17, 18, 19, 20 These small molecules neutralize toxic RNA and ameliorate splicing abnormalities to some extent. However, most of these compounds are not pharmaceuticals, and the safety of these compounds in humans is still unknown. Pentamidine is the only exception, being currently used to treat Pneumocystis carinii pneumonia. However, the dose of pentamidine required for effective splicing rescue in DM1 mice is much higher than that used for humans, and such high‐dose treatment showed certain toxicity in mice.15 Thus, in spite of the present high demand for DM1 treatment, these small molecules remain far from clinical use. In addition, the time required to develop a new drug de novo is estimated to be >10 years because of regulatory requirements regarding safety, efficacy, and quality.21 A strategy to find new uses for existing drugs, known as “drug repositioning,” is now being used to bring drugs to more patients without delay. In this study, we conducted a drug‐repositioning screen to identify potential therapeutic agents for treating DM1, and found that not only systemic delivery but also oral administration of erythromycin, a widely used antibiotic in humans, reduced RNA toxicity, and improved splicing abnormalities in DM1 model mice.
Methods
MBNL sequestration assays
To evaluate the effect of tested small compounds on the inhibition of CUGexp/MBNL1 complex formation, we used modified DiscoveRx PathHunter ProLabel Enzyme Fragment Complementation Assay as we described previously.22
Fluorescence titration assay
Recording of emission spectra was carried out for fluorescently labeled (CUG)10 RNA incubated with different concentrations of antibiotics. Five‐nanomolar RNA (GCG‐(CUG)10‐CGC) labeled at the 5′‐end with Cy‐3 (Future Synthesis, Poznan, Poland) was incubated at 37°C for 5 min with erythromycin lactobionate, streptomycin, and paramomycin of indicated concentrations (ranging from 0 to 100 μmol/L). The binding reaction was performed in a volume of 2 mL in 1× binding buffer (250‐mmol/L NaCl, 15‐mmol/L KCl, 50‐mmol/L Tris–HCl, 1‐mmol/L MgCl2, pH 8.0). The emission spectra of RNA and RNA/antibiotic complexes were recorded using a spectrophotometer (UV1602; Shimadzu, Kyoto, Japan) at 520‐nm excitation wavelength. Raw data were corrected for dilution‐dependent changes, and Em560 was plotted against antibiotic concentrations and fit to the one site‐specific binding equation y = (b max × x)/(K d + x).
Cell model
Fibroblasts derived from DM1 patients (cell line GM03989 expressing a DMPK transcript with ~2000 CUG repeats) and control fibroblasts derived from non‐DM1 patients (cell line GM07492) were purchased from the Coriell Cell Repositories (Camden, NJ). A conditional cell model for analysis of MBNL1 splicing regulatory activity has been established as reported previously.23 Briefly, C2C12 mouse muscle cells were co‐transfected with pLC16 containing 800 CTG repeats24 and plasmid PhiC31o encoding PhiC31 integrase (Addgene, Cambridge, MA). Transfection was performed, using Nucleofector technology (Lonza, Basel, Switzerland) according to the manufacturer's program B‐32. Stably transfected clones were selected with puromycin (1.25 μg/mL). Transcription across the expanded repeat was activated by Cre recombinase‐mediated excision of a transcription terminator cassette. C2C12 cells with recombination were selected, using hygromycin B (300 μg/mL). RNA was harvested after 2 days of incubation with erythromycin lactobionate.
Treatment in mice
Mouse handling and experimental procedures were performed in accordance with the Osaka University guidelines for the welfare of animals, and were approved by the institutional review board. Homozygous HSA LR transgenic mice of line 20b (FVB inbred background) have been described previously.25 Gender‐matched and age‐matched (<3 months old) mice were treated with phosphate‐buffered saline (PBS) or erythromycin lactobionate or ethylsuccinate at the indicated dose and period by daily intraperitoneal injection or oral administration. After the treatments, mice were sacrificed, and the rectus femoris (quadriceps) muscle was obtained for splicing analysis. At least six mice were used in each experimental group. Electromyography was performed under general anesthesia as described previously.16 Briefly, at least 10 needle insertions were performed in the vastus muscle, and myotonic discharges were graded on a four‐point scale: 0, no myotonia; 1, occasional myotonic discharge in ≤50% of needle insertions; 2, myotonic discharge in >50% of insertions; and 3, myotonic discharge with nearly all insertions.
RNA extraction and splicing analysis
Total RNA extraction from model cells and mouse muscles, cDNA synthesis, and polymerase chain reaction (PCR) amplification were performed as described previously.16, 26 The PCR products were separated by agarose gel electrophoresis, and the gel was stained with GelRed (Biotium, Hayward, CA). The gel was imaged using a Typhoon laser fluorimager (GE Healthcare, Pittsburgh, PA) and the products quantified using ImageQuant (GE Healthcare). Splicing analyses for DM1 and non‐DM1 fibroblasts treated with erythromycin were performed as described previously.14 A two‐tailed t‐test was used to determine the statistical significance of differences between two groups.
mRNA quantification
Quantitative reverse transcriptase‐PCR was performed, using TaqMan Gene Expression assays on an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems, Carlsbad, CA). The level of endogenous mouse Dmpk, and transgene‐derived mRNA was normalized to 18S rRNA or Gtf2b as described previously.11
Fluorescence in situ hybridization
Fluorescence in situ hybridization (FISH) for C2C12 cells and DM1 fibroblasts was performed as previously described.11, 14 Fluorescence images were obtained using an Olympus FV1000D confocal laser scanning microscope (Olympus, Tokyo, Japan). The number of foci was counted from at least 200 nuclei in two independent experiments.
