

Abstract
Background
Mosquitoes host diverse microbial communities that influence many aspects of their biology including reproduction, digestion, and ability to transmit pathogens. Unraveling the composition, structure, and function of these microbiota can provide new opportunities for exploiting microbial function for mosquito-borne disease control.
Methods
MiSeq® sequencing of 16S rRNA gene amplicons was used to characterize the microbiota of adult females of Culex pipiens L. and Cx. restuans Theobald collected from nine study sites in central Illinois.
Results
Out of 195 bacterial OTUs that were identified, 86 were shared between the two mosquito species while 16 and 93 OTUs were unique to Cx. pipiens and Cx. restuans, respectively. The composition and structure of microbial communities differed significantly between the two mosquito species with Cx. restuans hosting a more diverse bacterial community compared to Cx. pipiens. Wolbachia (OTU836919) was the dominant bacterial species in Cx. pipiens accounting for 91 % of total microbiota while Sphingomonas (OTU817982) was the dominant bacterial species in Cx. restuans accounting for 31 % of total microbiota. Only 3 and 6 OTUs occurred in over 60 % of individuals in Cx. pipiens and Cx. restuans, respectively. There was little effect of study site on bacterial community structure of either mosquito species.
Conclusion
These results suggest that the two mosquito species support distinct microbial communities that are sparsely distributed between individuals. These findings will allow investigations of the role of identified microbiota on the spatial and temporal heterogeneity in WNV transmission and their potential application in disease control.
Electronic supplementary material
The online version of this article (doi:10.1186/s13071-016-1299-6) contains supplementary material, which is available to authorized users.
Keywords: Culex pipiens, Culex restuans, Microbiota
Background
The urgent need to develop novel tools for controlling mosquito-borne diseases such as malaria, dengue, Chikungunya, and West Nile Virus (WNV) has stimulated research interest in understanding the structure and function of mosquito microbiota. These studies have ranged from field surveys on the composition and diversity of mosquito microbiota [1–5] to investigations of their impact on host fitness and response to parasitic and viral infections [6–9]. Results from these studies have revealed that adult mosquitoes host a community of natural microbiota [1–3] that contributes to host survival, reproduction, nutrition, and immunity [7, 8, 10–13]. Some endogenous microbes have also been shown to reduce vector lifespan [14, 15] and susceptibility to parasitic and viral agents [7, 16–18]. In addition, certain mosquito microbial symbionts can be genetically modified to express molecules that inhibit pathogen transmission [19–21]. These findings have provided the impetus for controlling mosquito-borne diseases through manipulation of mosquito-associated microbiota [22]. However, our limited understanding of the composition, diversity, and function of mosquito microbiota continues to be a critical barrier to practical application of microbes for the control of mosquito-borne diseases. Specifically, while the natural microbiota of malaria vectors (Anopheles spp.) and dengue and Chikungunya (Aedes spp.) vectors [1, 3, 6, 12, 23, 24] are well characterized, little is known about the natural microbiota of mosquitoes from other genera such as Culex and Mansonia [2, 4, 25, 26], despite their role as vectors of human and animal diseases such as WNV and lymphatic filariasis.
In this study we employ high throughput MiSeq® sequencing of 16S rRNA to investigate the diversity and structure of bacterial communities of adult females of Culex pipiens and Culex restuans mosquitoes collected in central Illinois from three of the dominant land-use categories across the Midwestern USA: urban woodlots, rural woodlots, and agricultural land [27]. Despite the role of the two mosquito species as the primary vectors of WNV in northern United States, little is known about the composition and structure of their microbiota. Understanding the microbiota of these two vector species will allow investigations of their influence on WNV transmission and provide new insights into how these microbiota may be exploited for disease control.
Methods
Study sites
The studies were conducted in Champaign County, Illinois where WNV is well established [27]. Nine study sites representing three urban woodlots (Weaver Park, Busey Woods, South Farms), three rural woodlots (Collins Woods, Trelease Woods, Brownfield) and three agricultural sites (sites 1-3) were selected for this study (Fig. 1). These sites were chosen to reflect the dominant land-use categories common to the Midwestern United States. GPS coordinates for these sites are presented in Additional file 1: Table S1. Agricultural sites are row crop farms rotated with corn and soybeans. At the time of sampling, these sites were occupied by rows of corn plantations. Weaver Park is a 24.28-ha land located approximately 4.35 km NE of University of Illinois at Urbana-Champaign (UIUC) campus. The park contains an in-progress 2.02-ha woodland/savanna restoration, 14.16-ha planted with prairie and native grasses and a watershed management wetland. Busey Woods is a 23.88-ha bottomland Oak-Hickory forest located at the north end of Crystal Lake Park approximately 1.6 km NE of UIUC campus. South Farms is a 8.15-ha forest located approximately 2.41 km SE of UIUC campus. The site is characterized by high canopy trees composed of maple, sycamore, pine, oak, and a dense understory of grasses and invasive Amur honeysuckle (Lonicera maackii).
Fig. 1.
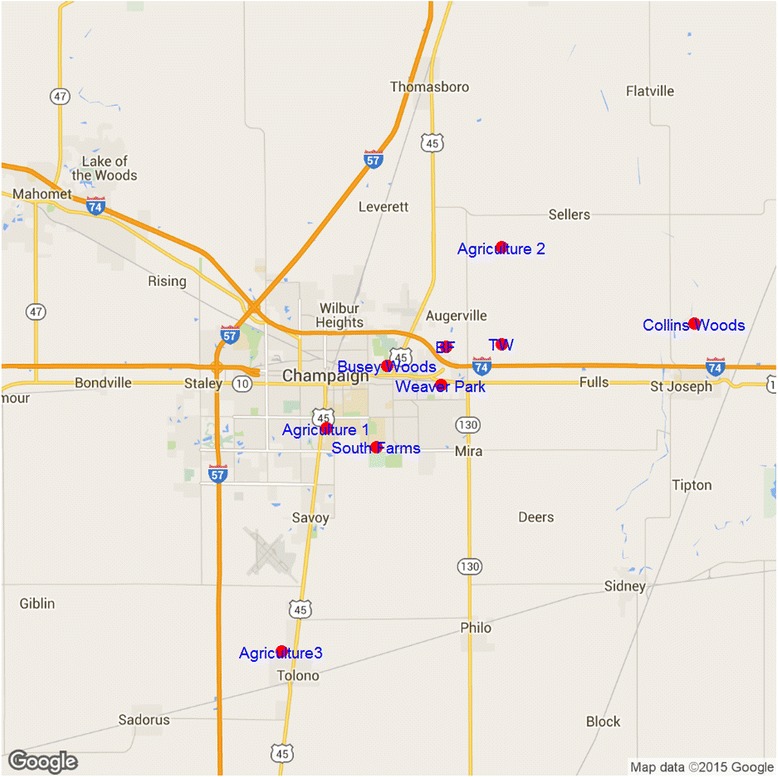
Map of Champaign County showing the location of the nine study sites. TW and BF represent the Trelease Woods and Brownfield Woods study sites respectively
Collins Woods is a 5.3-ha second growth deciduous forest with a livestock grazing history. The site is located about 19.31 km NE of the UIUC campus. The eastern half is a mix of older “wolf” trees and woody successional species. The western half is a more mature oak/hickory/ash/osage orange woods with a small area of old river oxbow bottom land. Bush honeysuckle (Lonicera spp.) dominates the understory in many areas.
Trelease Woods is a 28.8-ha old growth deciduous forest located approximately 9.66 km northeast of the UIUC campus. The site is characterized by a high closed canopy with a moderately dense understory and primarily composed of mature oak, ash, hackberry, and maple species. A detailed description of this study site is provided elsewhere [28].
