


Abstract
Detailed mechanistic understanding of L1 retrotransposition is sparse, particularly with respect to ORF1p, a coiled coil-mediated homotrimeric nucleic acid chaperone that can form tightly packed oligomers on nucleic acids. Although the coiled coil motif is highly conserved, it is uniquely susceptible to evolutionary change. Here we studied three ORF1 proteins: a modern human one (111p), its resuscitated primate ancestor (555p) and a mosaic modern protein (151p) wherein 9 of the 30 coiled coil substitutions retain their ancestral state. While 111p and 555p equally supported retrotransposition, 151p was inactive. Nonetheless, they were fully active in bulk assays of nucleic acid interactions including chaperone activity. However, single molecule assays showed that 151p trimers form stably bound oligomers on ssDNA at <1/10th the rate of the active proteins, revealing that oligomerization rate is a novel critical parameter of ORF1p activity in retrotransposition conserved for at least the last 25 Myr of primate evolution.
INTRODUCTION
The non-LTR L1 (LINE-1) retrotransposon has been replicating and evolving in mammals for the last 80–120 Myr and has generated ∼40% of the human genome (1). It replicates (retrotransposes) by copying its transcript (and those of other genes) into genomic DNA, and although seriously deleterious (2,3) with the potential for causing catastrophic effects (4-6), L1 replication and evolution persist in modern humans (7-9). L1 generates genetic diversity, defects and rearrangements, and can be activated in tumors and other somatic cells (9-17). However, our understanding of the regulation and biochemistry of L1 replication and how it persists in mammalian lineages is incomplete.
Mammalian L1 elements are 6–7 kb, contain a regulatory 5′ UTR, two protein-encoding sequences, ORF1 and ORF2, and a 3′ UTR of unknown function (18,19). ORF1p and ORF2p are essential for retrotransposition (20) and preferentially associate with their encoding transcript (cis-preference) to form a ribonucleoprotein (RNP) retrotransposition intermediate (21-25). ORF2p contains highly conserved endonuclease and reverse transcriptase domains and functions as the L1 replicase (20,26,27) wherein endonuclease-nicked genomic DNA primes the synthesis of a DNA copy of the L1 transcript, referred to as target site-primed reverse transcription, TPRT (28).
The function of ORF1p is less clear. It is the major protein component of the L1RNP and present in large molar excess over the presumed single molecule of ORF2p (23,29). Mouse and human ORF1p form stable coiled coil mediated trimers and can be roughly divided into distinct functional halves, which evolved under different selective constraints (Figure 1A and B and Supplementary Figure S1). The carboxy-terminal half (amino acids 153–338) is highly conserved and comprises two structurally distinct domains: a non-canonical RNA recognition motif (RRM) and a carboxy-terminal domain (CTD). Residues in these domains endow the protein with high affinity nucleic acid (NA) binding and chaperone activity in vitro, but only in the context of the trimer. However, the mechanistic relationship between these activities and retrotransposition is not known (18,20,25,29-36).
Figure 1.

ORF1p variants. (A) ORF1p domains: N-terminal domain (NTD), coiled coil domain; RNA recognition motif (RRM) and C-terminal domain (CTD). The inverted triangles indicate the location of the proline directed protein kinase sites at S18, S27, T203 and T 213 (38). The diagram of the trimer is a representation of the structure revealed by atomic force microscopy of the mouse ORF1p (37). The ovals corresponding to the NTD and the carboxy-terminal half of the protein are scaled to the relative masses of these regions. (B) Modern (111p), ancestral (555p) and mosaic (151p) ORF1 proteins. The numbers in the 555p cartoon indicate the number of amino acid substitutions between the corresponding regions of 555p and 111p. Thus, 151p contains nine ancestral amino acids (confined to the coiled coil domain) compared to 111p. In contrast to 151p, other mosaic constructs, 551p (ancestral sequences up to the BsmF1 site), 511p (ancestral sequences up to the BsmI site) exhibited ≥80% of the activity of 111p or 555p (results not shown). (C) The retrotransposition reporter. Upon transfection, a full-length L1 transcript is synthesized and spliced (removes the inactivating intron in the neo gene). Retrotransposition competent elements support subsequent cDNA synthesis of this transcript at a DNA target site and ultimately the insertion of an active copy of the neo gene, which when expressed from its promoter (Pr, in red) generates colonies of G418 resistant cells or foci (see ‘Materials and Methods’ section). (D) Retrotransposition activity of ORF1p variants. (E) Western blot of 75 μg of extracts of control HeLa cells or those transfected with vectors that express the indicated ORF1p fused at the carboxy-terminus with the FLAG epitope. These vectors were prepared and the transfections were carried out as described in Methods. (F) Denaturing gel electrophoresis under reducing conditions of ∼200 ng each of 111p, 151p and 555p purified from insect cells as described in ‘Materials and Methods’ section and ref. (35).
The carboxy-terminal half also mediates the formation of ORF1p polymers, which are an active conformer of the protein that can rapidly bind single-stranded-DNA (ssDNA) or RNA (35). Because 40–50 bases are occupied by a single trimer (29,35), multimers of trimers (trimer2, trimer3) assemble at high protein/NA ratios on NAs of sufficient length (e.g. 120 nt). These can be captured by protein cross-linking reagents, indicating that NA binding and protein multimerization are not mutually exclusive (35). RNA-bound mouse ORF1p clusters were also observed by atomic force microscopy under similar conditions in the absence of cross-linkers (29).
In contrast, the DNA sequence corresponding to the amino-terminal half of ORF1p is highly variable, although both an N-terminal domain (NTD) and coiled coil domain are conserved (34,35,37). The NTD is exemplified by two highly conserved proline directed phosphokinase sites (Figure 1A, arrow heads), shown to be essential for retrotransposition in the case of human ORF1p (38). Although a coiled coil motif is conserved in ORF1p from fish to mammals, its sequence can undergo rampant amino acid substitutions and even complete replacement (22,39-44). But paradoxically for such a variable sequence, ORF1p activity can be exquisitely sensitive to even a single amino acid substitution (32). Thus the persistence of L1 activity apparently requires periodic remodeling of the coiled coil to maintain ORF1p activity.
To address the biochemical consequences of coiled coil evolution we resuscitated an ORF1p that was encoded by the ancestral primate L1Pa5 family, which went extinct about 25 MYA (43,44). The emergence of this family coincided with a major episode of amino acid substitutions primarily in the coiled coil that ultimately generated the modern human L1Pa1 family ORF1p (hereafter called 111p). This protein differs from L1Pa5 ORF1p (555p) at 42 positions, 30 of which are in the coiled coil, which accounts for 101 residues of the 381 amino acid protein (43) (Figure 1B). We also constructed several mosaic proteins that contained mixtures of ancestral and modern coiled coil residues.
Although 555p and 111p were equally active in retrotransposition, one mosaic protein, 151p (9 ancestral coiled coil amino acids), was completely inactive. But the purified proteins were similar in their interactions with NAs including binding affinity and chaperone activity when tested with oligonucleotide substrates (≤120 nt) traditionally used in these assays. However, by using single molecule stretching studies with λ phage DNA (45,46) we substantially advanced our understanding of ORF1p/NA interactions. In particular, we detected and quantified distinct populations of ORF1p–NA complexes that differed in their dissociation kinetics. And thereupon we uncovered a significant difference between the proteins—the retrotransposition incompetent 151p formed stably bound oligomers on ssDNA at an order of magnitude lower rate than the active proteins. Thus, rapid formation of stable NA-bound ORF1p polymers is positively correlated with retrotransposition. Not only is the rate of oligomerization determinative for retrotransposition activity, it is sensitive to the amino acid composition of the coiled coil. Therefore, we discuss these results in the context of both the mechanistic and evolutionary properties of ORF1p.
MATERIALS AND METHODS
The structures of all of the recombinant DNA clones and ORF1 constructs described here were verified by DNA sequencing.
ORF1 constructs
111—The modern ORF1 (111) sequence was obtained from the highly active L1.3 element (47), a member of the Ta-1 subfamily of the human-specific L1Pa1 family (8) and kindly provided to us by Dr John Moran on the JCC8 plasmid (20). We subcloned ORF1 as a NotI/XmaI fragment into MB18 (pUC18 modified to contain BamHI, NotI, XmaI and MluI sites between SphI and EcoRI) to generate p111-mb18. The encoded protein is 111p, the corresponding retrotransposition reporter is p111-rtc (see ‘Retrotransposition assays’ section below).