Results
Drug‐repositioning screen for DM1
Previous reports showed that small molecules can efficiently interrupt toxic RNA–MBNL interaction.17, 18, 19 Many of these compounds are antibiotics‐like molecules with an ability to bind rRNA. Therefore, we screened 20 FDA‐approved antibiotics that repress translation by binding RNA and are well characterized with respect to tolerability, safety, and toxicity for human use. To find potential inhibitors of interactions between toxic RNA and MBNLs, we first performed in vitro sequestration assays using surface‐attached (CUG)100 RNA and labeled recombinant MBNL1 at three concentrations of each compound (Fig. S1). Of these compounds, neomycin and erythromycin showed significant inhibition of protein aggregation (up to 50%) at 10 and 50 μmol/L (Fig. S1). To confirm the initial screening results, we performed electrophoretic mobility shift assays (EMSA) with a higher range of concentrations of neomycin and erythromycin. A maximum of 50–60% reduction in (CUG)100 RNA molecules bound to MBNL1 was observed, with K i being ~2–10 μmol/L (Fig. 1A). To determine whether an inhibition observed in two assays was caused by direct interaction between the antibiotics and RNA, we carried out the fluorescence titration assay for internal fluorescein‐labeled (CUG)100 for selected compounds. Neomycin and erythromycin showed K d < 1 μmol/L (streptomycin as a negative control with much higher K d; data not shown). We then calculated K d values using shorter 5′‐Cy3‐labeled (CUG)10 and found that erythromycin lactobionate showed the strongest fluorescence change with K d = 0.78 μmol/L (Fig. 1B and C).
Figure 1.
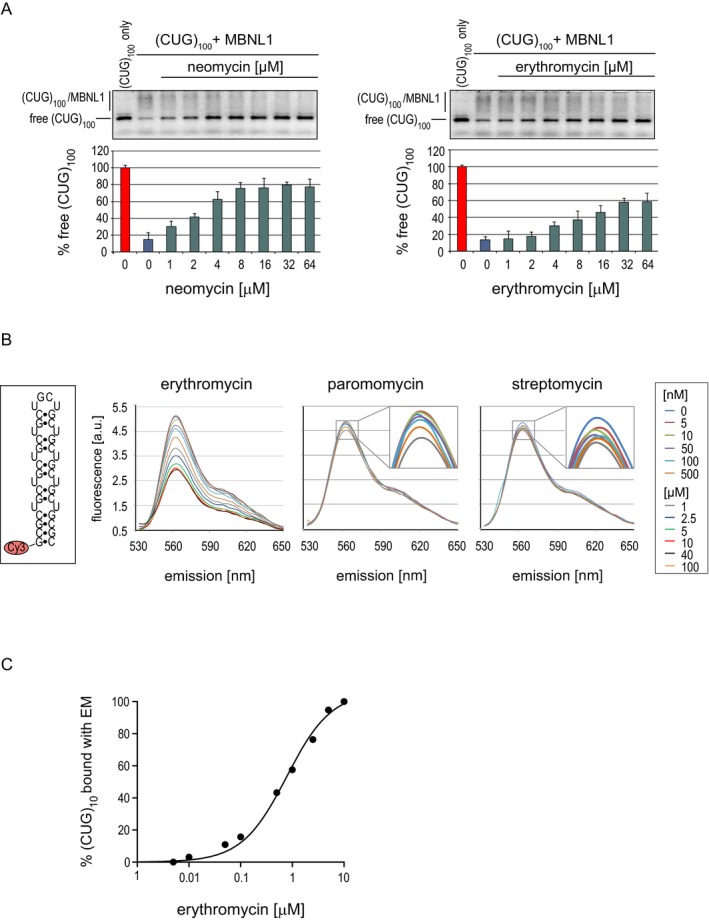
Erythromycin and neomycin inhibit CUG exp/MBNL1 interaction in vitro. (A) EMSA on a native agarose gel showing the inhibitory property of indicated concentrations of neomycin and erythromycin lactobionate on MBNL1 interaction with fluorescently labeled (CUG)100. Complexes formed by (CUG)100 and MBNL1 are indicated. Unbound RNA is marked as free (CUG)100. (B) Boxed: 5′‐Cy3‐labeled 5′‐CCG‐(CUG)10‐CGG‐3′ RNA structure. Graphs show an emission spectra of Cy3‐(CUG)10 in the presence of increasing concentrations of erythromycin lactobionate, paromomycin, and streptomycin at 520‐nm excitation wavelength. (C) The binding curve of erythromycin and (CUG)10 based on changes of emission spectra. MBNL, muscleblind‐like; EMSA, electrophoretic mobility shift assays.