Brownfield Woods is a 26.1-ha “virgin” deciduous upland forest located in Urbana, Illinois, approximately 6.44 km NE of the UIUC campus. It is primarily a mature oak/ash/maple forest with a high, closed canopy and fairly open understory. Sugar maple has rapidly become the dominant tree species. The woods are a remnant of a much larger prairie grove that was present at settlement times. A small creek that is fed by runoff and field tiles, runs diagonally through the woods. Agricultural land borders the west side and across the road on part of the east side.
Mosquito collection
Adult females of Cx. pipiens and Cx. restuans were collected from the nine study sites once every two weeks between June 19, 2014 and September 10, 2014. The collections were made using Centers for Disease Control (CDC) gravid traps (John W. Hock Co., Gainesville, FL) baited with 5-day old grass infusion. Attempts were made to distinguish Cx. pipiens and Cx. restuans mosquitoes based on the presence of two pale spots that are present on the scutum of Cx. restuans but are absent in Cx. pipiens [29]. However, this effort was abandoned because most samples had lost their scales and therefore mosquito samples identified as Cx. pipiens/Cx. restuans were classified as Culex spp. (see Additional file 1: Table S1) and stored at -80 °C until further processing.
DNA extraction and 16 s rRNA library preparation
One hundred and forty seven (147) randomly selected non-blood fed mosquito samples representing all the nine study sites were retrieved from -80 °C freezer, rinsed three times in sterile water and surface disinfected in 70 % ethanol for 10 min. The ethanol was then removed by rinsing the mosquitoes five times in sterile water and once in dilute (0.8 %) saline solution [3]. DNA from mosquito samples was extracted using MoBio PowerSoil isolation kit (MO BIO, Carlsbad, CA). Briefly, individual mosquitoes were ground in 500 μL of solution from PowerBead tubes of MoBio PowerSoil kit and the entire homogenate was used for DNA extraction as per manufacturer’s recommendation. DNA was quantified using NanoDrop 1000 (Thermo Scientific, Pittsburg, PA) and its quality assessed using Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). A portion of DNA samples was used for mosquito species identification using real-time polymerase chain reaction [30]. These yielded 54 and 93 Cx. pipiens and Cx. restuans samples respectively. For bacterial characterization, we targeted the V3-V5 hypervariable region of the 16S rRNA gene (694 base pairs) using the following primer set: forward 5`-CCTACGGGAGGCAGCAG-3` and reverse 5`-CCGTCAATTCMTTTRAGT-3` [31].
Library preparation and sequencing were conducted at the W. M. Keck Center for Comparative and Functional Genomics at the University of Illinois at Urbana-Champaign (Additional file 2: Appendix S1). Briefly, DNA of each mosquito sample was amplified using the above primer set on the Fluidigm® microfluidics quantitative PCR platform with appropriate linkers and sample barcodes. The final Fluidigm® libraries were pooled in equimolar ratio for sequencing. The final denatured library pool was spiked with 10 % non-indexed PhiX control library (Illumina®) and sequenced by 2 x 300 nt paired-end sequencing on the Illumina® MiSeq® V3 Bulk system. The PhiX control library provides a balanced genome for calculation of matrix, phasing and pre-phasing, which are essential for accurate base-calling. The libraries were sequenced from both ends of the molecules to a total read length of 300 nt from each end. Cluster density was 964 k/mm2 with 85.9 % of clusters passing filter.
We present the results for the forward reads only because reverse reads, which are not independent from the forward reads, showed the same patterns among samples (see Additional file 3: Figure S1, Additional file 4: Figure S2 and Additional file 5: Table S2). This is expected because single-end reads are known to be sufficient to observe the same relationships among samples that are revealed with paired-end reads [32].
Illumina OTU analysis and statistics
The QIIME version 1.9.0 pipeline [33] was used to demultiplex and quality filter the forward and reverse reads using defaults [34]. Barcodes and primer sequences were removed using Fastx toolkit operated in QIIME. The operational taxonomic units (OTUs) were picked and identified at 97 % similarity using Greengenes reference [35] sequence database (http://greengenes.lbl.gov/cgi-bin/nph-index.cgi) downloaded on May 2015. Due to variations in the number of sequences between samples (range 5-210,445 sequences), read depth was rarefied to 2000 reads per sample to retain adequate samples for statistical analysis and to standardize the sampling effort [36]. OTUs accounting for <0.005 % of the total sequences were discarded to reduce the problem of spurious OTUs which may result from random sequencing errors and are likely to overestimate the overall diversity [6, 34]. Alpha diversity metrics including Shannon diversity index, observed species, chao1, and evenness were generated in QIIME and analysis of variance (ANOVA) with Tukey adjustments was used to test the effect of site and mosquito species on these indices using SPSS statistical package (IBM Inc.). A subset of the data containing equal numbers of Cx. pipiens and Cx. restuans was also analyzed, and overall results were similar to those of data containing all samples indicating that sample size disparities did not contribute to observed differences in microbial diversity and richness between the two mosquito species. Bacterial communities were visualized using non-metric multi-dimensional scaling (NMDS) plots generated using vegan package [37] in R [38]. Non-parametric multivariate community analysis including indicator species analysis and Multi-Response Permutation Procedures (MRPP) were conducted using PC-ORD version 6.08 [39]. MRPP was used to test for differences in microbial communities between mosquito species and study sites while indicator species analysis was used to identify bacterial species that are strongly associated with each of the two mosquito species. Indicator values range from 0 to 1 with a value of 1 indicating that the species occurs in all samples of a treatment and are specific to those samples [40].
Results
Bacterial species composition differs between Cx. pipiens and Cx. restuans
We analyzed the V3-V5 hyper-variable region of 16S rRNA gene to estimate the composition and structure of bacterial communities of field-collected adult females of Cx. pipiens and Cx. restuans. A total of 50 and 84 Cx. pipiens and Cx. restuans females, respectively had at least 2000 16S rRNA sequences and were retained for further analyses. After rarefaction and quality filtering, 268,000 16S rRNA sequences were retained and classified into Operational Taxonomic Units (OTUs). In total, there were 195 bacterial OTUs belonging to 9 phyla (including one unclassified phyla) (Table 1) and 54 families. Of the 195 OTUs, 188 (96.4 %) were classified to the family level while only 135 (69.2 %) OTUs were assigned to the genera rank. Only 3 (OTU836919-Wolbachia, OTU817982-Sphingomonas and OTU6118-Wolbachia) and 6 OTUs (OTU836919-Wolbachia, OTU817982-Sphingomonas, OTU4323871-Methylobacterium komagatae, OTU573035-Alicyclobacillus, OTU4363508-Sphingomonadaceae and OTU17329- Sphingomonadaceae) occurred in over 60 % of individuals in Cx. pipiens and Cx. restuans respectively. Overall, OTU836919-Wolbachia and OTU817982-Sphingomonas occurred in 98 % and 90 % of all samples. The phylum Proteobacteria was the most dominant in both mosquito species accounting for 99 % of total microbiota in Cx. pipiens and 81 % of total microbiota in Cx. restuans (Table 1). Alphaproteobacteria and Gammaproteobacteria were the most dominant subdivisions of phylum Proteobacteria accounting for 94 % and 4 % of total microbiota respectively in Cx. pipiens and 56 % and 21 % of total microbiota respectively in Cx. restuans (Table 1). Firmicutes was the second most common phylum in both mosquito species accounting for 0.57 % of total bacteria in Cx. pipiens and 13 % of total bacteria in Cx. restuans (Table 1).
Table 1.
