

Abstract
The Gram-positive actinomycete Williamsia sp. ARP1 was originally isolated from the Arabidopsis thaliana phyllosphere. Here we describe the general physiological features of this microorganism together with the draft genome sequence and annotation. The 4,745,080 bp long genome contains 4434 protein-coding genes and 70 RNA genes. To our knowledge, this is only the second reported genome from the genus Williamsia and the first sequenced strain from the phyllosphere. The presented genomic information is interpreted in the context of an adaptation to the phyllosphere habitat.
Electronic supplementary material
The online version of this article (doi:10.1186/s40793-015-0122-x) contains supplementary material, which is available to authorized users.
Keywords: Draft genome, Phyllosphere, Williamsia sp. ARP1, Adaption, Whole genome sequencing, Next generation sequencing, Assembly, Annotation, Arabidopsis thaliana
Introduction
The genus Williamsia was originally proposed by Kämpfer et al. in 1999 [1] to accommodate an unusual mycolic-acid containing actinomycete. Members of the genus Williamsia are Gram-positive, non-spore forming, and form round, orange colonies. Their cell shape is coccoid- or rod-like [2]. The genus Williamsia forms a distinct group within actinomycetes of the suborder Corynebacterineae [3], which also comprises the genera Corynebacterium, Dietzia, Gordonia, Mycobacterium, Nocardia, Rhodococcus, Skermania, TsukamurellaandTuricella. Based on the mycolic-acid profile with carbon chain lengths ranging from 50 to 56, the genus Williamsia is likely to be placed between the genera Gordonia and Rhodococcus [1]. At the time of writing, only one other draft genome of Williamsia sp. D3 was publicly available [4] and nine species of this taxon were recognized with valid scientific names: Williamsia deligens [5], Williamsia faeni [6], Williamsia limnetica [7], Williamsia marianensis [8], Williamsia maris [9], Williamsia muralis [1], Williamsia phyllosphaerae [10], Williamsia serinedens [11] and Williamsia sterculiae [12]. Further this genus has been linked with the degradation of hexahydro-1,3,5-trinitro-1,3,5-triazine in soils as a sole nitrogen source [13], the degradation of carbonyl sulfide in soils [14] and polychlorinated biphenyls in tree habitats [15]. Williamsia was isolated from various sources, including indoor building material [1], human blood [5] and following pulmonary infections [16], oil-contaminated and Antarctic soils [4, 11], extreme environments as glacier ice [17], deep sea sediments of the Mariana Trench [8], hay meadows [6], and the rare soil biosphere [18]. Besides, Williamsia was also reported as an endophyte of grey box eucalyptus tree roots [19] and as an epiphytic bacterium residing in the phyllosphere of white clover [20].
The phyllosphere, known as the aerial surface of plant leaves, is a short-lived environment [21] to diverse microorganisms of various taxonomic groups comprising bacteria, filamentous fungi, yeasts, viruses and protists. The phyllosphere presents a challenging environment for microbial colonizers with respect to climatic conditions, UV radiation, desiccation, water availability, reactive oxygen species, and in terms of antimicrobial compounds produced by the plant or possibly also microbes [21–25]. Additionally, the wax composition of the cuticle, surface characteristics such as stomata and veins affect nutrient availability and leaching, as they are likely to retain more water [23, 26].
Here, we present a summary, classification and general physiological features of the strain Williamsia sp. ARP1 together with the genomic sequencing, assembly, annotation, and its putative adaptions to the phyllosphere.
Organism information
Classification and features
The genus Williamsia belongs to the suborder Corynebacterineae [3] of actinomycetes owing to the presence of mycolic acid in the cell wall [2]. Since 2009, it was assigned to the family Nocardiaceae [27, 28]. Williamsia and other genera of this family form a distinct clade in a 16S rRNA phylogenetic tree as well as by using a combination of phenotypic markers [29]. In order to resolve the taxonomic position of Williamsia sp. ARP1, a 16S rRNA sequence (length of 1504 bp) derived from the assembled genome was compared with the NCBI non-redundant and 16S microbial database using BLASTn [30]. The five nearest sequences with the highest identity (all <100 %), the nine validly described Williamsia species, as well as representative sequences of the suborder Corynebacterineae – Gordonia, Rhodococcus, Dietzia, Mycobacterium, Tsukamurella and Turicella - were used for phylogenetic analysis. A strain of the family Frankineae was chosen as the outgroup. All 16S rRNA sequences were aligned using the SINA web aligner (variability profile: Bacteria) [31] and the phylogenetic tree was assessed using PhyML [32] with a generalised time reversible (GTR) substitution model, gamma distribution and 1000 bootstrap replications. All genera formed distinct clades (except Rhodococcus) and were well supported by bootstrap values ≥50 %. Williamsia formed two well supported distinct clades consisting of five and nine sequences, respectively. Within these clades, however, bootstrap values were weaker, due to low variation between 16S sequences. Closest sequences to Williamsia sp. ARP1 were Williamsia sp. 7B-582, A2-614 and A2-437 (all three originating from sediment), and phylogeny in this subclade could not be resolved better due to a multifurcation (Fig. 1). All three 16S rRNA gene sequences showed a sequence identity of 99.93 % for strain 7B-582, 99.93 % for strain A2-614, 99.64 % for strain A2-437 to Williamsia ARP1. Minimum information about the genome sequence of Williamsia sp. ARP1 (MIGS) is provided in Table 1.