Validation in DM1 model cells
From our initial screen, we found that neomycin and erythromycin lactobionate effectively interrupted MBNL1 sequestration. However, neomycin is less used in humans because of its nephrotoxicity and ototoxicity27 and did not improve misregulated splicing in a previous study.15 Indeed, in our study, subcutaneous injection of neomycin into the DM1 model mice did not significantly improve splicing, even with a dosage equivalent to LD50 (100 mg/kg per day; data not shown). Therefore, we focused on erythromycin and investigated its effects on DM1 model cells. First, we used C2C12 murine myogenic cells that were stably transfected with a construct for conditional expression of (CUG)800 RNA. After induction of toxic RNA expression, these cells displayed ribonuclear foci (62.8% positive for nuclear foci) and showed a splicing abnormality in Atp2a1 (36.8% inclusion of exon 22) similar to that observed in DM1 (Fig. 2A and B). After 2 days of treatment with 25‐ or 50‐μmol/L erythromycin, the frequency of cells showing nuclear foci decreased to 14.3% or 5.9%, respectively (Fig. 2A). The missplicing of Atp2a1 was mostly rescued by erythromycin in a dose‐dependent manner, returning exon 22 inclusion up to 82.8% (Fig. 2B). Several small molecules such as pentamidine have been reported to decrease toxic RNA by suppressing transcription.16 Although our in vitro data suggest erythromycin neutralizes toxic RNA by directly inhibiting MBNL1 sequestration, erythromycin may have the potential to suppress transcription of repeat‐containing transcripts. To exclude the possibility that erythromycin affects transcription of toxic RNA, we assessed the expression of a transgene containing 800 CUG repeats by qPCR. However, the expression level of toxic RNA was not affected by erythromycin (Fig. 2C). We then investigated whether treatment with erythromycin can change molecular phenotypes of fibroblasts derived from DM1 patients. Two days of treatment with erythromycin significantly decreased the number of CUGexp foci (Fig. 3A). The focus size also decreased significantly, with 50% of the examined foci ≤0.08 and ≤0.05 μm3 in cells treated with 100‐ and 500‐μmol/L erythromycin, respectively, compared to untreated cells, which were half their size at most. We also observed significant correction of splicing of several exons regulated by MBNLs (MBNL1 ex5, MBNL1 ex7, MBNL2 ex5, and NCOR2 ex45a) in cells treated with 50–100‐μmol/L erythromycin (Fig. 3B). As was expected, the antibiotic did not perturb the splicing of MBNL‐independent transcripts (APLP2 ex14 and KIF13A ex26) (Fig. 3C).
Figure 2.
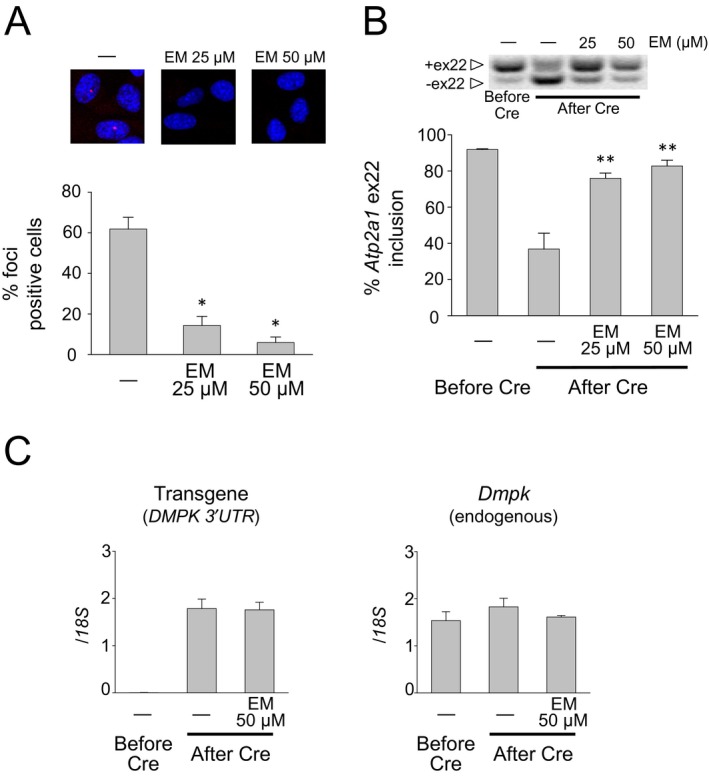
Erythromycin decreases the number of RNA foci and rescues splicing defects in a C2C12 cell model. (A) Top: FISH showing foci of CUG exp RNA (red) in nuclei (blue) of stably transfected C2C12 cells. Bottom: histogram showing the percentage of cells with nuclear foci of CUG exp RNA. The number of cells counted was 485 for no treatment, 359 for 25‐μmol/L erythromycin treatment, and 313 for 50‐μmol/L erythromycin treatment. Mean ± SD, n = 3 or more. *P < 0.001. (B) Top: representative gel images of RT‐PCR assay for inclusion of Atp2a1 exon 22. Bottom: Bar graph representation of partial rescue of Atp2a1 exon 22 splicing. Exon 22 inclusion was decreased by Cre‐mediated activation. Treatment of the (CUG)800 expressing cells with 25‐ or 50‐μmol/L erythromycin caused partial rescue of exon 22 splicing. **P < 0.01. Error bars indicate SDM. (C) RNA transcript levels of transgene and Dmpk in the C2C12 cells treated with or without erythromycin. Quantitative RT–PCR was performed using TaqMan Gene Expression assays. Erythromycin did not affect transcription of CUG‐repeat‐containing sequences. Error bars indicate SDM. FISH, fluorescence in situ hybridization; EM, erythromycin; RT‐PCR, reverse transcription polymerase chain reaction.
Figure 3.