555—We resuscitated the ancestral ORF1 (555) sequence of the 25 Myr old extinct L1Pa5 family by first constructing a 60% majority consensus sequence from 840 L1Pa5 ORF1 sequences in the human genome data base (UCSC hg18, NCBI Build 36.1). We converted TpGs to CpG if the alignment contained CpG or CpA at this position, and likewise for any CpA if the alignment contained CpG or TpG. Ambiguities were resolved by reference to the antecedent L1Pa6 or descendant L1Pa4 families. We synthesized the sequence that included the NotI site just 5′ of the ORF1 ATG through the highly conserved BsmFI site (incises the codon for K154, Supplementary Figure S1) by ligating overlapping, complementary gel-purified 50-nt oligonucleotides. The sequence of the BsmF1 site through XmaI downstream of the TAA was synthesized by Retrogen, Inc. (San Diego, CA, USA). We ligated these sequences (NotI/BsmFI, BsmFI/XmaI) into the NotI/XmaI sites of MB18 to generate 555-mb18. The base sequences between NotI/ATG, and TAA/XmaI are the same in p111-mb18 and p555-mb18. The encoded protein is 555p, and the retrotransposition reporter is p555-rtc (see ‘Retrotransposition assays’ section below).
151—We generated the mosaic ORF1 sequence (151) by inserting the BsmI/BsmFI fragment of 555-mb18 (Figure 1B) in the corresponding sites of 111-mb18 to generate 151-mb18. BsmI incises the DNA after the first base, A, for either the R codon in 111-mb18 or the T codon in 555-mb18. Therefore this ligation preserves the ancestral T in the mosaic 151p as shown in Supplementary Figure S1. The encoded protein is 151p, the retrotransposition reporter is 151-rtc (see ‘Retrotransposition assays’ section below).
Expression vectors
FLAG-tagged 111p, 151p and 555p expression vectors—to compare expression of 111p, 151p and 555p in HeLa_JM cells we fused the FLAG epitope (DYKDDDDK) to their C-termini. We used polymerase chain reaction with the Phusion® High-Fidelity DNA Polymerase (New England Biolabs) and oligonucleotides F-BamHI-111 and R-Flag-EcorI-111 to recover the ORF1 sequences from templates p111-rtc or p151-mb18, and oligonucleotides F-BamHI-555 and R-Flag-EcorI-555 for p555-mb18 (Supplementary Table S1, #8-11). The 5′ BamHI-Kozak and FLAG-EcoRI-3′ bounded amplicons were cloned into the corresponding sites in pcDNA3.1(+)-puro obtained from the Don Ganem laboratory, UCSF.
ORF1p expression and purification
As described previously for the expression and purification of 111p (35) we inserted the coding sequences for a hexa-histidine (HIS) affinity tag and tobacco etch virus (TEV) protease site (generated by annealing oligonucleotides histev_t and histev_b, Supplementary Table S1, #12 and #13) into the BsiWI and NotI sites of p555-mb18 or p151-mb18 (i.e. HIS-TEV-ORF1) followed by deletion of the NotI site just 5′ of ORF1 (48). Cloning of the fusions into the pFastBac1 vector (Invitrogen) for baculovirus infection, generation of the respective recombinant baculoviruses using the Bac-to-Bac® based baculovirus expression system (Invitrogen) and infection of Hi5 insect cells, were carried out by the Protein Expression Laboratory, Advanced Technology Program, SAIC-Frederick, as previously described (35). We purified the proteins from the insect cell pellets as described (35). The amino termini of all of the ORF1p proteins contain an N terminal glycine followed by the normal initiating methionine that resulted from TEV cleavage step.
Retrotransposition assays
We used HeLa cells (HeLa JM, kindly provided by Dr John Moran, University of Michigan, Ann Arbor) and a retrotransposition reporter, pRTC2, an L1.3-containing version of the retrotransposition reporter plasmid (20) that we extensively modified as described in detail in the Supporting Information that accompanies Cook et al. (38) except that the version used here lacks the puromycin N-acetyl-transferase gene (Figure 1C). HeLa cells were plated in a 6-well dish at 2 × 105 cells/well and within 24 h a mixture of 3 μl FuGene®6 Transfection Reagent (Roche) and 1 μg of p111rtc, p151rtc or p555rtc were applied to the cells (per the supplier's suggestions). After 3 days, 400 μg/ml G418 antibiotic (Gibco) was added to select for G418 resistant foci and incubation was continued for an additional 10–14 days with media change as needed. The cells were then washed twice with 1× PBS, fixed (2% formaldehyde, 0.2% glutaraldehyde in 1× PBS) and stained with Karyo Max® Giemsa Stain (Gibco). After 30 min the stain was removed and the cells were washed repeatedly with 1× PBS until the background was colorless.
Filter-binding assay
We employed a previously described modification of a dual membrane filter-binding assay (35). Binding reactions (20 mM Tris, 10% glycerol (w/v), 0.5 mM MgCl2, 0.1 mM DTT and 100 μg/ml BSA, on ice) contained 0.1 nM pre-formed mismatched duplex of d29 and [32P]-d29_c-mm (Supplementary Table S1, #2-3). After adding 1/10th volume of the appropriately diluted ORF1p in 0.5 M NaCl (final [NaCl] = 0.05 M), the reaction was incubated at 37°C for 1 h whereupon duplicate 5 μl samples were added under vacuum to the membrane layer of nitro-cellulose (binds protein-bound DNA) atop of zeta-probe GT (Bio-Rad, binds free DNA). We washed the membrane stack with binding buffer and determined the radioactivity of the dried membranes using a Fuji FLA-5000 series Image Analyzer (Fuji Medical Systems) and Image Gauge software (version 3.0, Fuji Medical Systems). We fit the fraction bound (FB) [radioactivity bound to nitrocellulose/(radioactivity bound to nitrocellulose + radioactivity bound to zeta probe)] as a function of protein concentration using a logistic function (KaleidaGraph, 4.1): y (FB) = m1 + (m2 − m1)/(1 + (X/m3)m4), where X = [ORF1p]; m1 = [ORF1p] at maximal ligand bound; m2 = fraction ligand bound at 0 [ORF1p], m3 = [ORF1p] where half of the ligand is bound, i.e. [ORF1p0.5FB]; m4 = slope of the binding curve plotted as a function of the natural log of [ORF1p] (49). We also determined the fraction of ss- and ds-[32P]DNA in some samples (Figure 2A, heavy tick marks): 10 μl samples were diluted two-fold with ice-cold 0.2% sodium dodecyl sulphate, 3% glycerol, 400 ng/μl tRNA, bromophenol blue followed by electrophoresis at 4°C (6% 29:1 cross-linked polyacrylamide gels, 20 mM HEPES, 10 mM sodium acetate, 200 V). Dried gels (Figure 2B) were scanned with the Fuji Image Analyzer. A portion of the reaction without ORF1p was kept at 0°C to serve as a control for melting at 37°C in the absence of protein. The fraction ssDNA = [ssDNA / (ssDNA + dsDNA)] – (fraction ssDNA at 0 protein) and is plotted in Figure 2C.
Figure 2.

Binding of ORF1p variants to mismatched duplex oligonucleotide. Binding, protection and eventual melting of mismatched double stranded 29-nt by the indicated ORF1p as a function of protein concentration. (A) ORF1p variants were incubated with 0.1 nM radioactive mismatched duplex 29-nt for 1 h at 37°C using the filter-binding assay described in ‘Materials and Methods’ section. (B) Autoradiogram of dried polyacrylamide gel of selected samples (heavy tick marks in panel (A) labeled a to e) of the NA binding reaction shown in (A) and the 0 protein controls that were incubated for 1 h at either 0 or 37°C. Samples were electrophoresed as described in the ‘Materials and Methods’ section. (C) Fraction ssDNA of the selected samples relative to zero protein control. These values were determined by quantifying the ds and ss NA species shown in (B) as described in ‘Materials and Methods’ section.