Fig. 1.
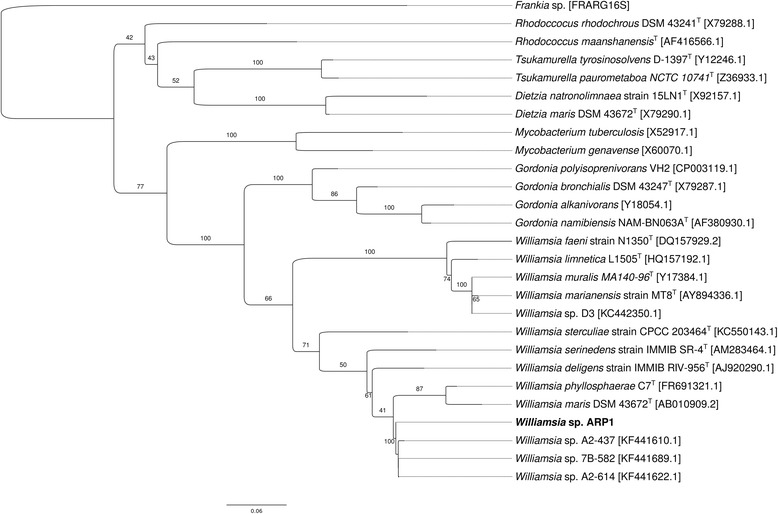
16S rRNA gene based maximum likelihood phylogenetic tree highlighting the position of Williamsia sp. ARP1 within the suborder Corynebacterineae. The tree is based on 16 s rRNA sequences comprising the genera Williamsia, Gordonia, Mycobacterium, Dietzia, Tsukamurella, Rhodococcus and Frankia as an outgroup. The Williamsia sp. ARP1 is highlighted in bold text to show its position. The maximum-likelihood phylogenetic tree was generated using PhyML with the GTR substitution model. Numbers at the nodes are percentages of 1000 bootstrap replicates. Genbank accession numbers are indicated in parentheses; type strains are tagged with a superscripted T. The scale bar represents 0.06 substitutions per nucleotide position
Table 1.
Classification and general features of Williamsia sp. ARP1 [34]
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Classification | Domain Bacteria | TAS [73] | |
| Phylum Actinobacteria | TAS [74] | ||
| Class Actinobacteria | TAS [3] | ||
| Order Actinomycetales | TAS [3, 28, 75, 76] | ||
| Family Nocardiaceae | TAS [3, 28, 75, 76] | ||
| Genus Williamsia | TAS [1] | ||
| Species Williamsia sp. | IDA | ||
| (Type) strain: ARP1 | IDA | ||
| Gram stain | Positive | IDA | |
| Cell shape | Coccoid to rod-like | IDA | |
| Motility | Non-motile | IDA | |
| Sporulation | Non-sporulating | IDA | |
| Temperature range | 4–36 °C | IDA | |
| Optimum temperature | 25–30 °C | IDA | |
| pH range; Optimum | Not reported | NAS | |
| Carbon source | organic carbon | IDA | |
| MIGS-6 | Habitat | Phyllosphere | IDA |
| MIGS-6.3 | Salinity | 1.0–6.0 % | IDA |
| MIGS-22 | Oxygen requirement | Aerobic | IDA |
| MIGS-15 | Biotic relationship | Commensal | IDA |
| MIGS-14 | Pathogenicity | Non-pathogenic | NAS |
| MIGS-4 | Geographic location | Würzburg, Germany | IDA |
| MIGS-5 | Sample collection | 2012 | IDA |
| MIGS-4.1 | Latitude | 49.766556 | IDA |
| MIGS-4.2 | Longitude | 9.931768 | IDA |
| MIGS-4.3 | Depth | Plant surface | IDA |
| MIGS-4.4 | Altitude | 198 m above sea level | IDA |
aEvidence codes - IDA Inferred from Direct Assay, TAS Traceable Author Statement (i.e., a direct report exists in the literature), NAS Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [77]
The colonies of Williamsia sp. ARP1 were orange to red in color on LB agar medium (Fig. 2a). Strain ARP1 was shown to be Gram-positive by Gram staining (data not shown). The cells of the strain were coccoid to rod-like with a diameter of about 1.0–1.5 μm (Fig. 2b). Further, the strain showed positive oxidase and catalase reaction and an aerobic respiratory metabolism. Cells were growing at a temperature range between 4 and 36 °C. Optimal growth was observed between 25 and 30 °C after 3 days on tryptic