
Table 1. Linear combinations of shifts and reduction of measurement time in GFT NMR experiments.
| Group* | Experiment† | Linear combinations of chemical shifts‡ | Minimal measurement time, h§ | Reduction in measurement time¶ |
|---|---|---|---|---|
| I.1∥ | (4,3)D CαβCα(CO)NHN and (4,3)D HNN(CO)CαβCα | 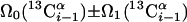 |
3.7 | 14 |
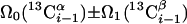 |
||||
| I.2∥ | L-(4,3)D HNN(CO)CαβCα | as in I.1 | 1.5 | 14 |
| I.3** | (5,3)D HαβCαβCα (CO)NHN | 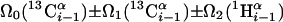 |
12.7 | 75 |
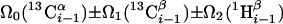 |
||||
| I.4†† | (6,3)D HαβCαβCαCONHN | 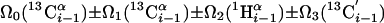 |
29.0 | 600 |
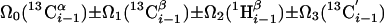 |
||||
| II.1∥‡‡ | (4,3)D HNNCαβCα | 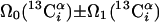 |
3.7 | 14 |
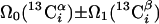 |
||||
| II.2∥‡‡ | L-(4,3)D HNNCαβCα | as in II.1 | 1.5 | 14 |
| III.1§§ | (5,3)D HCC-CH | Ω0(13C(2)) ± Ω1(13C(2)) ± Ω2(1H(2)) “diagonal peak” | 24.0 | 423 |
| Ω0(13C(2)) ± Ω1(13C(1)) ± Ω2(1H(1)) “cross peak” | ||||
| III.2¶¶ | (4,2)D HCCH | as in III.1 | 0.3 | 30 |
The roman number indicates the group of the experiment (see text)
Nuclei for which (i) shifts are sampled in a single GFT dimension are underlined, (ii) shifts are measured in a separate indirect dimension are italicized, and (iii) shifts are not measured are given in parentheses. The nucleus for which the shift is detected in quadrature in the GFT dimension is shown in bold type. Longitudinal 1H relaxation optimization is indicated as “l-”
Linear combinations are measured in the GFT dimension of the respective basic spectra, with Ω0 being the chemical shift detected in quadrature. The lines of the chemical shift multiplets are centered about this shift, and subscript i — 1 and i indicate two neighboring amino acid residues. Linear combinations measured in first-order central peaks are obtained by omitting the right-most shift of a given linear combination, e.g. Ω2(1H) for (5,3)D HαβCαβCα(CO)NHN. Accordingly, linear combinations measured in higher-order central peak spectra are obtained by successive omission of shifts (9)
Minimal measurement times are calculated for the radio-frequency pulse schemes shown in Figs. 7—11, assuming that the experiments are recorded at 600-MHz 1H resonance frequency with the following acquisition parameters: 0.7-s relaxation delay between scans, one scan per free induction decay with 0.05-s acquisition time each, and t1max(GFT) = 6.5 ms or 4.2 ms for aromatic (4,2) HCCH (III.2). Spectral widths and maximal evolution times of other indirect dimensions are provided in the footnotes of the individual experiments
The reduction in minimal measurement time is given relative to the parent ND experiment. The value given for each experiment corresponds to the ε value defined in ref. 9 and represents the ratio of the number of free induction decays of the parent experiment divided by the number of free induction decays of the GFT experiment
No first-order central peak detection is required (see text). SW(GFT) = 12,000 Hz; t2max(15N) = 12 ms, SW(15N) = 2,000 Hz
SW(GFT) = 15,000 Hz; t2max(15N) = 12 ms, SW(15N) = 2,000 Hz
SW(GFT) = 16,500 Hz; t2max(15N) = 12 ms, SW(15N) = 2,000 Hz
First-order central peak detection is not required (see text). Only correlations arising from one-bond scalar couplings are considered; smaller two-bond 2JNCα scalar couplings (14) give rise to additional sequential signals as detected in I.1
The chemical shifts of H(1)C(1)—C(2)H(2) moieties are correlated. The carbons indicated in bold and italic type represent the same nucleus (see text). Therefore, second-order central peak detection is not required. SW(GFT) = 25,000 Hz; t2max(13C) = 4.5 ms, SW(13C) = 9,000 Hz
The chemical shifts of H(1)C(1)—C(2)H(2) moieties are correlated. SW(GFT) = 11,000 Hz