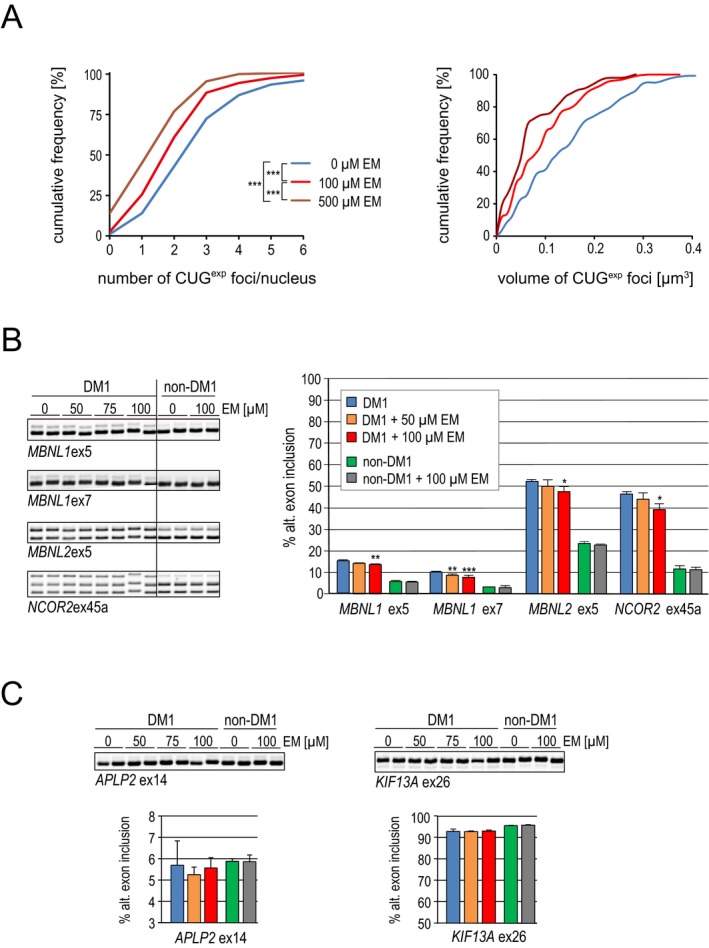
Erythromycin diminishes CUG exp foci size and corrects splicing defects in DM1 cells. (A) Left: Graph showing the distribution of nuclei containing different number of CUG exp foci. The number of cells counted was 200 for both untreated and erythromycin (100‐ or 500‐μmol/L) treated fibroblasts. Right: The distribution of CUG exp foci volume (μm3). The number of foci measured for volume was 120 for both untreated and erythromycin treated fibroblasts. P‐value was determined using the Mann–Whitney U test; ***P < 0.001. (B) Analysis of alternative splicing of MBNL1 ex 5, MBNL1 ex7, MBNL2 ex5, and NCOR2 ex45 by RT‐PCR in DM1 and non‐DM1 fibroblasts after incubation for 2 days with 50‐ or 100‐μmol/L erythromycin. *P < 0.05. **P < 0.01. Error bars indicate SDM. (C) As in (B), but shows the analysis of alternative splicing of two MBNL‐independent exons, APLP2 ex14 and KIF13A ex26. DM1, myotonic dystrophy type 1; RT‐PCR, reverse transcription polymerase chain reaction; MBNL, muscleblind‐like.
In vivo treatment with erythromycin
Next, we tested the effect of erythromycin on RNA toxicity in the HSA LR transgenic DM1 mouse model. These mice express 220 CUG repeats in skeletal muscle and exhibit several DM1‐like characteristics, including myotonia and misregulated splicing.25, 28 To test activity and acute toxicity, we administrated erythromycin (150 mg/kg per day) to the HSA LR mice by daily intraperitoneal (i.p.) injection for 8 days. Although the dosage of erythromycin was 2–3 times as much as used for humans, no mice showed signs of toxicity. We assessed splicing of Clcn1 and Atp2a1, both of which are regulated by MBNL1, in skeletal muscle. Missplicing of Clcn1 has been suggested to cause myotonia.28 The wild‐type adult mice had a Clcn1 exon 7a exclusion rate of 94%, whereas the HSA LR mice showed an exclusion rate of 57% (Fig. 4A). Treatment of the HSA LR mice with i.p. erythromycin significantly rescued the exclusion rate to 83% (P = 0.000005). The splicing misregulation of Atp2a1 is also a robust biomarker for DM1.26 In the wild‐type mice, the inclusion rate for exon 22 is 100%, whereas the inclusion rate in the HSA LR mice was 31% (Fig. 4A). After erythromycin treatment, the inclusion rate improved significantly to 76% (P = 0.0000001). In addition to Clcn1 and Atp2a1, misregulation of other splicing events related to muscle wasting (Bin1 and Cacna1s),29, 30 early transition (Camk2b and Ryr1),26 and regulation by MBNL1 (Nfix and Ldb3) was rescued by systemic erythromycin treatment (Fig. S2). Furthermore, splicing misregulation of both Clcn1 and Atp2a1 was significantly improved by the dose usually used in humans (50 mg/kg per day for 8 days; P = 0.003 and 0.004, respectively). We also graded the severity of myotonia in mice using electromyography. In the HSA LR mice treated with saline, we observed grade 3 myotonia in the vastus muscle, indicating abundant repetitive discharges with nearly all electrode insertions. When treated with i.p. injection of erythromycin, the myotonia decreased from grade 3 to grade 2 (myotonic discharge in >50% but not full of insertions) or grade 1 (occasional myotonic discharge) (Fig. 4B).
Figure 4.