FRET NA chaperone assay
This assay was carried out as described in ref. (50) using the relative concentrations of ORF1p and NA wherein a mismatched duplex is stabilized (caged), before being eventually melted (Figure 2A, gray rectangle, and ref. (35)). Reactions (20 μl) initially contained 1 μM ORF1p (in terms of monomer), 10 nM 5′-Cy3-ACTGCcAGAGAcTTcCCACAT (Supplementary Table S1, #6) and 2× FRET buffer (100 mM Tris–Cl, pH 7.4, 100 mM NaCl, 6 mM MgCl2 and 4 mg/ml BSA), incubated at room temperature for ∼5 min in a 96-well half-area, low binding black plate (Corning #3993). Annealing was started by injection of 20 μl 10 nM Cy5-ATGTGGAAAATCTCTAGCAGT (Supplementary Table S1, #7) in water for a final concentration of 500 nM ORF1p and 5 nM of each oligonucleotide in 1× FRET buffer. Cy3 was excited every 0.7 seconds at 535 nm (25 nm band pass) and emissions were read at 590/25 (Cy3) and 680/30 (Cy5). At ∼3 min, we added 5 μl of 500 nM of the perfect complement (Supplementary Table S1, #5, 21r_dna) of Cy5- ATGTGGAAAATCTCTAGCAGT (Supplementary Table S1, #7) and the reading was continued for another 3 min. We used a Synergy2 Microplate Reader running Gen5 data analysis software (Biotek Instruments, Inc.), which had been fitted with a red photomultiplier tube and a xenon flash lamp.
Protein cross-linking
As described previously (35), 10–20 μl of an ORF1p solution in 20 mM Hepes, pH 7.5, 10% w/v glycerol, 0.05 M NaCl was incubated for 30 min at room temperature in the presence or absence of a 120-nt single-stranded oligonucleotide DNA (Supplementary Table S1, #1, d120_c) and then for an additional 30 min with either 0.05 mM or 1 mM ethylene glycobis (succinimidylsuccinate) (EGS, Pierce Biotechnology) freshly made in dimethyl sulfoxide (DMSO, Sigma). These respective concentrations partially or completely cross-link the protein. The concentrations of protein, oligonucleotide and EGS are given in the legend to Figure 4. The reactions were quenched with one-tenth volume of 1 M Tris–Cl, pH 8.0, subjected to denaturing sodium dodecyl sulphate-polyacrylamide gel electrophoresis on either Bis-Tris 10% (Figure 4, insert) or 4–12% gradient gels (Novex Life Technologies). The protein bands were visualized with silver stain (Pierce).
Figure 4.

Trimer oligomerization on 125-nt ssDNA. Silver stained polyacrylamide gel of half of the indicated reactions electrophoresed under reducing and denaturing conditions. Except for lane 4 and the insert, the indicated amounts of ORF1p trimer were incubated in 15 μl with either 0.125 μM of a single stranded-oligonucleotide (Supplementary Table S1, #1) in 50 mM NaCl or in 0.5 M NaCl for 20 min at room temperature and then cross-linked for 30 min with 1.0 mM of the bifunctional EGS reagent and then processed for electrophoresis as described in the ‘Materials and Methods’ section. The protein in lane 4 and those in the insert, at 0.189 μM, were incubated with 0.015 mM EGS, which partially cross-links the trimer or polymers thereof giving a ladder of products on denaturing gels equivalent in size to monomern—indicated by the numbers to the left of lane 4 and the insert. Each lane of the insert contains the entire sample. The gels were imaged as described in Ref. 35 and the scans are shown in Supplementary Figure S2.
Western blots
Each well of a 6-well plate was plated with 6 × 105 HeLa cells, and after 24 h transfected with 1 μg pORF1-Flag vector using 3 μl FuGENE®6 (Promega). After 48 h the cells were washed twice with cold PBS and lysed in cold 50 mM Tris-Cl, pH 7.4, 650 mM NaCl, 1 mM EDTA, 1% Triton X-100, cOmplete™ EDTA-free protease inhibitor cocktail (Roche), 100 μM leupeptin and 1 mM PMSF. Sonicated lysates were centrifuged (17 000 × g, 15 min, 4°C) and 75 μg of supernatant protein (Bradford reagent—BioRad) was electrophoresed under denaturing conditions, transferred to nitrocellulose membranes using iBlot® (Invitrogen), which were then incubated with ANTI-FLAG® M2 monoclonal antibody (Sigma-Aldrich®). Bands were detected with SuperSignal™ West Pico Chemiluminescent Substrate (Thermo Scientific) and exposed on HyBlot ES™ autoradiography film (Denville Scientific, Inc.). Blots were then treated with Restore™ PLUS Western Blot Stripping Buffer (Thermo Scientific) according to manufacturer's protocol and re-probed with anti-γ-tubulin (Sigma-Aldrich®).
Single molecule methods
Each experiment was conducted on a biotinylated bacteriophage λ DNA molecule, tethered from its opposite ends between two streptavidin-coated polystyrene beads in 10 mM HEPES, 50 mM Na+ at pH 7.5. While one bead was fixed on a micropipette tip the other was held in an optical trap. By gradually moving the fixed bead, the force-extension curve of a DNA or DNA–ORF1p complex was obtained. The experiment to quantify ssDNA fractions bound by distinct ORF1p kinetics states is described in the text and Supplementary Methods. To directly measure the dissociation time constants, an overstretched dsDNA was incubated with ORF1p for 360 s. During the return after incubation, the ORF1p–DNA complex was stopped and maintained a constant force (Fst = 53, 43 or 33 pN) for 360 s, via a force feedback loop. The change in the extension during the constant force feedback was recorded every 50 ms and fit to a double exponential function in time to obtain the characteristic dissociation time constants of the fast and intermediate ssDNA-bound ORF1p kinetic states (Supplementary Table S3).
RESULTS
Comparisons of modern, ancestral and mosaic ORF1p
Figure 1B summarizes the amino acid differences between the modern, mosaic and ancestral versions of ORF1p: 111p, 151p and 555p (amino acid alignment in Supplementary Figure S1). Modern 111p is encoded by the L1.3 element (47), a member of the currently active Ta-1 L1Pa1 subfamily (8). Ancestral 555p is encoded by the resuscitated ORF1 that we derived from a consensus sequence of 840 L1Pa5 sequences as described in ‘Materials and Methods’ section. The mosaic 151p was created by swapping in the ancestral coding region encompassed by the conserved BsmI and BsmFI sites as described in the ‘Materials and Methods’ section. The structure and relative retrotransposition activity of other mosaic ORF1ps are given in the legend to Figure 1B.
Using the retrotransposition assay depicted in Figure 1C we found that 555p and 111p are equally active. However, the 151p mosaic protein cannot support retrotransposition (Figure 1D). This result is not due to an inherent inability of 151p to be stably expressed in HeLa cells, as all three are expressed to about the same extent (Figure 1E). To investigate the basis of this inactivity we purified 111p, 555p and 151p that had been expressed in baculovirus infected insect cells as described in ‘Materials and Methods’ section and ref. (35). Denaturing gel electrophoresis (Figure 1F) shows that the proteins were essentially homogenous. The bands at ∼80 kDa are likely not fully-denatured trimers (i.e. dimers) and the band between the 66 and 55 kDa markers is an apparent staining artifact as it was not seen on other gels (e.g. insert in Figure 4).
Interactions of modern, ancestral and mosaic ORF1p with oligonucleotides
Earlier work showed that while purified 111p bound a 29-nt duplex DNA with 1/10th the affinity as its single-stranded counterpart, it bound a mismatched version of the duplex with the same affinity as the single strand i.e., the protein ‘sensed’ the mismatched duplex as an ssDNA. This was not due to the protein melting the mismatched duplex and then binding the released single strand. Rather, the protein protected the mismatched duplex from melting (which we termed caging) before eventually melting it at higher molar excesses of ORF1p to oligonucleotide (35). Figure 2 shows that 111p, 151p and 555p are indistinguishable in this regard. These reactions were carried out at 0.05 M NaCl, which is optimal for NA binding (35). Such complexes would also be a substrate for the strand exchange phase of NA chaperone activity. Therefore, we compared the activities of the proteins in both annealing and strand exchange with a Förster resonance energy transfer (FRET) based chaperone assay as described in the Methods. We used the relative amounts of protein and NAs indicated by the gray rectangle in Figure 2A. As Figure 3 and Supplementary Table S2 show, the proteins showed similar activities in both phases of this assay. Thus, none of the results in Figures 2 and 3 would account for the inactivity of 151p.
Figure 3.

NA chaperone activity of ORF1p variants. (A) Schematic of the FRET assay to measure the annealing and exchange phases of NA chaperone activity using the assay conditions described in the ‘Materials and Methods’ section. (B) Reactions (40 μl, 0.5 μM ORF1p, 5 nM 21-nt mismatched duplex) were incubated at room temperature, irradiated every 0.7 s at 535 nm and fluorescence was measured at 590 and 680 nm (solid circles). At the indicated time (vertical arrow) 50 nM perfect complement was added. The relative concentrations of duplex and ORF1p during the annealing phase of these reactions correspond to those that produce maximal protection of the mismatched duplex (caging, Figure 2A, gray rectangle). The annealing and exchange phases data are fit to single and double exponential functions in time (solid lines and Supplementary Table S2) respectively, using the minimization of χ2 method.