Phylum-level classification of bacterial communities from Cx. pipiens and Culex restuans
| Cx. pipiens | Cx. restuans | ||||
|---|---|---|---|---|---|
| Phylum | Class | #OTUs | Relative abundance (%) | #OTUs | Relative abundance (%) |
| Acidobacteria | Solibacteres | 0 | 0.00 | 2 | 0.02 |
| Actinobacteria | Actinobacteria | 3 | 0.24 | 9 | 2.90 |
| Thermoleophilia | 1 | 0.01 | 0 | 0.00 | |
| Bacteroidetes | Bacteroidia | 0 | 0.00 | 2 | 0.21 |
| Cytophagia | 1 | 0.01 | 2 | 0.03 | |
| Flavobacteriia | 3 | 0.19 | 2 | 1.48 | |
| Sphingobacteriia | 0 | 0.00 | 2 | 0.61 | |
| Chloroflexi | Ellin6529 | 1 | 0.01 | 0 | 0.00 |
| Cyanobacteria | Chloroplast | 0 | 0.00 | 2 | 0.54 |
| Oscillatoriophycideae | 0 | 0.00 | 1 | 0.20 | |
| Firmicutes | Bacilli | 11 | 0.53 | 15 | 11.23 |
| Clostridia | 5 | 0.04 | 13 | 1.56 | |
| Proteobacteria | Alphaproteobacteria | 32 | 94.37 | 45 | 55.70 |
| Betaproteobacteria | 16 | 0.17 | 31 | 3.81 | |
| Deltaproteobacteria | 1 | 0.30 | 0 | 0.00 | |
| Gammaproteobacteria | 28 | 4.14 | 51 | 21.13 | |
| Spirochaetes | Spirochaetes | 0 | 0.00 | 1 | 0.45 |
| WPS-2 | Unclassified | 0 | 0.00 | 1 | 0.14 |
| Total | 102 | 100.00 | 179 | 100.00 | |
At the family level, Rickettsiaceae accounted for 91 % of total microbiota in Cx. pipiens followed by Enterobacteriaceae (4 %) and Sphingomonadaceae (3 %). In Cx. restuans, the three most abundant families were Sphingomonadaceae (34 %), Enterobacteriaceae (19 %), and Methylobacteriaceae (14 %). The top 20 bacterial families across study sites represented 97–100 % of total microbiota in Cx. pipiens and 84–99 % of total microbiota in Cx. restuans (Fig. 2). The relative abundance of the 20 families varied markedly by mosquito species and study site (Fig. 2). Overall, only 9 families had a relative abundance equal to or greater than 1 % (Fig. 2).
Fig. 2.
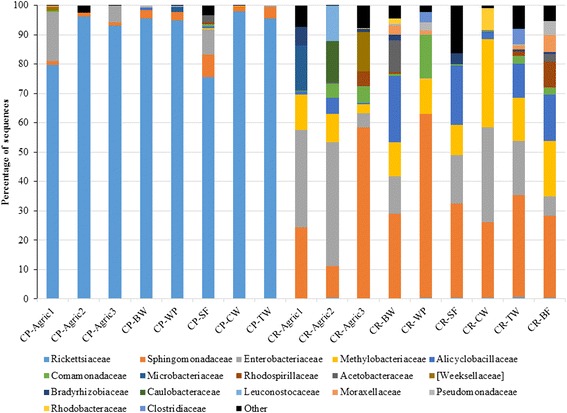
Relative abundance of the top 20 bacterial families in Cx. pipiens and Cx. restuans samples from different study sites. (CP = Cx. pipiens, CR = Cx. restuans, Agric = Agriculture, BW = Busey Wood, WP = Weaver Park, SF = South Farms, CW = Collins Woods, TW = Trelease Woods, and BF = Brownfield Woods)
Eighty six bacterial OTUs were shared between the two mosquito species but none were present in all samples. These shared bacterial OTUs were from four phyla: Proteobacteria (67 OTUs), Firmicutes (14 OTUs), Bacteroidetes (3 OTUs), and Actinobacteria (2 OTUs) (Additional file 6: Table S3). Sixteen bacterial OTUs were unique to Cx. pipiens and 93 bacterial OTUs were unique to Cx. restuans (Additional file 6: Table S3).
Bacterial species diversity and richness is higher in Cx. restuans than in Cx. pipiens
To determine the expected richness in each of the two mosquito species, we calculated Chao1 estimator based on OTUs abundance (Table 2). Overall, we were able to detect 80 % (range 62–100 %) and 69 % (54–79 %) of expected number of OTUs in Cx. pipiens and Cx. restuans, respectively indicating an underestimation of gut microbial diversity. On average, we estimated that Cx. pipiens contains 11 bacterial OTUs (range 5–15) while Cx. restuans contains 22 bacterial OTUs (range 18–29). In addition to the total number of OTUs and the Chao1 richness estimator to compare sample microbial diversity, we calculated the Shannon index to measure OTU richness and homogeneity in mosquito samples. These estimates indicated that bacterial OTUs were significantly more diverse and equitably distributed in Cx. restuans compared to Cx. pipiens in all study sites (Table 2). The top 20 bacterial OTUs were heterogeneously distributed across study sites accounting for 93–99.5 % of total microbiota in Cx. pipiens and 69–87 % of total microbiota in Cx. restuans (Fig. 3). Lower OTU diversity and evenness in Cx. pipiens was driven by the dominance of Wolbachia spp. (OTU836919), which accounted for between 76 % and 98 % of total bacteria in this mosquito species (Fig. 3). Overall, each of the remaining OTUs accounted for less than 0.75 % of total bacteria in Cx. pipiens with exception of OTU817982 (Sphingomonas spp.) and OTU3101394 (Enterobacteriaceae), which accounted for 2.8 % (range 1.1–7.3 %) and 2.3 % (0–12 %) of total bacteria respectively. In contrast, the most dominant OTU in Cx. restuans was OTU817982-Sphingomonas spp. (31 %) and was more abundant in two study sites, Weaver (61 %) and agriculture site 3 (51 %), and less common in agriculture site 2 (10 %, Fig. 3). The second and third most abundant OTUs in Cx. restuans were OTU4323871 (Methylobacterium spp.) and OTU573035 (Alicyclobacillus spp.), which accounted for 10 % (range 0.4–29 %) and 9 % (range 0.1–23 %) of total bacteria, respectively (Fig. 3). Overall, only 12 OTUs had an overall relative abundance equal to, or greater than, 1 % (Fig. 3).
Table 2.