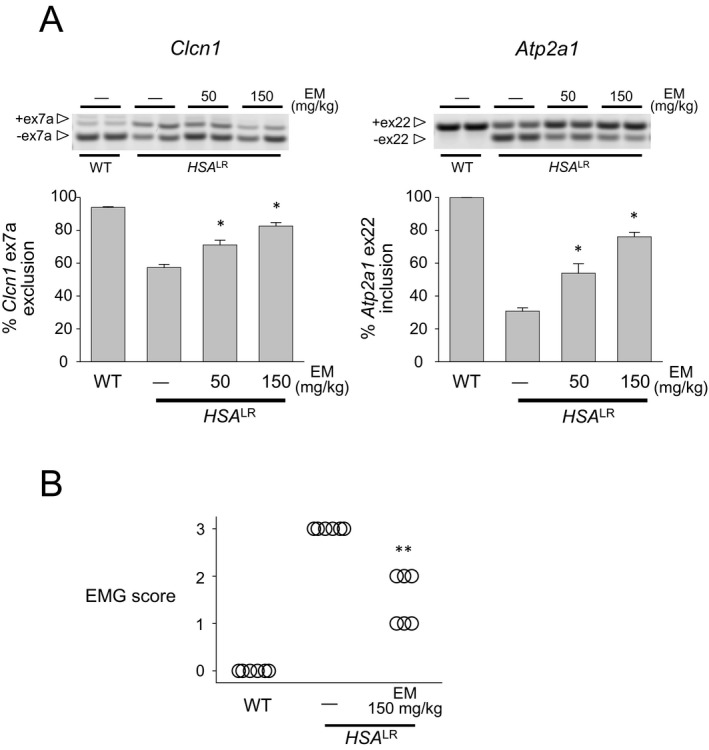
Intraperitoneal injection of erythromycin improves splicing defects in Clcn1 and Atp2a1 in a DM1 mouse model. (A) Analysis of Clcn1 (left) and Atp2a1 (right) alternative splicing by RT‐PCR in mice treated with intraperitoneal injection of erythromycin (50 or 150 mg/kg for 8 days; n = 6 for experimental, control, and wild type). *P < 0.01. Error bars indicate SEM. (B) Electromyographic myotonia analysis in HSALR mice treated with intraperitoneal erythromycin. Mice treated with saline showed myotonic discharge with nearly all electrode insertions (grade 3), whereas those treated with erythromycin had decreased myotonic discharges. **P < 0.001. DM1, myotonic dystrophy type 1; RT‐PCR, reverse transcription polymerase chain reaction.
In addition to systemic delivery, oral administration of erythromycin is widely used for human diseases.31 Continuous oral administration rather than periodic systemic administration is more convenient for patients and is feasible for the treatment of chronic progressive diseases such as DM1. Therefore, we next studied the effect of oral erythromycin administration in the HSA LR mice. Short‐term high‐dose per oral (p.o.) erythromycin treatment (300, 600, or 900 mg/kg per day for 5 days) rescued missplicing of Clcn1, Atp2a1, and other events (Bin1, Cacna1s, Camk2b, Ryr1, Nfix, and Ldb3) similar to i.p. injection (Fig. 5A and Fig. S2). The myotonia in treated mice was also decreased from grade 3 to grade 2 (Fig. 5B). To verify that erythromycin did not affect the transcription of toxic RNA, we assessed the expression of the transgene (HSA) containing expanded CUG repeats by qPCR. Similar to the C2C12 cell model, the expression level of toxic RNA in the HSA LR mice was not changed by erythromycin treatment (Fig. 5C), suggesting that erythromycin blocks binding between CUGexp and MBNL1 in vivo. Then, to determine whether erythromycin is effective at the dosage used in humans, we tested low‐dose long‐term treatment (50 or 100 mg/kg per day p.o. erythromycin for 21 days) in the HSA LR mice. The 50‐mg/kg per day treatment group showed a modest but statistically significant (P = 0.006) improvement in Clcn1 splicing relative to saline‐injected controls (Fig. 6). We also observed statistically significant rescues of splicing misregulation in Clcn1, Atp2a1 (P = 0.0002 and 0.0005, respectively), and other events (Bin1, Cacna1s, Camk2b, Ryr1, Nfix, and Ldb3) with 100‐mg/kg per day treatment (Fig. 6 and Fig. S2). Furthermore, another form of erythromycin approved for oral use in humans (erythromycin ethylsuccinate ester; 100 mg/kg per day p.o. for 21 days) also significantly improved missplicing of Clcn1 and Atp2a1 (P = 0.003 and 0.042, respectively).
Figure 5.
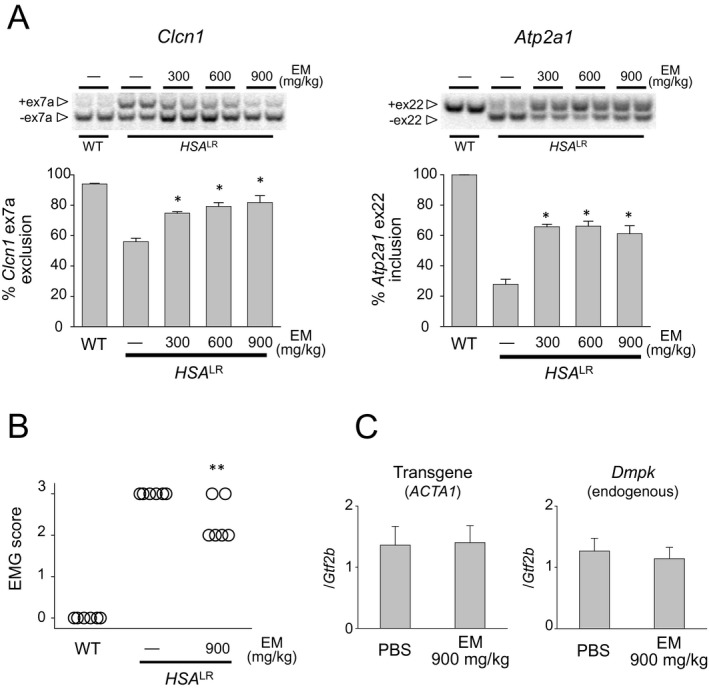
Oral administration (high‐dose, short‐term) of erythromycin improves splicing defects in Clcn1 and Atp2a1 in a DM1 mouse model. (A) Analysis of Clcn1 (left) and Atp2a1 (right) alternative splicing by RT‐PCR in mice treated with per oral erythromycin (300, 600, or 900 mg/kg for 5 days; n = 6 for experimental, control, and wild type). *P < 0.001. Error bars indicate SEM. (B) Electromyographic myotonia analysis in HSALR mice treated with per oral erythromycin. Mice treated with per oral 900 mg/kg per day erythromycin decreased myotonic discharges. **P < 0.05. (C) qRT‐PCR analysis of ACTA1 (HSA) and Dmpk levels in HSALR mice treated with saline or 900‐mg/kg erythromycin for 5 days. No significant difference was observed between the saline‐treated and erythromycin‐treated mice. Error bars indicate SEM. DM1, myotonic dystrophy type 1; RT‐PCR, reverse transcription polymerase chain reaction.