The lengths of the oligonucleotides used in the foregoing assays could only accommodate a single trimer. But, as noted above, 1.0 mM EGS can cross-link multimers of trimers (e.g. trimer2, trimer3) that can assemble on longer oligonucleotides in 0.05 M NaCl. To determine whether 151p can be differentiated from 111p and 555p on this basis we compared the distribution of trimer and multimers at two concentrations of the proteins on 0.125 μM of a 120-nt oligonucleotide (Supplementary Table S1, #1). Figure 4 and Supplementary Figure S2 show that the distribution of trimer and multimers is similar for all three proteins at both concentrations. In the absence of NA all of the proteins form cross-linkable polymers in 1.0 mM EGS that are too large to enter the gel (results not shown and 35).
However, 0.5 M NaCl, which does not support NA binding, essentially inhibited polymer formation by the three proteins as shown previously (35). The insert in Figure 4 shows that partial cross-linking (0.05 mM EGS) of the three proteins in the absence of NA at 0.05 M NaCl generated similar monomern ladders when electrophoresed under denaturing conditions. These lanes were obtained from the same gel from which the intervening non-relevant lanes were cropped. The slower migration of the monomer, dimer and trimer bands in the 111p lane is due to its location at the gel edge where migration is retarded by distortion of the electrophoretic field Thus, none of the foregoing assays were sensitive to the biochemical defect of 151p that renders it inactive in retrotransposition. Therefore we employed an assay capable of extending analysis of protein/NA interactions beyond those possible using bulk assays with oligonucleotide substrates (51,52).
Single-molecule measurements of ORF1p-ssDNA binding kinetics
In this assay a double-stranded (ds) λ phage DNA molecule is tethered between two polystyrene beads. One is held in an optical trap while the other is immobilized on a micropipette tip (46). The force-extension profile of dsDNA is obtained by gradually moving the fixed bead while recording the extension and the force exerted on the single DNA molecule (Figure 5A–F, solid black line) without DNA-binding ligands. The plateau, or the rapid increase of the extension in the narrow 60–65 pN force regime, represents force-induced melting, wherein dsDNA is converted to ssDNA, by either peeling from the ends or forming melted bubbles (53). Although the plateau may represent transition from B-form dsDNA into another form referred to as S-DNA (45,54) in high salt conditions (>0.15 M), it is well established that at pH 7.5 and 0.05 M Na+ (the conditions used here) the plateau represents force-induced melting (46,53). If the ssDNA strands could not anneal, the return curve would be the same as the ssDNA curve (Figure 5A, purple line). However, here, in the absence of ligands, the DNA strands rapidly anneal indicated by the almost complete reversal of the dsDNA extension curve, which exhibits minimal hysteresis—i.e. the discrepancy between the dsDNA-stretch and return curves (Figure 5A, dashed black line). Ligands that bind to ssDNA could inhibit annealing, and by preventing dsDNA formation result in increased DNA length at forces below the melting plateau. This provides an accurate quantitative measure for the ligand-bound ssDNA fraction, and was used previously to characterize the binding of viral restriction factor APOBEC3G to ssDNA (51). Here we adapted this method to accommodate the more complex binding characteristics of ORF1p to ssDNA.
Figure 5.

Single molecule analysis reveals and quantifies three populations of ORF1p bound to ssDNA. (A) Stretch (solid black line) and return (dashed black line) curves of a dsDNA molecule in the absence of ORF1p. Purple line is the force-extension curve of an ssDNA. (B) Return of the ORF1p-DNA complex (blue circles) after incubating an overstretched dsDNA for 360 s in 2 nM ORF1p (111p). (C) Subsequent stretch (red circles) of the 111p–DNA complex shown in (C). (D) Quantifying the 111p-bound ssDNA fraction (f) bound by ORF1p exhibiting slow dissociation kinetics. The subsequent stretch is fit (red line, f = fslow) to a linear combination of dsDNA (gray line, f = 0) and the 111p-saturated ssDNA (solid gold, f = 1) curves. (E) Quantification of the total fraction (fT) of 111p-bound ssDNA. Dashed blue curve is the linear combination intersecting the force (F0) at which the 111p–DNA complex begins to approach the force regime below the melting plateau, which yields fT. Return data is fit by allowing ffast to vary with force in order to correct for the rapid 111p dissociation during the return to find ffast and fint. (F) Summary of the analysis method. The green dashed line is the summed linear combination of fslow and fint. (G) Schematic of the model used to interpret the data from (A–F), representing a single event from each kinetic class of ssDNA-bound ORF1p (not to scale and does not reflect the total fraction in each state). DsDNA (i) is initially stretched in the optical tweezers to convert it into ssDNA through force-induced melting. The stretched ssDNA is incubated in the solution containing ORF1p (ii) resulting in a combination of three types of bound protein: fast, intermediate and slow. The bound fast component equilibrates quickly with the protein in solution, but is also converted into small oligomers (intermediate component) and then into large oligomers (slow component) on ssDNA (ii). As the DNA–protein complex is released, the fast component quickly dissociates (iii). When the DNA is completely relaxed, just before the subsequent stretch, the intermediate component dissociates, leaving only the slow component bound (iv). (See also Supplementary Methods and Supplementary Figure S3).
To analyze ORF1p–ssDNA interactions we first overstretch the dsDNA up to ∼75 pN, converting most of the dsDNA into ssDNA. We then replace the surrounding solution with 2 nM retrotransposition competent 111p or 555p, or the defective 151p. After incubating the ssDNA with ORF1p for 360 s, we release the force on the DNA and obtain a return curve of the DNA–ORF1p complex (Figure 5G and B, blue circles) that reflects the fraction of protein-bound ssDNA. We generate a second curve (Figure 5C, red circles) by re-stretching the DNA–ORF1p complex immediately after the initial return. The extension produced by the subsequent stretch is substantially smaller than the extension observed during the initial return (Figure 5B, blue circles) but larger than that of dsDNA (Figure 5A–F, solid black line). This indicates that while a population of ssDNA-bound ORF1p dissociated during the first return allowing concomitant duplex formation, another population still remains bound, suggesting multiple dissociation kinetics for ORF1p. Further subsequent stretches trace the first subsequent stretch (data not shown for clarity), indicating a ssDNA-bound ORF1p population with negligible dissociation.
At any given force below the melting plateau, the extension of the DNA attained during either the return after incubation or the subsequent stretch represents the sum of dsDNA and ORF1p-bound ssDNA fractions. One can quantify the ORF1p-bound ssDNA fraction (f), by modeling the data as a linear combination (Supplementary Figure S3 and Supplementary Methods) of the curves for dsDNA, with no protein bound, f = 0 (Figure 5D–F, gray line), and ORF1p-saturated ssDNA curves, for which all the possible binding sites of ssDNA are stably occupied by ORF1p, f = 1 (Figure 5D–F, gold line). The ORF1p-saturated ssDNA is experimentally achieved by incubating the overstretched dsDNA in high (>15 nM) ORF1p concentrations for long times (>30 min). However, assuming a simple linear combination of two kinetic states (slow and fast) yielded a poor fit due to the concave nature of the return curve, an effect that decreases with incubation time (see below and Figure 5B). Therefore we postulated three ORF1p-bound ssDNA fractions, each populated with ORF1p exhibiting distinct dissociation kinetics: fast (ffast), intermediate (fint) and slow (fslow). We hypothesized that the fast fraction is due to the binding of single ORF1p trimers to ssDNA, which equilibrate quickly, exhibiting rapid bimolecular association and dissociation. During the return at forces below that of the melting plateau, rapid dissociation of ORF1p trimers is accompanied by concomitant duplex formation (Figure 5G). Continuous net dissociation of ORF1p from this fraction during the return would account for the concave nature of the return data (Figure 5B). The intermediate fraction could consist of intermediate-sized ORF1p oligomers (trimer2, trimer3, trimern), which dissociate when the DNA is completely relaxed. The remaining fraction is bound by ORF1p that does not dissociate from ssDNA during the timescale of our experiments, which we assume constitutes fslow and likely consists of large protein polymers or aggregates, similar to those observed for APOBEC3G (51).