Biodiversity (Shannon, evenness) and richness estimators of Cx. pipiens and Cx. restuans microbiota (± standard error)
| Mosquito species | Study site | Shannon | Equitability | Observed species | Chao1 |
|---|---|---|---|---|---|
| Cx. pipiens | Agriculture1 | 0.65 ± 0.14 | 0.23 ± 0.05 | 7.36 ± 0.62 | 9.45 ± 0.88 |
| Agriculture2 | 0.24 ± 0.11 | 0.09 ± 0.04 | 6.33 ± 1.20 | 8.00 ± 2.65 | |
| Agriculture3 | 0.36 ± 0.26 | 0.11 ± 0.07 | 6.20 ± 1.98 | 8.20 ± 2.82 | |
| Busey Woods | 0.32 ± 0.26 | 0.11 ± 0.08 | 6.50 ± 2.50 | 8.00 ± 4.00 | |
| Weaver Park | 0.29 ± 0.09 | 0.10 ± 0.03 | 7.20 ± 0.85 | 11.55 ± 2.23 | |
| South Farms | 0.75 ± 0.16 | 0.21 ± 0.04 | 10.41 ± 1.14 | 14.78 ± 2.33 | |
| Collins Woods | 0.19 ± 0.00 | 0.06 ± 0.00 | 9.00 ± 0.00 | 9.60 ± 0.00 | |
| Trelease Woods | 0.33 ± 0.00 | 0.14 ± 0.00 | 5.00 ± 0.00 | 5.00 ± 0.00 | |
| Total | 0.53 ± 0.07 | 0.17 ± 0.02 | 8.14 ± 0.54 | 11.33 ± 1.04 | |
| Cx. restuans | Agriculture1 | 2.23 ± 0.17 | 0.55 ± 0.01 | 17.33 ± 4.10 | 23.58 ± 8.25 |
| Agriculture2 | 1.32 ± 0.26 | 0.36 ± 0.07 | 12.57 ± 1.57 | 18.05 ± 2.20 | |
| Agriculture3 | 1.66 ± 0.42 | 0.39 ± 0.10 | 18.80 ± 1.07 | 28.37 ± 3.17 | |
| Busey Woods | 1.46 ± 0.16 | 0.38 ± 0.04 | 14.60 ± 1.17 | 18.37 ± 1.61 | |
| Weaver Park | 1.16 ± 0.09 | 0.29 ± 0.02 | 17.20 ± 1.46 | 25.27 ± 2.82 | |
| South Farms | 1.66 ± 0.15 | 0.42 ± 0.03 | 16.17 ± 1.54 | 20.45 ± 2.33 | |
| Collins Woods | 1.20 ± 0.20 | 0.31 ± 0.06 | 15.67 ± 1.52 | 28.92 ± 5.01 | |
| Trelease Woods | 1.72 ± 0.19 | 0.46 ± 0.05 | 14.64 ± 1.17 | 20.26 ± 2.96 | |
| Brownfield | 1.63 ± 0.18 | 0.41 ± 0.03 | 15.82 ± 1.68 | 25.64 ± 4.31 | |
| Total | 1.56 ± 0.07 | 0.40 ± 0.02 | 15.49 ± 0.55 | 22.37 ± 1.24 |
Fig. 3.
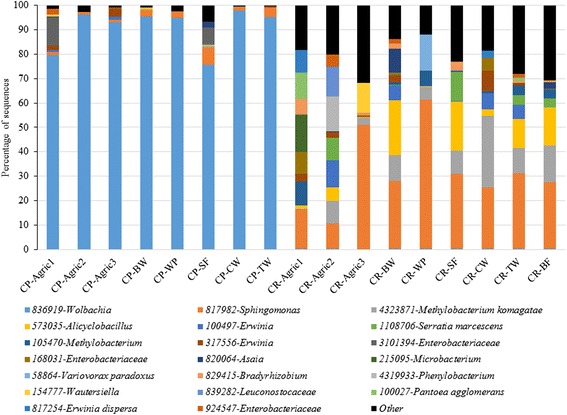
Relative abundance of the top 20 bacterial OTUs in Cx. pipiens and Cx. restuans samples from different study sites. (CP = Cx. pipiens, CR = Cx. restuans, Agric = Agriculture, BW = Busey Wood, WP = Weaver Park, SF = South Farms, CW = Collins Woods, TW = Trelease Woods, and BF = Brownfield Woods)
The two mosquito species have different bacterial community structure
To determine if bacterial communities differed between the two mosquito species and across study sites, nonparametric multivariate community analyses were conducted. MRPP results indicated that midgut bacterial communities of the two mosquito species across all study sites were significantly different with those of Cx. pipiens being more homogeneous compared to those of Cx. restuans (Table 3, Fig. 4). Indicator Species Analysis (ISA) was used to characterize the bacterial OTUs that were strongly associated with one mosquito species over the other. One and 8 indicator species were identified for Cx. pipiens and Cx. restuans respectively (Table 4). Wolbachia (OTU836919), the only indicator species identified for Cx. pipiens had an indicator value close to 1. Only three of 8 indicator species identified for Cx. restuans had an indicator value greater than 0.6 (Table 4). These included OTU817982 (Sphingomonas spp.), OTU573035 (Alicyclobacillus spp.), and OTU4323871 (Methylobacterium komagatae, Table 4).
Table 3.
MRPP results showing differences in bacterial communities between Cx. pipiens and Cx. restuans from each study site. Only 5 of 9 study sites are presented because the remaining sites had either few or no Cx. pipiens samples to facilitate meaningful comparisons
| Location | N(Cp vs Cr) | Ad (Cp vs. Cr) | T | A3 | P 4 |
|---|---|---|---|---|---|
| Agriculture 1 | 11 vs. 3 | 0.31 vs. 0.95 | -5.47 | 0.22 | 0.001 |
| Agriculture 2 | 3 vs. 7 | 0.61 vs. 0.97 | -5.78 | 0.24 | 0.001 |
| Agriculture 3 | 5 vs. 5 | 0.13 vs. 0.69 | -5.62 | 0.44 | 0.002 |
| Busey woods | 2 vs. 15 | 0.09 vs. 0.78 | -5.25 | 0.44 | 0.003 |
| South farms | 17 vs. 12 | 0.38 vs. 0.74 | -13.55 | 0.26 | 0.000002 |
| Weaver Park | 10 vs. 5 | 0.07 vs. 0.53 | -8.90 | 0.57 | 0.00007 |
N number of Cx. pipiens (Cp) and Cx. restuans (Cr) samples
Ad average within group distances for the mosquito species
T test statistic describing separation between groups
A chance-corrected within group agreement as log10
Fig. 4.
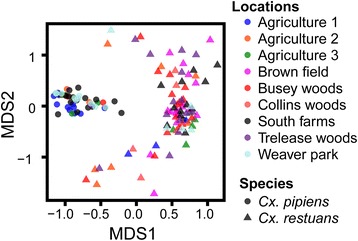
Nonmetric Multidimensional Scaling (NMDS) ordination displaying microbiome communities of Cx. pipiens and Cx. restuans. Microbiomes were distinct between the two mosquito species in each study site
Table 4.
Bacterial OTUs that characterize Cx. pipiens and Cx. restuans mosquitoes
| OTU ID | Phyluma | Classb | Species/Otherc | Indicator valued | P | |
|---|---|---|---|---|---|---|
| Cx. pipiens | 836919 | P | A | Wolbachia spp | 99.6 | 0.0002 |
| Cx. restuans | 817982 | P | A | Sphingomonas spp. | 82.5 | 0.0002 |
| 573035 | F | B | Alicyclobacillus spp. | 60.6 | 0.0002 | |
| 4323871 | P | A | Methylobacterium komagatae | 66.8 | 0.0002 | |
| 4363508 | P | A | Sphingomonadaceae | 54.0 | 0.0002 | |
| 17329 | P | A | Sphingomonadaceae | 54.2 | 0.0002 | |
| 4344371 | P | A | Sphingomonas spp. | 46.2 | 0.0002 | |
| 4378646 | P | A | Sphingomonadaceae | 45.0 | 0.0002 | |
| 4396025 | P | A | Sphingomonas spp. | 40.4 | 0.0002 |
aPhyla abbreviations: P Proteobacteria, F Firmicutes
bClass abbreviations: A Alphaproteobacteria, B Bacilli, G Gammaproteobacteria
c The lowest classification based on greengenes database
dIndicator value computed by Indicator Species Analysis
MRPP analyses revealed that bacterial communities did not differ significantly between Cx. pipiens from different study sites (data not shown). In addition, only 4 of 36 (11 %) study site combinations had significantly different bacterial communities for Cx. restuans indicating weak effects of study site on bacterial community structure (Table 5).
Table 5.