Figure 6.
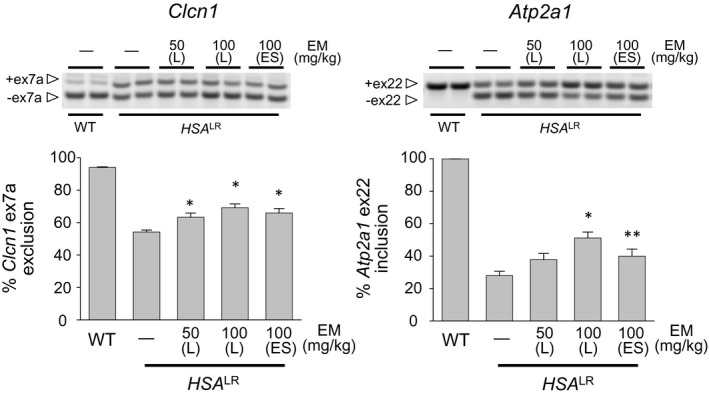
Oral administration (low‐dose, long‐term) of erythromycin improves splicing defects in Clcn1 and Atp2a1 in a DM1 mouse model. Analysis of Clcn1 (left) and Atp2a1 (right) alternative splicing by RT‐PCR in mice treated per oral with erythromycin (50 or 100 mg/kg for 21 days; n = 6 or more for experimental, control, and wild type). *P < 0.01. **P < 0.05. Error bars indicate SEM. DM1, myotonic dystrophy type 1; RT‐PCR, reverse transcription polymerase chain reaction; L; lactobionate, ES; ethylsuccinate.
Discussion
DM1 is a slowly progressive, multisystemic disorder for which no curative treatment presently exists. Previous studies identified several small molecules that specifically bind CUG repeats and neutralize the toxic RNA.17, 18, 19, 20 Some of these molecules partially rescued missplicing events in DM1 model mice. However, the toxicity and tolerability of these compounds have not been determined in humans, precluding their clinical use in the near future. Drug repositioning is the application of established drugs to new indications and represents a promising way to speed up the development of treatments by capitalizing on existing data and experience.32 The drug‐repositioning screen conducted in this study revealed that erythromycin binds CUGexp and competitively releases sequestered MBNL1, thereby inducing dispersal of RNA foci and reversing missplicing events in in vitro models of DM1. Furthermore, erythromycin administrated either orally or systemically rescued missplicing and improved myotonia in the DM1 model mice. Erythromycin has long been a widely used antibiotic. Erythromycin binds to the 50S ribosomal subunit, inhibiting bacterial protein synthesis by blocking the polypeptide exit tunnel.33 In addition to interacting with prokaryotic rRNA, our study demonstrates that erythromycin has a surprisingly high affinity for CUG repeat RNA. Erythromycin also has an anti‐inflammatory effect on the respiratory tract, and low‐dose long‐term oral erythromycin treatment has already been well established in chronic obstructive pulmonary disease (COPD) with excellent tolerability.31 Because a constant, steady‐state level of a therapeutic agent could provide continual inhibition of CUGexp‐MBNL1 interactions, long‐term oral administration of erythromycin would be a more relevant and convenient therapy for DM1 rather than periodic parenteral injections. Indeed, we observed a significant splicing correction in the DM1 model mice with administration of the dose of erythromycin approved for human use (50 or 100 mg/kg per day, p.o.). This is an encouraging result indicating future clinical trials of DM1, because the dose given to mice for long‐term per oral erythromycin ethylsuccinate treatment would equate to ~10 mg/kg per day for human adults, based on mouse‐to‐human equivalence calculations,34 which is within the recommended dose range for COPD patients.
Erythromycin is also reported to stimulate gastrointestinal motor activity via effects on motilin receptors.35 In a previous study, the erythromycin ethylsuccinate ester was tested to improve gastric emptying in DM1 patients when administered at a dose of 50–200 mg/day for 4 weeks.36 While no significant change in the gastric emptying rate was observed in the study, some gastrointestinal symptoms, including nausea, ability to eat, and diarrhea, were attenuated in DM1 patients. Although the dosage of erythromycin was relatively low and skeletal muscle symptoms were not evaluated, such improvements might have occurred through neutralization of toxic RNA. Interestingly, our in vitro study indicated that erythromycin binding to CUG repeats depends on whether it is in the salt or ester form. In fluorescence titration assays, erythromycin lactobionate showed the highest affinity to CUGexp of the erythromycin derivatives. Although per oral erythromycin ethylsuccinate ester significantly improved Clcn1 and Atp2a1 missplicing in the DM1 model mice, the effect was less than that of the lactobionate form. Because the lactobionate form is currently approved only for intravenous use, encapsulation or lipid‐coating will be required for per oral use, as previously attempted in humans.37, 38
Erythromycin has been shown to induce QT‐interval prolongation in some patients by blocking delayed‐rectifier potassium channels.39 Cardiac conduction abnormalities are critical symptoms in DM1, sometimes leading to sudden death.40 Although the major manifestation is prolongation of the PR interval and QRS duration, several studies report a prolonged QT interval in DM1.41 Therefore, careful consideration of the indication for treatment and cautious observation are required for administration of erythromycin in DM1 patients.