We directly obtain fslow by modeling the subsequent stretch as a linear combination of dsDNA and ORF1p-saturated ssDNA curves (Figure 5D, red line). Next, we find the linear combination that intersects F0, where F0 is the force at which the return curve begins to approach the force regime below the melting plateau (Figure 5F). This linear combination yields the instantaneous total fraction (fT = ffast + fint + fslow) of ORF1p-bound ssDNA that emerged during the incubation (Figure 5E, dashed blue line). The discrepancy between the dashed blue line and the observed return data (Figure 5E, blue circles) is due to continuous duplex formation driven by the fast dissociating ssDNA-bound ORF1p, which continuously decreases the fast fraction, ffast during the return. To account for the fast dissociating protein we modify ffast to be varied with a phenomenological force (F) dependence 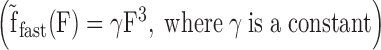 and model the return curve after incubation (Figure 5E, blue line). Collectively our analytical method quantifies the three ORF1p-bound ssDNA fractions fslow, fint and ffast, in agreement with the observed data. The summary of the analysis and the hypothetical curve that represents the combined fractions of fslow and fint (dashed green line), are shown in Figure 5F. Because F0 is determined directly from the data, there are a total of three fitting parameters for all of the data, and each curve is fit to one parameter (see also Supplementary Figure S3 and Supplementary Methods).
and model the return curve after incubation (Figure 5E, blue line). Collectively our analytical method quantifies the three ORF1p-bound ssDNA fractions fslow, fint and ffast, in agreement with the observed data. The summary of the analysis and the hypothetical curve that represents the combined fractions of fslow and fint (dashed green line), are shown in Figure 5F. Because F0 is determined directly from the data, there are a total of three fitting parameters for all of the data, and each curve is fit to one parameter (see also Supplementary Figure S3 and Supplementary Methods).
ssDNA-bound ORF1p exhibits three distinct kinetic states
We tested the hypothesis of three ssDNA-bound ORF1p populations by directly measuring the dissociation times of ssDNA-bound ORF1p variants. After incubating overstretched dsDNA in 2 nM ORF1p, we stopped the return of the DNA–ORF1p complex at different forces (Fst≈33, 43 and 53 pN) below the melting plateau, which we maintained for 360 s (Figure 6A). To maintain constant force the DNA–protein complex was gradually released to compensate for any increase in force caused by duplex formation that would accompany dissociation of ssDNA-bound ORF1p. Hence the temporal decrease in the extension directly measures the net dissociation of the ssDNA-bound ORF1p as a function of time. During the constant force feedback loop the extension reaches a local equilibrium, converging at the subsequent stretch (Figure 6A, red circles). This is greater than the extension of dsDNA (Figure 6A, black line and Supplementary Table S3) at the corresponding force, and results from the stably bound fraction of ORF1p oligomers that exhibit negligible or slow dissociation from ssDNA (fslow). The change in the extension during the constant force feedback loop yielded an exponential function in time with two time constants (τfast and τint) that differed by an order of magnitude (Figure 6B and C, and Supplementary Figure S4), in agreement with the fast (ffast) and intermediate (fint) ssDNA- ORF1p kinetic states. Dissociation time constants τfast and τint were similar for the three ORF1p variants at each stopped force Fst (Figure 6C and Supplementary Table S3). The overall dissociation time constants of all three ORF1p variants averaged over Fst are fit to an exponential function of force (Figure 6C, dashed gray lines). The corresponding fits yield τ0,fast = 2.7 ± 0.4 s and τ0,int = 57 ± 4 s, the zero force fast and intermediate dissociation time constants, respectively. The τ0,fast results are consistent with the behavior of 111p, 555p and 151p in the oligonucleotide-based assays shown in Figures 2 and 3, as the oligonucleotides used in these assays can only accommodate a single trimer.
Figure 6.

Direct single molecule measurements of fast and intermediate dissociation time constants. (A) Representative data for measuring protein dissociation at constant force. Overstretched dsDNA (solid black) is incubated in 2 nM 111p for 360 s. The returning 111p–DNA complex after incubation (open blue circles) is stopped and maintained at a constant force of 43 pN via a force feedback loop for 360 s. (B) The change in extension with time during the constant force feedback loop (green circles) is fit to a double exponential function of time (solid black). Two time constants τint and τfast represent the characteristic dissociation time constants of the ssDNA-bound 111p populations exhibiting respectively intermediate and fast dissociation kinetics. (C) Variation of the time constants with stopped force for ORF1p variants. Intermediate (circles) and fast (triangles) dissociation time constants are measured as a function of a stopped force (Fst). Green, blue and red data points correspond to: 111p, 555p and 151p respectively. Overall variant averages are fit to an exponential function of force, τ(F) = τ0eF/Ω, using the minimization of χ2 method (dashed gray lines), where Ω is a factor that describes the scale of time constant variation with force. Corresponding fits for τf and τi yield τ0,fast = 2.7 ± 0.4 s, Ωfast = 28 ± 1 pN and τ0,int = 57 ± 4 s, Ωint = 75 ± 10 pN respectively. Error bars are standard errors for at least three measurements (also see Supplementary Figure S4 and Table S3).
Quantifying oligomerization rates of ORF1p variants on ssDNA
In order to quantify the three ssDNA-bound ORF1p populations 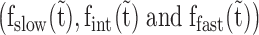 as a function of incubation time (
as a function of incubation time (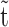 ), we repeated the first experiment by incubating the overstretched dsDNA for different durations in 2 nM ORF1p (Figure 7A and Supplementary Figure S5). For the three ORF1p variants,
), we repeated the first experiment by incubating the overstretched dsDNA for different durations in 2 nM ORF1p (Figure 7A and Supplementary Figure S5). For the three ORF1p variants, 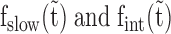 increase with incubation time and reach saturation following a simple exponential function (Figure 7B, blue and green lines and Supplementary Table S4). However,
increase with incubation time and reach saturation following a simple exponential function (Figure 7B, blue and green lines and Supplementary Table S4). However, 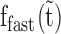 first increases and then exponentially decays (Figure 7B, red lines and see also Supplementary Table S4). Taken together, the results indicate that
first increases and then exponentially decays (Figure 7B, red lines and see also Supplementary Table S4). Taken together, the results indicate that 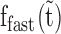 , the ssDNA fraction bound by rapidly dissociating ORF1p, is converted with time to the more stable ssDNA-ORF1p fractions fint and fslow. This result supports the hypothesis that, as was the case for APOBEC3G (51), ORF1p trimers oligomerize to the more stably bound oligomeric forms on ssDNA with increasing incubation time. Therefore, the rate for this process, or oligomerization time constant, (Toligo), can be evaluated in terms of the decay time of
, the ssDNA fraction bound by rapidly dissociating ORF1p, is converted with time to the more stable ssDNA-ORF1p fractions fint and fslow. This result supports the hypothesis that, as was the case for APOBEC3G (51), ORF1p trimers oligomerize to the more stably bound oligomeric forms on ssDNA with increasing incubation time. Therefore, the rate for this process, or oligomerization time constant, (Toligo), can be evaluated in terms of the decay time of 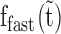 . Toligo for 111p, 555p and 151p were 356 ± 2 s, 1620 ± 124 s and 18000 ± 8190 s respectively (Supplementary Table S4). Thus, the Toligo of 151p, which is completely inactive in retrotransposition, is one to two orders of magnitude higher than both 111p and 555p, which support retrotransposition. This inverse correlation between Toligo and retrotransposition efficiency strongly suggests that a rapid rate of ORF1p oligomerization is essential for retrotransposition.
. Toligo for 111p, 555p and 151p were 356 ± 2 s, 1620 ± 124 s and 18000 ± 8190 s respectively (Supplementary Table S4). Thus, the Toligo of 151p, which is completely inactive in retrotransposition, is one to two orders of magnitude higher than both 111p and 555p, which support retrotransposition. This inverse correlation between Toligo and retrotransposition efficiency strongly suggests that a rapid rate of ORF1p oligomerization is essential for retrotransposition.
Figure 7.

Single molecule measurements to quantify slow, intermediate and fast ORF1p-bound ssDNA fractions as a function of incubation time. (A) Representative data (open circles) and corresponding fits (solid lines) for returns of ssDNA–ORF1p complexes after incubating an overstretched dsDNA (ssDNA) in 2 nM ORF1p variants for 180 s (pink) and 2880 s (blue). Data are fit to a linear combination of dsDNA (gray line) and ORF1p-saturated ssDNA curves (gold line, see Supplementary Methods and Supplementary Figure S5). (B) ssDNA fractions bound by ORF1p exhibiting slow (blue), intermediate (green), fast (red) dissociation kinetics and the total ORF1p-bound fractions (purple) are quantified as a function of incubation time. The fits of slow, intermediate and total fractions (blue, green and purple lines, respectively) are single exponential functions of incubation time and the fast fractions are fit (red lines) to a sum of increasing and decaying exponential functions of incubation time (Supplementary Table S4). Error bars are standard errors for at least three measurements.