MRPP results showing differences in bacterial communities of Cx. restuans mosquitoes from different study sites
| Location | Ad | N | T | A | P |
|---|---|---|---|---|---|
| Agriculture 2 vs. Agriculture 3 | 0.97 vs. 0.69 | 7 vs. 5 | -2.27 | 0.05 | 0.03 |
| Agriculture 2 vs. Weaver Park | 0.97 vs. 0.54 | 7 vs. 5 | -2.93 | 0.10 | 0.02 |
| Weaver Park vs. South farms | 0.54 vs. 0.79 | 5 vs. 12 | -2.45 | 0.06 | 0.03 |
| Weaver vs. Brownfield | 0.54 vs. 0.77 | 5 vs. 17 | -2.80 | 0.04 | 0.02 |
Ad average within group distances for the mosquito species
N sample size
T test statistic describing separation between groups
A chance-corrected within group agreement as log10
Results are only shown for groups that were significantly different at P ≤ 0.05
Discussion
This study provides the first comprehensive analysis of the microbial consortia of field-collected populations of Cx. pipiens and Cx. restuans, the primary vectors of WNV in northeastern and Midwestern United States [41]. Although both mosquito species were dominated by members of phylum Proteobacteria as reported for other mosquito species [2, 4, 6, 25], their bacterial composition and community structure differed significantly with Cx. restuans hosting more diverse and evenly distributed microbial communities compared to Cx. pipiens. In particular, we found Wolbachia (OTU836919) and Sphingomonas (OTU817982) to be the most abundant and the best indicator species for Cx. pipiens and Cx. restuans, respectively.
Wolbachia (OTU836919) was present in 98 % of all mosquito samples and accounted for 76–98 % of total microbiota in Cx. pipiens and 0.2–0.4 % total microbiota in Cx. restuans. Wolbachia are maternally inherited bacterial endosymbionts that infect numerous invertebrates [42–45] and are known to cause reproductive alterations in their hosts including feminization, parthenogenesis, male killing, and cytoplasmic incompatibility [46]. In Cx. pipiens, Wolbachia causes partial or complete cytoplasmic incompatibility (CI) between infected males and uninfected females or females infected by incompatible strains of Wolbachia [47, 48]. This manipulation confers relative fitness advantage to infected females allowing Wolbachia to rapidly invade host populations [49]. These properties combined with recent reports that certain strains of Wolbachia can shorten the mosquito lifespan [50], reduce their blood feeding success [51], and inhibit their ability to transmit a range of pathogens including dengue, Chikungunya, and malaria [52–56] has generated interest in utilizing Wolbachia for the control of mosquito-borne diseases. However, the utility of this endosymbiont for the control of Culex-borne viruses such as WNV requires further investigations because certain Wolbachia strains may enhance or inhibit replication of WNV in some mosquito species [57, 58]. Therefore, it is likely that Wolbachia infection contributes to the spatial and temporal heterogeneity in WNV transmission that is common in endemic areas.
Wolbachia occurs in most populations of Cx. pipiens [44, 59, 60] and were recently detected in three Cx. restuans individuals [61]. Thus our results suggest that Wolbachia is more widespread in Cx. restuans than previously reported, and is a widespread and dominant bacterial species in Cx. pipiens. It is unclear why the relative abundance of this endosymbiont was low in Cx. restuans. Potential explanations include differences in the physiology of the two mosquito species where environmental conditions in Cx. restuans limit proliferation of Wolbachia or competition with the diverse bacterial communities that were identified in this mosquito species. The latter has been demonstrated in Anopheles gambiae Giles and An. stephensi Liston where native microbiota impeded replication and maternal transmission of Wolbachia [62].
Unlike Cx. pipiens, the most abundant bacterial species in Cx. restuans was Sphingomonas spp. (OTU817982), which accounted for 31 % of total microbiota. This OTU occurred in 90 % of all mosquito samples albeit at low abundance in Cx. pipiens. These findings suggest that this bacterium has either established a symbiotic association with the two mosquito species or is widespread in nature. Existing literature indicates that members of the genus Sphingomonas are widely distributed in both terrestrial and aquatic environments primarily due to their unique abilities to survive under low concentrations of nutrients and to metabolize a wide variety of carbon sources [63]. Sphingomonas spp. are also common in insects and have been isolated in mosquito species from the genera Anopheles, Culex, Aedes and Mansonia [2, 64, 65]. Future studies should investigate their role in mosquito biology.
The majority of bacterial OTUs were sparsely distributed among individuals of the two mosquito species likely due to genetic factors and/or inter-individual variations in diet [1, 2, 66]. This variation may partly account for population and species level variation in vector competence that is commonly observed in nature because certain bacterial species are known to increase [58, 67] or reduce [7, 16–18] vector susceptibility to pathogens. Further studies are needed to investigate the role of identified microbiota on the biology of the two mosquito species including susceptibility to pathogens such as WNV.
The two mosquito species had distinct bacterial profiles although they were collected from the same study sites, are known to utilize the same aquatic habitats for larval development, and tend to blood feed primarily on avian hosts [68–70]. The mechanisms underlying these variations are unclear but we can offer some hypotheses. First, differences in larval foraging and adult sugar feeding may lead to differential exposure of the two mosquito species to microbial communities. Although the larval feeding behavior of Cx. pipiens and Cx. restuans is not well understood, variation in larval feeding behavior of Culex mosquitoes has been reported before [71]. Moreover, all mosquitoes feed on plant sugars and each mosquito species may show preference for specific plants [72, 73]. Previous studies have documented a marked difference in bacterial composition between plant species [74, 75]. Therefore, if the two mosquito species differ in their preferred sugar-meal sources, they may be exposed to different bacterial communities. Second, the two mosquito species may possess different physiologies and thereby inherent differences in the ability to support proliferation and survival of different bacterial species. Third, Cx. pipiens and Cx. restuans show temporal variation in their distribution with Cx. restuans populations peaking earlier (June-July) than those of Cx. pipiens [76]. Temporal variations in the collection of the two mosquito species may partly account for differences in microbial communities given that the relative abundance of bacterial species may vary seasonally [77].
Lastly, although gravid traps are specifically designed to collect gravid females seeking suitable oviposition sites, they also collect a small proportion of host-seeking females [78]. These may consist of newly emerged females as well as older females that may have previously acquired a sugar meal and/or a blood meal. Previous research suggests that newly emerged adults have a more diverse microbial community relative to sugar fed or blood fed mosquitoes [1]. Although we used mosquito samples that were not visibly blood-engorged, we did not record their physiological status. We also did not have prior knowledge of whether the small fraction of host-seeking females that may have been collected in our study had acquired a blood meal or a sugar meal before they were captured. Nevertheless, there is no documented evidence that gravid traps are more effective at collecting gravid females of Cx. pipiens compared to those of Cx. restuans and vice versa. Thus we cannot attribute the differences in microbiota between the two mosquito species to trap bias in the collection of gravid versus host-seeking individuals of the two species.
Conclusions
Our study is the first to provide a comprehensive description of microbial communities associated with two of the most important vectors of WNV in the United States. We found that the two mosquito species harbor distinct microbial communities that are heterogeneously distributed between individuals. Wolbachia was the dominant bacterial species in Cx. pipiens while more diverse and variable microbial communities were observed in Cx. restuans. The results expand the range of mosquito species whose microbial communities have been characterized and will allow further studies to characterize the function of different bacterial species on the biology of the two vector species and their contribution to vectorial capacity. This knowledge will inform identification of microbial communities that can be used to block transmission of Culex-borne pathogens such as WNV.
Data availability
All relevant data are either within the paper and its Additional files or held in a public repository at NCBI under project number SUB1053826. The accession numbers for 16S amplicons from whole Culex pipiens and Culex restuans mosquitoes are SAMN03968624-SAMN03968758.
Acknowledgements
We thank Andrew Donelson, Khamisha Wilson, and Michael Kozak for their assistance in mosquito collection and identification. This study was supported by the Used Tire Fund and the Emergency Public Health Fund from the State of Illinois.