Our study revealed erythromycin as the first FDA‐approved small molecule that improves the splicing abnormality in DM1 at the usual dose for human use. In addition to its potential for combination therapy with antisense oligonucleotides that degrade the toxic RNA, erythromycin treatment has distinct clinical advantages, as it is effective by oral administration, inexpensive, and remarkably safe as indicated over its long history of use for the treatment of infections and pulmonary diseases.31, 42 Our results indicate promising prospects for future DM1 therapy, using erythromycin. Furthermore, the availability of detailed information regarding erythromycin safety hastens clinical trial readiness.
Author Contributions
M. N. conceived and designed the study, performed the experiments, analyzed the data, and wrote the manuscript. K. T. performed the experiments and analyzed the data. K. S. designed the study, analyzed the data, and wrote the manuscript. H. M. helped to analyze the data and write the manuscript. M. P. T. helped to write the manuscript.
Conflict of Interest
M. N. and H. M. have a patent (2015‐140599) pending relevant to this work.
Supporting information
Figure S1. Results of screening of 20 antibiotics in MBNL1 sequestration assay. Experiments were done for three concentrations of each compound and each experiment was performed in triplicate. The results are mean + SD.
Figure S2. Analysis of alternative splicing events (Bin1, Cacna1s, Camk2b, Ryr1, Nfix, and Ldb3) by RT‐PCR in mice treated with erythromycin (150 mg/kg per day i.p. for 8 days, 900 mg/kg per day p.o. for 5 days, or 100 mg/kg per day p.o. for 21 days; n = 6 for experimental, control, and wild type). *P < 0.01. Error bars indicate SEM.
Acknowledgments
The authors thank Dr. Charles Thornton for providing the HSA LR mouse model and his advice, Ms. Kimie Hayashi and Ms. Rika Manabe for technical assistance, and Dr. Andonis Karachitos for help with spectrofluorometric measurements. This work was supported by Grant‐in‐Aid for Young Scientists (Start‐up) and (A), Grant‐in‐Aid for Specially Promoted Research and Grant‐in‐Aid for Challenging Exploratory Research from Japan Society for the Promotion of Science (KAKENHI Grant Number 24890110, 25713034, 26000007 and 15K15339 to M. N.), Translational Research Network Program from Japan Agency for Medical Research and Development, AMED to M.N., the Ministry of Health, Labour and Welfare, Intramural research grant (26‐8 to M. P. T.) of the National Center of Neurology and Psychiatry, the Polish National Science Centre grant (2011/01/B/NZ1/01603 to K. S.), and the Foundation for Polish Science‐TEAM program (TEAM/2011‐7/10 to K. S.).
References
- 1. Harper PS. Myotonic dystrophy. London: W.B. Saunders Company, 2001. [Google Scholar]
- 2. Nakamori M, Thornton C. Epigenetic changes and non‐coding expanded repeats. Neurobiol Dis 2010;39:21–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Miller JW, Urbinati CR, Teng‐Umnuay P, et al. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J 2000;19:4439–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kanadia RN, Johnstone KA, Mankodi A, et al. A muscleblind knockout model for myotonic dystrophy. Science 2003;302:1978–1980. [DOI] [PubMed] [Google Scholar]
- 5. Wang ET, Cody NA, Jog S, et al. Transcriptome‐wide regulation of pre‐mRNA splicing and mRNA localization by muscleblind proteins. Cell 2012;150:710–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rau F, Freyermuth F, Fugier C, et al. Misregulation of miR‐1 processing is associated with heart defects in myotonic dystrophy. Nat Struct Mol Biol 2011;18:840–845. [DOI] [PubMed] [Google Scholar]
- 7. Batra R, Charizanis K, Manchanda M, et al. Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA‐mediated disease. Mol Cell 2014;56:311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ho TH, Charlet BN, Poulos MG, et al. Muscleblind proteins regulate alternative splicing. EMBO J 2004;23:3103–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mulders SA, van den Broek WJ, Wheeler TM, et al. Triplet‐repeat oligonucleotide‐mediated reversal of RNA toxicity in myotonic dystrophy. Proc Natl Acad Sci USA 2009;106:13915–13920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wheeler TM, Sobczak K, Lueck JD, et al. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science 2009;325:336–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nakamori M, Gourdon G, Thornton CA. Stabilization of expanded (CTG)*(CAG) repeats by antisense oligonucleotides. Mol Ther 2011;19:2222–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wheeler TM, Leger AJ, Pandey SK, et al. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 2012;488:111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sobczak K, Wheeler TM, Wang W, Thornton CA. RNA interference targeting CUG repeats in a mouse model of myotonic dystrophy. Mol Ther 2013;21:380–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wojtkowiak‐Szlachcic A, Taylor K, Stepniak‐Konieczna E, et al. Short antisense‐locked nucleic acids (all‐LNAs) correct alternative splicing abnormalities in myotonic dystrophy. Nucleic Acids Res 2015;43:3318–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Warf MB, Nakamori M, Matthys CM, et al. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc Natl Acad Sci USA 2009;106:18551–18556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Coonrod LA, Nakamori M, Wang W, et al. Reducing levels of toxic RNA with small molecules. ACS Chem Biol 2013;8:2528–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Childs‐Disney JL, Parkesh R, Nakamori M, et al. Rational design of bioactive, modularly assembled aminoglycosides targeting the RNA that causes myotonic dystrophy type 1. ACS Chem Biol 2012;7:1984–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ofori LO, Hoskins J, Nakamori M, et al. From dynamic combinatorial ‘hit’ to lead: in vitro and in vivo activity of compounds targeting the pathogenic RNAs that cause myotonic dystrophy. Nucleic Acids Res 2012;40:6380–6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Parkesh R, Childs‐Disney JL, Nakamori M, et al. Design of a bioactive small molecule that targets the myotonic dystrophy type 1 RNA via an RNA motif‐ligand database and chemical similarity searching. J Am Chem Soc 2012;134:4731–4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Siboni RB, Bodner MJ, Khalifa MM, et al. Biological efficacy and toxicity of diamidines in myotonic dystrophy type 1 models. J Med Chem 2015;58:5770–5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tobinick EL. The value of drug repositioning in the current pharmaceutical market. Drug News Perspect 2009;22:119–125. [DOI] [PubMed] [Google Scholar]
- 22. Gareiss PC, Sobczak K, McNaughton BR, et al. Dynamic combinatorial selection of molecules capable of inhibiting the (CUG) repeat RNA‐MBNL1 interaction in vitro: discovery of lead compounds targeting myotonic dystrophy (DM1). J Am Chem Soc 2008;130:16254–16261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hoskins JW, Ofori LO, Chen CZ, et al. Lomofungin and dilomofungin: inhibitors of MBNL1‐CUG RNA binding with distinct cellular effects. Nucleic Acids Res 2014;42:6591–6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nakamori M, Pearson CE, Thornton CA. Bidirectional transcription stimulates expansion and contraction of expanded (CTG)*(CAG) repeats. Hum Mol Genet 2011;20:580–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mankodi A, Logigian E, Callahan L, et al. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 2000;289:1769–1773. [DOI] [PubMed] [Google Scholar]
- 26. Nakamori M, Sobczak K, Puwanant A, et al. Splicing biomarkers of disease severity in myotonic dystrophy. Ann Neurol 2013;74:862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Masur H, Whelton PK, Whelton A. Neomycin toxicity revisited. Arch Surg 1976;111:822–825. [DOI] [PubMed] [Google Scholar]
- 28. Mankodi A, Takahashi MP, Jiang H, et al. Expanded CUG repeats trigger aberrant splicing of ClC‐1 chloride channel pre‐mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell 2002;10:35–44. [DOI] [PubMed] [Google Scholar]
- 29. Fugier C, Klein AF, Hammer C, et al. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat Med 2011;17:720–725. [DOI] [PubMed] [Google Scholar]
- 30. Tang ZZ, Yarotskyy V, Wei L, et al. Muscle weakness in myotonic dystrophy associated with misregulated splicing and altered gating of Ca(V)1.1 calcium channel. Hum Mol Genet 2012;21:1312–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ramos FL, Criner GJ. Use of long‐term macrolide therapy in chronic obstructive pulmonary disease. Curr Opin Pulm Med 2014;20:153–158. [DOI] [PubMed] [Google Scholar]
- 32. Pantziarka P, Cairns L. Recycling existing drugs for cancer therapy: delivering low cost cancer care. Ecancermedicalscience 2014;8:ed40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schlunzen F, Zarivach R, Harms J, et al. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature 2001;413:814–821. [DOI] [PubMed] [Google Scholar]
- 34. Reagan‐Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J 2008;22:659–661. [DOI] [PubMed] [Google Scholar]
- 35. Weber FH Jr, Richards RD, McCallum RW. Erythromycin: a motilin agonist and gastrointestinal prokinetic agent. Am J Gastroenterol 1993;88:485–490. [PubMed] [Google Scholar]
- 36. Ronnblom A, Andersson S, Hellstrom PM, Danielsson A. Gastric emptying in myotonic dystrophy. Eur J Clin Invest 2002;32:570–574. [DOI] [PubMed] [Google Scholar]
- 37. Patel SP, Jarowski CI. Oral absorption efficiency of acid‐labile antibiotics from lipid‐drug delivery systems. J Pharm Sci 1975;64:869–872. [DOI] [PubMed] [Google Scholar]
- 38. McDonald PJ, Mather LE, Story MJ. Studies on absorption of a newly developed enteric‐coated erythromycin base. J Clin Pharmacol 1977;17:601–606. [DOI] [PubMed] [Google Scholar]
- 39. Rubart M, Pressler ML, Pride HP, Zipes DP. Electrophysiological mechanisms in a canine model of erythromycin‐associated long QT syndrome. Circulation 1993;88:1832–1844. [DOI] [PubMed] [Google Scholar]
- 40. Groh WJ, Groh MR, Saha C, et al. Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. N Engl J Med 2008;358:2688–2697. [DOI] [PubMed] [Google Scholar]
- 41. Petri H, Vissing J, Witting N, et al. Cardiac manifestations of myotonic dystrophy type 1. Int J Cardiol 2012;160:82–88. [DOI] [PubMed] [Google Scholar]
- 42. Brittain DC. Erythromycin. Med Clin North Am 1987;71:1147–1154. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Results of screening of 20 antibiotics in MBNL1 sequestration assay. Experiments were done for three concentrations of each compound and each experiment was performed in triplicate. The results are mean + SD.
Figure S2. Analysis of alternative splicing events (Bin1, Cacna1s, Camk2b, Ryr1, Nfix, and Ldb3) by RT‐PCR in mice treated with erythromycin (150 mg/kg per day i.p. for 8 days, 900 mg/kg per day p.o. for 5 days, or 100 mg/kg per day p.o. for 21 days; n = 6 for experimental, control, and wild type). *P < 0.01. Error bars indicate SEM.