DISCUSSION
Despite the prominent role of L1 retrotransposons in shaping mammalian genomes and the persistence of L1 activity in most mammals, including humans, we have little mechanistic understanding of the evolutionary and biochemical processes that underlie the success of L1 elements. This is particularly true for ORF1p, a uniquely trimeric NA chaperone that is essential for retrotransposition and which frequently undergoes major evolutionary changes. Here we present two observations that advance our understanding of its biochemical properties that are relevant to its role in retrotransposition and to the function and evolutionary dynamics of its coiled coil domain.
To address the biochemical consequences of coiled coil evolution we resuscitated the ancestral L1Pa5 ORF1 (encodes 555p), which differs from the currently active L1Pa1 ORF1 (encodes 111p) at 42 positions, 30 of which were located in the coiled coil domain (Figure 1B). To assess the effect of these coiled coil substitutions on ORF1p function in retrotransposition, we exchanged different sets of the modern coiled coil amino acids for their ancestral counterparts to generate mosaic ORF1 sequences. We then inserted either the resuscitated L1Pa5 or mosaic ORF1 sequences in place of the modern L1Pa1 ORF1 sequence of a retrotransposition reporter vector and found that whereas 555p and 111p support retrotransposition equally, the mosaic ORF1 sequence (151, encodes 151p) is completely inactive (Figure 1D). To determine the biochemical basis for this inactivity we purified and compared the in vitro properties of insect-expressed 111p, 151p and 555p.
Interactions with nucleic acids
All of the corresponding purified proteins were similarly active in their interactions with NAs with respect to binding affinity, stabilization of mismatched duplex and NA chaperone activity when tested with oligonucleotide substrates (≤29 nt) traditionally used to assess these activities (Figures 2 and 3). These substrates are only long enough to accommodate one ORF1p trimer. Additionally, cross-linking studies showed no differences between the proteins in their assembly of the cross-linkable short oligomers, trimer2 and trimer3, on a 120-mer oligonucleotide (Figure 4). Therefore these assays were not sensitive to the biochemical defect of 151p.
However, single molecule stretching experiments with λ-DNA did reveal a defect in 151p; namely, this protein polymerized to stably bound oligomers on ssDNA at <1/10th the rate of retrotransposition-proficient 111p and 555p (Figure 7 and Supplementary Table S4). In particular, these experiments identified three populations of ssDNA-bound ORF1p with distinct dissociation timescales: fast (ffast, seconds), intermediate (fint, tens of seconds) and slow or negligible (fslow, Figures 5 and 6). The ffast kinetic state is consistent with association/dissociation of trimers; fint or intermediate kinetic state, is populated by more stably bound oligomers of trimers; the negligibly dissociating fslow population is composed of presumably large polymers or aggregates (51). The dissociation time constants for the populations ffast (τfast, fitted value, 2.7 ± 0.4 s) and fint (τint, fitted value, 57 ± 4 s) were similar for the three proteins (Figure 6C). Therefore, 151p is defective only in the conversion rate of DNA-bound trimers to the stably bound fint and fslow oligomers. Thus, retrotransposition requires fast conversion of NA bound trimers to more stably bound oligomers.
A role for the coiled coil in ORF1p oligomerization rate on NAs
The carboxy-terminal half of ORF1p mediates the inter-trimer interactions responsible for oligomerization (35). Figure 7 shows that the amino acid substitutions in the 151p coiled coil resulted in a reduced formation rate of stably bound oligomers. This finding indicates that the sequence of the coiled coil can determine the intra-trimer configuration that is conducive to oligomerization, an idea consistent with our conclusion that 0.5 M NaCl inhibits ORF1p oligomerization through its structural effect on the coiled coil (35,55). Whether all coiled coil mutations that inactivate or strongly reduce retrotransposition, such as the single substitution in the mouse coiled coil (32), do so by retarding rapid conversion of ORF1p to stably bound oligomers remains to be determined. However, others showed that the L93,100,107,113V set of coiled coil substitutions, which strongly inhibit retrotransposition, decrease the amount of ORF1p incorporated into L1RNP (23). This could be explained by a decreased rate of ORF1p oligomerization that we demonstrated here with purified 151p. Notably the substitution at L107 corresponds to the location of one of the ancestral replacements in 151p (Figure 1B and Supplementary Figure S1).
Because of its length and sensitivity to inactivating mutations, the coiled coil presents a relatively large target for deleterious mutations. Additionally, given the extensive intra- and inter-strand crosstalk within this motif (56), the occurrence of compensatory substitutions or other changes that ameliorate rather than reverse the original mutation would not be infrequent. Taken together, these factors could account for the periodic concerted changes in the coiled coil that typify ORF1p evolution.
ORF1p oligomerization kinetic classes and the L1RNP
Not only did the single molecule stretching assay provide a biochemical explanation for the inactivity of 151p in retrotransposition but it also revealed aspects of ORF1p/NA interaction that seem directly relevant to the L1RNP retrotransposition intermediate: namely its formation upon translation from the L1 transcript, its stability, and its ultimate dissolution during TPRT, necessary to expose the template for cDNA synthesis and the eventual generation of a new L1 insert.
The fast dissociating fraction would allow the protein translation machinery to compete with ORF1p binding for the L1 transcript and enable synthesis of enough ORF1p to coat it. However, at sufficient protein concentration, rapid conversion of the fast dissociating population to stably-bound fint and fslow would contribute to cis preference (40,57), by limiting diffusion of ORF1p away from its encoding transcript (35,58) and possible fruitless interactions with non-L1 NAs. In addition, its rapid oligomerization to more stably bound ORF1p–RNA complexes would protect the L1 transcript from degradative nucleases or components of the innate immunity pathways such as siRNA and the APOBEC3 family of enzymes (59,60). The fact that the ffast population is transient suggests that this kinetic class likely has no role in retrotransposition beyond L1RNP formation.
However, both fint and fslow, which increase for ∼20 min at the expense of ffast, persist unchanged at close to equal amounts for time of the experiment (∼50 min). We do not know the molecular distinction between the fint and fslow populations of ORF1p. However, the fint population exhibits several features that would be critical for the L1RNP: they are bound strongly enough to form a stable L1RNP but can be driven off the single strand under force conditions that promote double strand formation. This latter reaction could be considered a proxy for the double-strandedness that would result from cDNA synthesis during TPRT. The dissolution of the L1RNP would make the L1 transcript accessible as a cDNA template and the released trimer NA chaperones could facilitate the various other NA interactions at the integration site that are required to complete the L1 insertion event (18). One such reaction would be the in vivo counterpart of the ‘caging’ reaction that we demonstrated with the purified protein (Figure 2B and 35)—namely stabilization of the duplex (even if it contains mismatches) between the 3′ end of the L1 transcript and the DNA flap which is generated at the nicked target site and serves as the primer for TPRT.
In conclusion, our demonstration that oligomerization kinetics of ORF1p on NA is predictive of its ability to support retrotransposition substantially enhances our biochemical understanding of ORF1p beyond that revealed by standard measurements of NA binding and chaperone activity. Although an L1 retrotransposition event is more complex than just the biochemical interaction between purified ORF1p and NAs, our current results show that several critical features expected of the L1RNP retrotransposition intermediate can directly result from the biochemistry of the interaction between the purified protein and NA. We also showed that the sequence of the coiled coil can profoundly affect polymerization rate and that rapid polymerization survived an episode of positive selection (i.e. more amino acid changes than can occur by chance) that resulted in the turnover of one-third of the coiled-coil amino acids. These results imply that rapid ORF1p oligomerization kinetics is essential for L1 survival.
Taken together, our findings provide a mechanistic explanation for the long-standing enigma that despite the very strong conservation of a coiled coil motif throughout all L1-containing phyla, the persistence of L1 activity can be accompanied by episodic large scale evolutionary change in the coiled coil domain. However, whether preservation of rapid polymerization kinetics is the only force that drives coiled coil evolution, while an intriguing possibility, remains unanswered.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Dr John Moran for providing JM-HeLa cells and the JCC8 clone that contained the L1.3 element, Dr Jean-Claude Walser for deriving L1Pa5 ORF1 consensus sequence and express our appreciation of the proficiency of the Protein Expression Laboratory, Advanced Technology Program, SAIC-Frederick. We thank Dr Micah McCauley for preparation of the DNA constructs used for the single molecule experiments.
Footnotes
Present addresses:
Kathryn E. Callahan, Naperville, IL, USA.