Additional files
GPS coordinates for the nine study sites and the number of Culex spp. mosquitoes collected from each site. (DOCX 12 kb)
Description of Fluidign Access Array Amplification. (DOCX 14 kb)
Relative abundance of the top 20 bacterial families in Cx. pipiens and Cx. restuans samples from different study sites. (CP = Cx. pipiens, CR = Cx. restuans, Agric = Agriculture, BW = Busey Wood, WP = Weaver Park, SF = South Farms, CW = Collins Woods, TW = Trelease Woods, and BF = Brownfield Woods). Results were generated using reverse primer reads. (TIF 72 kb)
Relative abundance of the top 20 bacterial OTUs in Cx. pipiens and Cx. restuans samples from different study sites. (CP = Cx. pipiens, CR = Cx. restuans, Agric = Agriculture, BW = Busey Wood, WP = Weaver Park, SF = South Farms, CW = Collins Woods, TW = Trelease Woods, and BF = Brownfield Woods). Results were generated using reverse primer reads. (TIF 73 kb)
Biodiversity (Shannon, evenness) and richness estimators of Cx. pipiens and Cx. restuans microbiota (± standard error). The data was generated using reverse reads. (DOC 50 kb)
Taxonomy of bacterial OTUs that were shared between Cx. pipiens and Cx. restuans or were unique to either species. (XLSX 17 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EJM and JB designed the study. EJM, CHK, EMB, and MHS processed and analyzed the data. EJM wrote the manuscript. All authors read and approved the final version of the manuscript.
Contributor Information
Ephantus J. Muturi, Email: ephajumu@yahoo.com
Chang-Hyun Kim, Email: maraychk@illinois.edu.
Jeffrey Bara, Email: jay.bara@gmail.com.
Elizabeth M. Bach, Email: ebach@illinois.edu
Madhura H. Siddappaji, Email: siddappaji.madhura@gmail.com
References
- 1.Wang Y, Gilbreath TM, 3rd, Kukutla P, Yan G, Xu J. Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya. PLoS One. 2011;6:e24767. doi: 10.1371/journal.pone.0024767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Osei-Poku J, Mbogo CM, Palmer WJ, Jiggins FM. Deep sequencing reveals extensive variation in the gut microbiota of wild mosquitoes from Kenya. Mol Ecol. 2012;21:5138–5150. doi: 10.1111/j.1365-294X.2012.05759.x. [DOI] [PubMed] [Google Scholar]
- 3.Zouache K, Raharimalala FN, Raquin V, Tran-Van V, Raveloson LH, Ravelonandro P, et al. Bacterial diversity of field-caught mosquitoes, Aedes albopictus and Aedes aegypti, from different geographic regions of Madagascar. FEMS Microbiol Ecol. 2011;75:377–389. doi: 10.1111/j.1574-6941.2010.01012.x. [DOI] [PubMed] [Google Scholar]
- 4.Duguma D, Rugman-Jones P, Kaufman MG, Hall MW, Neufeld JD, Stouthamer R, et al. Bacterial communities associated with Culex mosquito larvae and two emergent aquatic plants of bioremediation importance. PLoS One. 2013;8:e72522. doi: 10.1371/journal.pone.0072522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gusmao DS, Santos AV, Marini DC, Bacci M, Jr, Berbert-Molina MA, Lemos FJ. Culture-dependent and culture-independent characterization of microorganisms associated with Aedes aegypti (Diptera: Culicidae) (L.) and dynamics of bacterial colonization in the midgut. Acta Trop. 2010;115:275–281. doi: 10.1016/j.actatropica.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 6.Boissiere A, Tchioffo MT, Bachar D, Abate L, Marie A, Nsango SE, et al. Midgut microbiota of the malaria mosquito vector Anopheles gambiae and interactions with Plasmodium falciparum infection. PLoS Pathog. 2012;8:e1002742. doi: 10.1371/journal.ppat.1002742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramirez JL, Souza-Neto J, Torres Cosme R, Rovira J, Ortiz A, Pascale JM, et al. Reciprocal tripartite interactions between the Aedes aegypti midgut microbiota, innate immune system and dengue virus influences vector competence. PLoS Negl Trop Dis. 2012;6:e1561. doi: 10.1371/journal.pntd.0001561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong Y, Manfredini F, Dimopoulos G. Implication of the mosquito midgut microbiota in the defense against malaria parasites. PLoS Pathog. 2009;5:e1000423. doi: 10.1371/journal.ppat.1000423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zouache K, Michelland RJ, Failloux AB, Grundmann GL, Mavingui P. Chikungunya virus impacts the diversity of symbiotic bacteria in mosquito vector. Mol Ecol. 2012;21:2297–2309. doi: 10.1111/j.1365-294X.2012.05526.x. [DOI] [PubMed] [Google Scholar]
- 10.Chouaia B, Rossi P, Epis S, Mosca M, Ricci I, Damiani C, et al. Delayed larval development in Anopheles mosquitoes deprived of Asaia bacterial symbionts. BMC Microbiol. 2012;12:S2. doi: 10.1186/1471-2180-12-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaio Ade O, Gusmao DS, Santos AV, Berbert-Molina MA, Pimenta PF, Lemos FJ. Contribution of midgut bacteria to blood digestion and egg production in Aedes aegypti (Diptera: Culicidae) (L) Parasit Vectors. 2011;4:105. doi: 10.1186/1756-3305-4-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coon KL, Vogel KJ, Brown MR, Strand MR. Mosquitoes rely on their gut microbiota for development. Mol Ecol. 2014;23:2727–2739. doi: 10.1111/mec.12771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Minard G, Mavingui P, Moro CV. Diversity and function of bacterial microbiota in the mosquito holobiont. Parasit Vectors. 2013;6:146. doi: 10.1186/1756-3305-6-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suh E, Mercer, DR, Fu Y, Dobson SL. Pathogenicity of life-shortening Wolbachia in Aedes albopictus after transfer from Drosophila melanogaster. App Environ Microbiol. 2009;7783–7788. [DOI] [PMC free article] [PubMed]
- 15.Iturbe-Ormaetxe I, Walker T, ON SL. Wolbachia and the biological control of mosquito-borne disease. EMBO Rep. 2011;12:508–518. doi: 10.1038/embor.2011.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramirez JL, Short SM, Bahia AC, Saraiva RG, Dong Y, Kang S, et al. Chromobacterium Csp_P reduces malaria and dengue infection in vector mosquitoes and has entomopathogenic and in vitro anti-pathogen activities. PLoS Pathog. 2014;10:e1004398. doi: 10.1371/journal.ppat.1004398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pan X, Zhou G, Wu J, Bian G, Lu P, Raikhel AS, et al. Wolbachia induces reactive oxygen species (ROS)-dependent activation of the Toll pathway to control dengue virus in the mosquito Aedes aegypti. Proc Natl Acad Sci U S A. 2012;109:E23–31. doi: 10.1073/pnas.1116932108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cirimotich CM, Dong Y, Clayton AM, Sandiford SL, Souza-Neto JA, Mulenga M, et al. Natural microbe-mediated refractoriness to Plasmodium infection in Anopheles gambiae. Science. 2011;332:855–858. doi: 10.1126/science.1201618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang S, Ghosh AK, Bongio N, Stebbings KA, Lampe DJ, Jacobs-Lorena M. Fighting malaria with engineered symbiotic bacteria from vector mosquitoes. Proc Natl Acad Sci U S A. 2012;109:12734–12739. doi: 10.1073/pnas.1204158109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riehle MA, Moreira CK, Lampe D, Lauzon C, Jacobs-Lorena M. Using bacteria to express and display anti-Plasmodium molecules in the mosquito midgut. Int J Parasitol. 2007;37:595–603. doi: 10.1016/j.ijpara.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 21.Yoshida S, Ioka D, Matsuoka H, Endo H, Ishii A. Bacteria expressing single-chain immunotoxin inhibit malaria parasite development in mosquitoes. Mol Biochem Parasitol. 2001;113:89–96. doi: 10.1016/S0166-6851(00)00387-X. [DOI] [PubMed] [Google Scholar]