soy agar, Reasoner’s 2A agar, and nutrient agar (all Oxoid). NaCl tolerance was investigated at different concentrations of NaCl (0.5–8.0 (w/v) %) in tryptic soy broth (TSB, Oxoid) with the cells growing in the presence of 1.0–6.0 % NaCl. The strain lacked motility after 3 days of growth in TSB at 30 °C, as observed under the light microscope. In agreement with this observation, a flagellum was not observed which is further backed up by the lack of flagellar genes (i.e., fliX, flgX and motX genes) on its genome. These findings were consistent with previous descriptions for this genus.
Fig. 2.
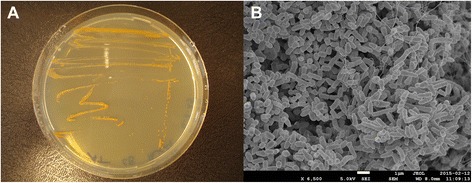
General characteristics of Williamsia sp. ARP1. a The morphology of the colonies after three days of growth on LB-agar at 30 °C. b Image of Williamsia sp. ARP1 using scanning electron microscopy
Genome sequencing information
Genome project history
The organism was selected for sequencing as part of ongoing Arabidopsis phyllosphere microbiology studies [33]. The sequencing project was completed in July 2014 and sequencing data was deposited as a Whole Genome Shotgun (WGS) project in Genbank under the BioProject PRJNA272726 and the accession number JXYP00000000 consisting of 50 contigs (≥1000 bp). The genome sequencing was carried out with a MiSeq (Illumina Inc.) located in-house at our University. A summary of the project information according to the MIGS version 2.0 is shown in Table 2 [34].
Table 2.
Project information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS 31 | Finishing quality | Draft genome |
| MIGS-28 | Libraries used | One Illumina paired-end library (400 bp insert size) |
| MIGS 29 | Sequencing platforms | Illumina MiSeq |
| MIGS 31.2 | Fold coverage | 65× |
| MIGS 30 | Assemblers | SPAdes 3.0, SSPACE 3.0 |
| MIGS 32 | Gene calling method | Prodigal 2.6.1 |
| Genbank ID | JXYP00000000 | |
| Locus Tag | TU34 | |
| GenBank Date of Release | July 1, 2015 | |
| GOLD ID | Gp0118481 | |
| BIOPROJECT | PRJNA272726 | |
| MIGS 13 | Source Material Identifier | DSM 46827 |
| Project relevance | Phyllosphere, Environmental |
Growth conditions and genomic DNA preparation
Several plants were collected from a Landsberg erecta (Ler) population of Arabidopsis thaliana from the Botanical Garden (University of Würzburg, June 2012). Leaf washings [35] were used for inoculation of minimal media with C16 alkane (Sigma-Aldrich) as the sole carbon source in order to enrich for bacteria with the ability to degrade long-chain hydrocarbons. Aliquots were streaked (in duplicate) on agar plates prepared with minimal media and supplemented with C22 alkane (Sigma-Aldrich). This procedure provided a total of 17 isolates, of which most belonged to the genus Rhodococcus and two to genus Williamsia [33].