Cesar E. Perez-Gonzalez, National Eye Institute, National Institutes of Health.
FUNDING
The Intramural Program of the National Institute of Diabetes, Digestive and Kidney Diseases; National Institutes of Health [HNK6H7 to K.E.C., P.R.C., C.E.P-G., A.V.F]; National Institutes of Health [R01GM072462 to M.C.W.]; National Science Foundation [MCB-1243883 to M.C.W.]. Funding for open access charge: National Institutes of Health [R01GM072462 to M.C.W., in part]; National Science Foundation [MCB-1243883 to M.C.W., in part]; The Intramural Program of the National Institute of Diabetes, Digestive and Kidney Diseases; National Institutes of Health [HNK6H7 to A.V.F., in part].
Conflict of interest statement. None declared.
REFERENCES
- 1.IHGS-Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 2.Boissinot S., Entezam A., Furano A.V. Selection against deleterious LINE-1-containing loci in the human lineage. Mol. Biol. Evol. 2001;18:926–935. doi: 10.1093/oxfordjournals.molbev.a003893. [DOI] [PubMed] [Google Scholar]
- 3.Boissinot S., Davis J., Entezam A., Petrov D., Furano A.V. Fitness cost of LINE-1 (L1) activity in humans. Proc. Natl. Acad. Sci. U.S.A. 2006;103:9590–9594. doi: 10.1073/pnas.0603334103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malki S., van der Heijden G.W., O'Donnell K.A., Martin S.L., Bortvin A. A role for retrotransposon LINE-1 in fetal oocyte attrition in mice. Dev. Cell. 2014;29:521–533. doi: 10.1016/j.devcel.2014.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bourc'his D., Bestor T.H. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature. 2004;431:96–99. doi: 10.1038/nature02886. [DOI] [PubMed] [Google Scholar]
- 6.Soper S.F.C., van der Heijden G.W., Hardiman T.C., Goodheart M., Martin S.L., de Boer P., Bortvin A. Mouse maelstrom, a component of nuage, is essential for spermatogenesis and transposon repression in meiosis. Dev. Cell. 2008;15:285–297. doi: 10.1016/j.devcel.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dombroski B.A., Mathias S.L., Nanthakumar E., Scott A.F., Kazazian H.H. Jr. Isolation of an active human transposable element. Science. 1991;254:1805–1808. doi: 10.1126/science.1662412. [DOI] [PubMed] [Google Scholar]
- 8.Boissinot S., Chevret P., Furano A.V. L1 (LINE-1) retrotransposon evolution and amplification in recent human history. Mol. Biol. Evol. 2000;17:915–928. doi: 10.1093/oxfordjournals.molbev.a026372. [DOI] [PubMed] [Google Scholar]
- 9.Boissinot S., Entezam A., Young L., Munson P.J., Furano A.V. The insertional history of an active family of L1 retrotransposons in humans. Genome Res. 2004;14:1221–1231. doi: 10.1101/gr.2326704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moran J.V., DeBerardinis R.J., Kazazian H.H. Jr. Exon shuffling by L1 retrotransposition. Science. 1999;283:1530–1534. doi: 10.1126/science.283.5407.1530. [DOI] [PubMed] [Google Scholar]
- 11.Gilbert N., Lutz-Prigge S., Moran J.V. Genomic deletions created upon LINE-1 retrotransposition. Cell. 2002;110:315–325. doi: 10.1016/s0092-8674(02)00828-0. [DOI] [PubMed] [Google Scholar]
- 12.Muotri A.R., Chu V.T., Marchetto M.C., Deng W., Moran J.V., Gage F.H. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature. 2005;435:903–910. doi: 10.1038/nature03663. [DOI] [PubMed] [Google Scholar]
- 13.Beck C.R., Collier P., Macfarlane C., Malig M., Kidd J.M., Eichler E.E., Badge R.M., Moran J.V. LINE-1 retrotransposition activity in human genomes. Cell. 2010;141:1159–1170. doi: 10.1016/j.cell.2010.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ewing A.D., Kazazian H.H. High-throughput sequencing reveals extensive variation in human-specific L1 content in individual human genomes. Genome Res. 2010;20:1262–1272. doi: 10.1101/gr.106419.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iskow R.C., McCabe M.T., Mills R.E., Torene S., Pittard W.S., Neuwald A.F., Meir E.G., Vertino P.M., Devine S.E. Natural mutagenesis of human genomes by endogenous retrotransposons. Cell. 2010;141:1253–1261. doi: 10.1016/j.cell.2010.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tubio J.M.C., Li Y., Ju Y.S., Martincorena I., Cooke S.L., Tojo M., Gundem G., Pipinikas C.P., Zamora J., Raine K., et al. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science. 2014;345:1251343. doi: 10.1126/science.1251343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Upton K., Gerhardt D., Jesuadian J.S., Richardson S., Sanchez Luque F., Sánchez Luque F., Bodea G., Ewing A., Salvador Palomeque C., Brennan P., et al. Ubiquitous l1 mosaicism in hippocampal neurons. Cell. 2015;161:228–239. doi: 10.1016/j.cell.2015.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin S.L. Nucleic acid chaperone properties of ORF1p from the non-LTR retrotransposon, LINE-1. RNA Biol. 2010;7:67–72. doi: 10.4161/rna.7.6.13766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beck C.R., Garcia-Perez J.L., Badge R.M., Moran J.V. LINE-1 elements in structural variation and disease. Annu. Rev. Genomics Hum. Genet. 2011;12:187–215. doi: 10.1146/annurev-genom-082509-141802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moran J.V., Holmes S.E., Naas T.P., DeBerardinis R.J., Boeke J.D., Kazazian H.H. Jr. High frequency retrotransposition in cultured mammalian cells. Cell. 1996;87:917–927. doi: 10.1016/s0092-8674(00)81998-4. [DOI] [PubMed] [Google Scholar]
- 21.Martin S.L. Ribonucleoprotein particles with LINE-1 RNA in mouse embryonal carcinoma cells. Mol. Cell. Biol. 1991;11:4804–4807. doi: 10.1128/mcb.11.9.4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hohjoh H., Singer M.F. Cytoplasmic ribonucleoprotein complexes containing human LINE-1 protein and RNA. EMBO J. 1996;15:630–639. [PMC free article] [PubMed] [Google Scholar]
- 23.Doucet A.J., Hulme A.E., Sahinovic E., Kulpa D.A., Moldovan J.B., Kopera H.C., Athanikar J.N., Hasnaoui M., Bucheton A., Moran J.V., et al. Characterization of LINE-1 Ribonucleoprotein Particles. PLoS Genet. 2010;6:e1001150. doi: 10.1371/journal.pgen.1001150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kulpa D.A., Moran J.V. Cis-preferential LINE-1 reverse transcriptase activity in ribonucleoprotein particles. Nat. Struct. Mol. Biol. 2006;13:655–660. doi: 10.1038/nsmb1107. [DOI] [PubMed] [Google Scholar]
- 25.Kulpa D.A., Moran J.V. Ribonucleoprotein particle formation is necessary but not sufficient for LINE-1 retrotransposition. Hum. Mol. Genet. 2005;14:3237–3248. doi: 10.1093/hmg/ddi354. [DOI] [PubMed] [Google Scholar]
- 26.Mathias S.L., Scott A.F., Kazazian H.H.J., Boeke J.D., Gabriel A. Reverse transcriptase encoded by a human transposable element. Science. 1991;254:1808–1810. doi: 10.1126/science.1722352. [DOI] [PubMed] [Google Scholar]
- 27.Feng Q., Moran J.V., Kazazian H.H. Jr, Boeke J.D. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell. 1996;87:905–916. doi: 10.1016/s0092-8674(00)81997-2. [DOI] [PubMed] [Google Scholar]
- 28.Luan D.D., Korman M.H., Jakubczak J.L., Eickbush T.H. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell. 1993;72:595–605. doi: 10.1016/0092-8674(93)90078-5. [DOI] [PubMed] [Google Scholar]
- 29.Basame S., Wai-lun Li P., Howard G., Branciforte D., Keller D., Martin S.L. Spatial assembly and RNA binding stoichiometry of a LINE-1 protein essential for retrotransposition. J. Mol. Biol. 2006;357:351–357. doi: 10.1016/j.jmb.2005.12.063. [DOI] [PubMed] [Google Scholar]
- 30.Kolosha V.O., Martin S.L. High-affinity, non-sequence-specific RNA binding by the open reading frame 1 (ORF1) protein from long interspersed nuclear element 1 (LINE-1) J. Biol. Chem. 2003;278:8112–8117. doi: 10.1074/jbc.M210487200. [DOI] [PubMed] [Google Scholar]