- 22.Ricci I, Valzano M, Ulissi U, Epis S, Cappelli A, Favia G. Symbiotic control of mosquito borne disease. Pathog Glob Hlth. 2012;106:380–385. doi: 10.1179/2047773212Y.0000000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ngo CT, Aujoulat F, Veas F, Jumas-Bilak E, Manguin S. Bacterial diversity associated with wild caught Anopheles mosquitoes from Dak Nong Province, Vietnam using culture and DNA fingerprint. PLoS One. 2015;10:e0118634. doi: 10.1371/journal.pone.0118634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chavshin AR, Oshaghi MA, Vatandoost H, Pourmand MR, Raeisi A, Enayati AA, et al. Identification of bacterial microflora in the midgut of the larvae and adult of wild caught Anopheles stephensi: a step toward finding suitable paratransgenesis candidates. Acta Trop. 2012;121:129–134. doi: 10.1016/j.actatropica.2011.10.015. [DOI] [PubMed] [Google Scholar]
- 25.Chandel K, Mendki MJ, Parikh RY, Kulkarni G, Tikar SN, Sukumaran D, et al. Midgut microbial community of Culex quinquefasciatus mosquito populations from India. PLoS One. 2013;8:e80453. doi: 10.1371/journal.pone.0080453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chandler JA, Liu RM, Bennett SN. RNA shotgun metagenomic sequencing of northern California (USA) mosquitoes uncovers viruses, bacteria, and fungi. Frontiers Microbiol. 2015;6:185. doi: 10.3389/fmicb.2015.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gardner AM, Lampman RL, Muturi EJ. Land use patterns and the risk of West Nile Virus transmission in central Illinois. Vector-Borne Zoonot Dis. 2014;14:338–345. doi: 10.1089/vbz.2013.1477. [DOI] [PubMed] [Google Scholar]
- 28.Muturi EJ, Allan BF, Ricci J. Influence of leaf detritus type on production and longevity of container-breeding mosquitoes. Environ Entomol. 2012;41:1062–1068. doi: 10.1603/EN11301. [DOI] [PubMed] [Google Scholar]
- 29.Harrington LC, Poulson RL. Considerations for accurate identification of adult Culex restuans (Diptera: Culicidae) in field studies. J Med Entomol. 2008;45:1–8. doi: 10.1093/jmedent/45.1.1. [DOI] [PubMed] [Google Scholar]
- 30.Sanogo YO, Kim CH, Lampman R, Novak RJ. A real-time TaqMan polymerase chain reaction for the identification of Culex vectors of West Nile and Saint Louis encephalitis viruses in North America. Am J Trop Med Hyg. 2007;77:58–66. [PubMed] [Google Scholar]
- 31.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A. 2011;108:4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, et al. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods. 2013;10:57–59. doi: 10.1038/nmeth.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gihring TM, Green SJ, Schadt CW. Massively parallel rRNA gene sequencing exacerbates the potential for biased community diversity comparisons due to variable library sizes. Environ Microbiol. 2012;14:285–290. doi: 10.1111/j.1462-2920.2011.02550.x. [DOI] [PubMed] [Google Scholar]
- 37.Oksanen J, Blanchet FG, Kindt R, Legandre P, Minchin PR, O'Hara RB. vegan: Community ecology package. 2013. [Google Scholar]
- 38.R Core Team . R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2015. [Google Scholar]
- 39.McCune B, Mefford MJ. PC-ORD: Multivariate Analysis of Ecological Data, version 6.08. MjM Software Design. OR: Gleneden Beach; 2011. [Google Scholar]
- 40.Dufrene M, Legendre P. Species assemblages and indicator species: the need for a flexible asymmetrical approach. Ecol Monog. 1997;67:345–366. [Google Scholar]
- 41.Turell MJ, Dohm DJ, Sardelis MR, Oguinn ML, Andreadis TG, Blow JA. An update on the potential of north American mosquitoes (Diptera: Culicidae) to transmit West Nile Virus. J Med Entomol. 2005;42:57–62. doi: 10.1093/jmedent/42.1.57. [DOI] [PubMed] [Google Scholar]
- 42.Taylor MJ, Bandi C, Hoerauf A. Wolbachia bacterial endosymbionts of filarial nematodes. Advances Parasitol. 2005;60:245–284. doi: 10.1016/S0065-308X(05)60004-8. [DOI] [PubMed] [Google Scholar]
- 43.Tijsse-Klasen E, Braks M, Scholte EJ, Sprong H. Parasites of vectors--Ixodiphagus hookeri and its Wolbachia symbionts in ticks in The Netherlands. Parasit Vectors. 2011;4:228. doi: 10.1186/1756-3305-4-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sanogo YO, Dobson SL, Bordenstein SR, Novak RJ. Disruption of the Wolbachia surface protein gene wspB by a transposable element in mosquitoes of the Culex pipiens complex (Diptera, Culicidae) Insect Mol Biol. 2007;16:143–154. doi: 10.1111/j.1365-2583.2006.00707.x. [DOI] [PubMed] [Google Scholar]
- 45.Cordaux R, Pichon S, Hatira HB, Doublet V, Greve P, Marcade I, et al. Widespread Wolbachia infection in terrestrial isopods and other crustaceans. Zookeys. 2012;176:123-131. [DOI] [PMC free article] [PubMed]
- 46.Stouthamer R, Breeuwer JAJ, Hurst GDD. Wolbachia pipientis: Microbial manipulator of arthropod reproduction. Ann Rev Microbiol. 1999;53:71–102. doi: 10.1146/annurev.micro.53.1.71. [DOI] [PubMed] [Google Scholar]
- 47.Sinkins SP, Walker T, Lynd AR, Steven AR, Makepeace BL, Godfray HCJ, et al. Wolbachia variability and host effects on crossing type in Culex mosquitoes. Nature. 2005;436:257–260. doi: 10.1038/nature03629. [DOI] [PubMed] [Google Scholar]
- 48.Walker T, Klasson L, Sebaihia M, Sanders MJ, Thomson NR, Parkhill J, et al. Ankyrin repeat domain-encoding genes in the wPip strain of Wolbachia from the Culex pipiens group. BMC Biol. 2007;5:39. doi: 10.1186/1741-7007-5-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Turelli M, Hoffmann AA. Rapid Spread of an inherited incompatibility factor in California Drosophila. Nature. 1991;353:440–442. doi: 10.1038/353440a0. [DOI] [PubMed] [Google Scholar]
- 50.McMeniman CJ, Lane RV, Cass BN, Fong AW, Sidhu M, Wang YF, et al. Stable introduction of a life-shortening Wolbachia infection into the mosquito Aedes aegypti. Science. 2009;323:141–144. doi: 10.1126/science.1165326. [DOI] [PubMed] [Google Scholar]
- 51.Turley AP, Moreira LA, O'Neill SL, McGraw EA. Wolbachia infection reduces blood-feeding success in the dengue fever mosquito, Aedes aegypti. PLoS Negl Trop Dis. 2009;3:e516. doi: 10.1371/journal.pntd.0000516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moreira LA, Iturbe-Ormaetxe I, Jeffery JA, Lu GJ, Pyke AT, Hedges LM, et al. A Wolbachia symbiont in Aedes aegypti limits infection with Dengue, Chikungunya, and Plasmodium. Cell. 2009;139:1268–1278. doi: 10.1016/j.cell.2009.11.042. [DOI] [PubMed] [Google Scholar]