- 31.Martin S.L., Cruceanu M., Branciforte D., Wai-Lun Li P., Kwok S.C., Hodges R.S., Williams M.C. LINE-1 retrotransposition requires the nucleic acid chaperone activity of the ORF1 protein. J. Mol. Biol. 2005;348:549–561. doi: 10.1016/j.jmb.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 32.Martin S.L., Bushman D., Wang F., Li P.W.L., Walker A., Cummiskey J., Branciforte D., Williams M.C. A single amino acid substitution in ORF1 dramatically decreases L1 retrotransposition and provides insight into nucleic acid chaperone activity. Nucleic Acids Res. 2008;36:5845–5854. doi: 10.1093/nar/gkn554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Januszyk K., Li P.W.-l., Villareal V., Branciforte D., Wu H., Xie Y., Feigon J., Loo J.A., Martin S.L., Clubb R.T. Identification and solution structure of a highly conserved C-terminal domain within ORF1p required for retrotransposition of long interspersed nuclear element-1. J. Biol. Chem. 2007;282:24893–24904. doi: 10.1074/jbc.M702023200. [DOI] [PubMed] [Google Scholar]
- 34.Khazina E., Weichenrieder O. Non-LTR retrotransposons encode noncanonical RRM domains in their first open reading frame. Proc. Natl. Acad. Sci. U.S.A. 2009;106:731–736. doi: 10.1073/pnas.0809964106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Callahan K.E., Hickman A.B., Jones C.E., Ghirlando R., Furano A.V. Polymerization and nucleic acid-binding properties of human L1 ORF1 protein. Nucleic Acids Res. 2012;40:813–827. doi: 10.1093/nar/gkr728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khazina E., Truffault V., Buttner R., Schmidt S., Coles M., Weichenrieder O. Trimeric structure and flexibility of the L1ORF1 protein in human L1 retrotransposition. Nat. Struct. Mol. Biol. 2011;18:1006–1014. doi: 10.1038/nsmb.2097. [DOI] [PubMed] [Google Scholar]
- 37.Martin S.L., Branciforte D., Keller D., Bain D.L. Trimeric structure for an essential protein in L1 retrotransposition. Proc. Natl. Acad. Sci. U.S.A. 2003;100:13815–13820. doi: 10.1073/pnas.2336221100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cook P.R., Jones C.E., Furano A.V. Phosphorylation of ORF1p is required for L1 retrotransposition. Proc. Natl. Acad. Sci. U.S.A. 2015;112:4298–4303. doi: 10.1073/pnas.1416869112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Demers G.W., Matunis M.J., Hardison R.C. The L1 family of long interspersed repetitive DNA in rabbits: sequence, copy number, conserved open reading frames, and similarity to keratin. J. Mol. Evol. 1989;29:3–19. doi: 10.1007/BF02106177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Furano A.V. The biological properties and evolutionary dynamics of mammalian LINE-1 retrotransposons. Prog. Nucleic Acid Res. Mol. Biol. 2000;64:255–294. doi: 10.1016/s0079-6603(00)64007-2. [DOI] [PubMed] [Google Scholar]
- 41.Boissinot S., Furano A.V. The recent evolution of human L1 retrotransposons. Cytogenet. Genome Res. 2005;110:402–406. doi: 10.1159/000084972. [DOI] [PubMed] [Google Scholar]
- 42.Martin S.L. The ORF1 protein encoded by LINE-1: structure and function during L1 retrotransposition. J. Biomed. Biotechnol. 2006:45621. doi: 10.1155/JBB/2006/45621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boissinot S., Furano A.V. Adaptive evolution in LINE-1 retrotransposons. Mol. Biol. Evol. 2001;18:2186–2194. doi: 10.1093/oxfordjournals.molbev.a003765. [DOI] [PubMed] [Google Scholar]
- 44.Khan H., Smit A., Boissinot S. Molecular evolution and tempo of amplification of human LINE-1 retrotransposons since the origin of primates. Genome Res. 2006;16:78–87. doi: 10.1101/gr.4001406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith S.B., Cui Y., Bustamante C. Overstretching B-DNA: the elastic response of individual double-stranded and single-stranded DNA molecules. Science. 1996;271:795–799. doi: 10.1126/science.271.5250.795. [DOI] [PubMed] [Google Scholar]
- 46.Chaurasiya K.R., Paramanathan T., McCauley M.J., Williams M.C. Biophysical characterization of DNA binding from single molecule force measurements. Phys. Life Rev. 2010;7:299–341. doi: 10.1016/j.plrev.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sassaman D.M., Dombroski B.A., Moran J.V., Kimberland M.L., Naas T.P., DeBerardinis R.J., Gabriel A., Swergold G.D., Kazazian H.H. Jr. Many human L1 elements are capable of retrotransposition. Nat. Genet. 1997;16:37–43. doi: 10.1038/ng0597-37. [DOI] [PubMed] [Google Scholar]
- 48.Makarova O., Kamberov E., Margolis B. Generation of deletion and point mutations with one primer in a single cloning step. Biotechniques. 2000;29:970–972. doi: 10.2144/00295bm08. [DOI] [PubMed] [Google Scholar]
- 49.DeLean A., Munson P., Rodbard D. Simultaneous analysis of families of sigmoidal curves: application to bioassay, radioligand assay, and physiological dose-response curves. Am. J. Physiol. 1978;235:E97–E102. doi: 10.1152/ajpendo.1978.235.2.E97. [DOI] [PubMed] [Google Scholar]
- 50.Rajkowitsch L., Schroeder R.e. Dissecting RNA chaperone activity. RNA. 2007;13:2053–2060. doi: 10.1261/rna.671807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chaurasiya K.R., McCauley M.J., Wang W., Qualley D.F., Wu T., Kitamura S., Geertsema H., Chan D.S., Hertz A., Iwatani Y., et al. Oligomerization transforms human APOBEC3G from an efficient enzyme to a slowly dissociating nucleic acid-binding protein. Nat. Chem. 2014;6:28–33. doi: 10.1038/nchem.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Evans J.D., Peddigari S., Chaurasiya K.R., Williams M.C., Martin S.L. Paired mutations abolish and restore the balanced annealing and melting activities of ORF1p that are required for LINE-1 retrotransposition. Nucleic Acids Res. 2011;39:5611–5621. doi: 10.1093/nar/gkr171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.King G.A., Gross P., Bockelmann U., Modesti M., Wuite G.J., Peterman E.J. Revealing the competition between peeled ssDNA, melting bubbles, and S-DNA during DNA overstretching using fluorescence microscopy. Proc. Natl. Acad. Sci. U.S.A. 2013;110:3859–3864. doi: 10.1073/pnas.1213676110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lebrun A., Lavery R. Modelling extreme stretching of DNA. Nucleic Acids Res. 1996;24:2260–2267. doi: 10.1093/nar/24.12.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Burkhard P., Ivaninskii S., Lustig A. Improving coiled-coil stability by optimizing ionic interactions. J. Mol. Biol. 2002;318:901–910. doi: 10.1016/S0022-2836(02)00114-6. [DOI] [PubMed] [Google Scholar]
- 56.Grigoryan G., Keating A.E. Structural specificity in coiled-coil interactions. Curr. Opin. Struct. Biol. 2008;18:477–483. doi: 10.1016/j.sbi.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wei W., Gilbert N., Ooi S.L., Lawler J.F., Ostertag E.M., Kazazian H.H., Boeke J.D., Moran J.V. Human L1 retrotransposition: cis preference versus trans complementation. Mol. Cell. Biol. 2001;21:1429–1439. doi: 10.1128/MCB.21.4.1429-1439.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kroutter E.N., Belancio V.P., Wagstaff B.J., Roy-Engel A.M. The RNA polymerase dictates ORF1 requirement and timing of LINE and SINE retrotransposition. PLoS Genet. 2009;5:e1000458. doi: 10.1371/journal.pgen.1000458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Soifer H.S., Zaragoza A., Peyvan M., Behlke M.A., Rossi J.J. A potential role for RNA interference in controlling the activity of the human LINE-1 retrotransposon. Nucleic Acids Res. 2005;33:846–856. doi: 10.1093/nar/gki223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bogerd H.P., Wiegand H.L., Hulme A.E., Garcia-Perez J.L., O'Shea K.S., Moran J.V., Cullen B.R. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc. Natl. Acad. Sci. U.S.A. 2006;103:8780–8785. doi: 10.1073/pnas.0603313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.