- 53.Walker T, Johnson PH, Moreira LA, Iturbe-Ormaetxe I, Frentiu FD, McMeniman CJ, et al. The wMel Wolbachia strain blocks dengue and invades caged Aedes aegypti populations. Nature. 2011;476:450–453. doi: 10.1038/nature10355. [DOI] [PubMed] [Google Scholar]
- 54.Bian G, Joshi D, Dong Y, Lu P, Zhou G, Pan X, et al. Wolbachia invades Anopheles stephensi populations and induces refractoriness to Plasmodium infection. Science. 2013;340:748–751. doi: 10.1126/science.1236192. [DOI] [PubMed] [Google Scholar]
- 55.Bian G, Zhou G, Lu P, Xi Z. Replacing a native Wolbachia with a novel strain results in an increase in endosymbiont load and resistance to dengue virus in a mosquito vector. PLoS Negl Trop Dis. 2013;7:e2250. doi: 10.1371/journal.pntd.0002250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blagrove MSC, Arias-Goeta C, Failloux AB, Sinkins SP. Wolbachia strain wMel induces cytoplasmic incompatibility and blocks dengue transmission in Aedes albopictus. Proc Natl Acad Sci U S A. 2012;109:255–260. doi: 10.1073/pnas.1112021108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hussain M, Lu GJ, Torres S, Edmonds JH, Kay BH, Khromykh AA, et al. Effect of Wolbachia on replication of West Nile Virus in a mosquito cell line and adult mosquitoes. J Virol. 2013;87:851–858. doi: 10.1128/JVI.01837-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dodson BL, Hughes GL, Paul O, Matacchiero AC, Kramer LD, Rasgon JL. Wolbachia enhances West Nile Virus (WNV) infection in the mosquito Culex tarsalis. PLoS Negl Trop Dis. 2014;8:e2965. doi: 10.1371/journal.pntd.0002965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cornel AJ, McAbee RD, Rasgon J, Stanich MA, Scott TW, Coetzee M. Differences in extent of genetic introgression between sympatric Culex pipiens and Culex quinquefasciatus (Diptera : Culicidae) in California and South Africa. J Med Entomol. 2003;40:36–51. doi: 10.1603/0022-2585-40.1.36. [DOI] [PubMed] [Google Scholar]
- 60.Rasgon JL, Scott TW. An initial survey for Wolbachia (Rickettsiales: Rickettsiaceae) infections in selected california mosquitoes (Diptera: Culicidae) J Med Entomol. 2004;41:255–257. doi: 10.1603/0022-2585-41.2.255. [DOI] [PubMed] [Google Scholar]
- 61.Kang Y, Dempsey B. Investigating the presence of Wolbachia pipientis in various mosquito species. J Exp Sec Sci. 2011;1:1–4. [Google Scholar]
- 62.Hughes GL, Dodson BL, Johnson RM, Murdock CC, Tsujimoto H, Suzuki Y, et al. Native microbiome impedes vertical transmission of Wolbachia in Anopheles mosquitoes. Proc Natl Acad Sci U S A. 2014;111:12498–12503. doi: 10.1073/pnas.1408888111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.White DC, Sutton SD, Ringelberg DB. The genus Sphingomonas: physiology and ecology. Curr Opin Biotechnol. 1996;7:301–306. doi: 10.1016/S0958-1669(96)80034-6. [DOI] [PubMed] [Google Scholar]
- 64.Terenius O, Lindh JM, Eriksson-Gonzales K, Bussiere L, Laugen AT, Bergquist H, et al. Midgut bacterial dynamics in Aedes aegypti. FEMS Microbiol Ecol. 2012;80:556–565. doi: 10.1111/j.1574-6941.2012.01317.x. [DOI] [PubMed] [Google Scholar]
- 65.Djadid ND, Jazayeri H, Raz A, Favia G, Ricci I, Zakeri S. Identification of the midgut microbiota of An. stephensi and An. maculipennis for their application as a paratransgenic tool against malaria. PLoS One. 2011;6:e28484. doi: 10.1371/journal.pone.0028484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Perez-Cobas AE, Maiques E, Angelova A, Carrasco P, Moya A, Latorre A. Diet shapes the gut microbiota of the omnivorous cockroach Blattella germanica. FEMS Microbiol Ecol. 2015;91:fiv022. [DOI] [PubMed]
- 67.Apte-Deshpande A, Paingankar M, Gokhale MD, Deobagkar DN. Serratia odorifera a midgut inhabitant of Aedes aegypti mosquito enhances its susceptibility to Dengue-2 virus. PLoS One. 2012;7:e40401. doi: 10.1371/journal.pone.0040401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Molaei G, Andreadis TG, Armstrong PM, Anderson JF, Vossbrinck CR. Host feeding patterns of Culex mosquitoes and West Nile virus transmission, northeastern United States. Emerg Infect Dis. 2006;12:468–474. doi: 10.3201/eid1203.051004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patrican LA, Hackett LE, Briggs JE, McGowan JW, Unnasch TR, Lee JH. Host-feeding patterns of Culex mosquitoes in relation to trap habitat. Emerg Infect Dis. 2007;13:1921–1923. doi: 10.3201/eid1312.070275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Apperson CS, Harrison BA, Unnasch TR, Hassan HK, Irby WS, Savage HM, et al. Host-feeding habits of Culex and other mosquitoes (Diptera: Culicidae) in the Borough of Queens in New York City, with characters and techniques for identification of Culex mosquitoes. J Med Entomol. 2002;39:777–785. doi: 10.1603/0022-2585-39.5.777. [DOI] [PubMed] [Google Scholar]
- 71.Workman PD, Walton WE. Larval behavior of four Culex (Diptera: Culicidae) associated with treatment wetlands in the southwestern United States. J Vector Ecol. 2003;28:213–228. [PubMed] [Google Scholar]
- 72.Foster WA. Mosquito sugar feeding and reproductive energetics. Annu Rev Entomol. 1995;40:443–474. doi: 10.1146/annurev.en.40.010195.002303. [DOI] [PubMed] [Google Scholar]
- 73.Manda H, Gouagna LC, Kabiru EW, Foster WA, Beier JC, Hassanali A, et al. Discriminative feeding behavior of Anopheles gambiae s.s on different plant species and effects on its survival, fecundity, and vector competence in a malaria endemic area of western Kenya. Am J Trop Med Hyg. 2007;77:293–294. [Google Scholar]
- 74.Yang CH, Crowley DE, Borneman J, Keen NT. Microbial phyllosphere populations are more complex than previously realized. Proc Natl Acad Sci U S A. 2001;98:3889–3894. doi: 10.1073/pnas.051633898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Redford AJ, Bowers RM, Knight R, Linhart Y, Fierer N. The ecology of the phyllosphere: geographic and phylogenetic variability in the distribution of bacteria on tree leaves. Environ Microbiol. 2010;12:2885–2893. doi: 10.1111/j.1462-2920.2010.02258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lampman RL, Slamecka M, Krasavin N, Kunkel KE, Novak R. Culex population dynamics and West Nile virus transmission in east-central Illinois. J Am Mosq Control Assoc. 2006;22:390–400. doi: 10.2987/8756-971X(2006)22[390:CPDAWN]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 77.Remold SK, Purdy-Gibson ME, France MT, Hundley TC. Pseudomonas putida and Pseudomonas fluorescens species group recovery from human homes varies seasonally and by environment. PLoS One. 2015;10:e0127704. doi: 10.1371/journal.pone.0127704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McCardle P, Webbe R, Norden B, Aldrich J. Evaluation of five trapping systems for the surveillance of gravid mosquitoes in Prince Georges County, Maryland. J Am Mosq Control Assoc. 2004;20:254–260. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are either within the paper and its Additional files or held in a public repository at NCBI under project number SUB1053826. The accession numbers for 16S amplicons from whole Culex pipiens and Culex restuans mosquitoes are SAMN03968624-SAMN03968758.