Williamsia sp. ARP1 was grown in 10 ml Luria-Bertani broth medium (10 g peptone, 5 g yeast extract, 5 g NaCl in 1000 ml demineralized water) for 24 h at 30 °C and rotary shaking at 180 rpm. For genomic DNA isolation, 2 ml of overnight culture were centrifuged at 8000 rpm for 5 min at room temperature. The pellet was rinsed in 1 ml TNE (1 ml 1 M Tris pH 8, 0.2 ml 5 M NaCl, 2 ml 0.5 M EDTA pH8, and 100 ml demineralized water) and resuspended in 270 μl TNEx (TNE, 1 % v/v TritonX-100) and 25 μl lysozyme (10 mg/ml). After a 30 min incubation at 37 °C, 50 μl of proteinase K (20 mg/ml) were added. After an incubation of 2 h and 55 °C, 15 μl of 5 M NaCl and 500 μl of 100 % EtOH were added. The mixture was then centrifuged at 13,000 rpm for 15 min at room temperature, rinsed with 70 % EtOH, air dried and resuspended in 150 μl TE buffer. The quality and quantity of the extracted DNA was evaluated by 0.8 % (w/v) agarose gel electrophoresis, by measuring absorption ratios 260/280 and 260/230 with a Nanodrop 2000c Spectrophotometer (Thermo Fisher Scientific) and an additional Qubit dsDNA HS assay (Life Technologies).
Genome sequencing and assembly
High molecular weight DNA was cleaned with the DNA Clean & Concentrator kit (Zymo Research). The genomic DNA library for the Illumina platform was generated using Nextera XT (Illumina Inc.) according to the manufacturer’s instructions. After tagmentation, size-selection was performed using NucleoMag NGS Clean-up and Size Select (Macherey-Nagel) to obtain a library with median insert-size around 400 bp. After PCR enrichment, the library was validated with a high-sensitivity DNA chip and Bioanalyzer 2100 (both Agilent Technologies, Inc.) and additionally quantified using the Qubit dsDNA HS assay (Life Technologies). Sequencing was performed on a MiSeq device using v2 2 × 250 bp chemistry, and the genome was multiplexed together with ten other bacterial genomes from other sources. Multiplexing was done via dual indexing, with the official Nextera indices N706 and S503 for Williamsia sp. ARP1.
In total, 1,304,294 (mean length 237.86 bp) raw paired-end sequences were subjected to the Trimmomatic software [36] for adapter and quality trimming (mean Phred quality score ≥30), filtering of sequences containing ambiguous bases and a minimum length of 200 bp. Subsequently, human and viral decontamination was excluded using DeconSeq [37]. The 1,287,247 (mean length 236.95 bp) remaining paired-end sequences were assembled with five different tools: a5-miseq [38], IDBA-UD [39], MaSuRCA [40], SPAdes [41] and Velvet [42]. In order to obtain the most reliable contigs, all assemblies were evaluated with QUAST [43], REAPR [44], ALE [45] and Feature Response Curves [46]. According to those evaluations, we have selected SPAdes assembler with enabled pre-correction and k-mer sizes ranging from 15 to 125 (step size of 10) as the best assembly. Obtained contigs were extended with remaining reads where possible. This led to 50 large contigs (≥1000 bp, N50: 140,970 bp, longest contig: 428,355 bp) and an overall genome size of 4,745,080 bp (GC content: 68.63 %). As a final step, the contigs were ordered according to the nearest related complete genome by functional content using Mauve in 12 iterations [47]. As Williamsia sp. D3 was only available as a draft genome, Gordonia bronchialis was used for this step.
Genome annotation
Open reading frames were identified using Prodigal [48] followed by manual correction. The predicted coding sequences were translated into amino acid sequences and searched against COG position-specific scoring matrices obtained from the Conserved Domains Database [49] using RPS-BLAST [30]. Comparisons with TIGRFAM, Pfam, and PANTHER databases were performed with the InterProScan pipeline [50]. Only matches with an e-value ≤1∗10−2, ≥25 % identity and a minimum of 70 % alignment length to the target sequence were maintained. During this run, matches were also mapped to Gene Ontology terms. Additional gene prediction and functional annotation was performed with the Integrated Microbial-Genomes Expert Review [51] and the Rapid Annotation using Subsystem Technology webserver [52, 53]. Features as tRNA, rRNA, ncRNA, transmembrane helices, signal peptides, CRISPR elements and secondary metabolite gene clusters were predicted using tRNAscan-SE [54], RNAmmer [55], INFERNAL [56] and Prokka’s prokaryotic RNA covariance models [57], TMHMM [58], SignalP [59] PILER-CR [60] and antiSMASH [61]. Searching for essential genes [62] was performed using HMMER3 [63]. Ortholog detection between Williamsia sp. ARP1 and three other genomes were carried out with InParanoid [64] whereas the mean percentage of nucleotide identity among the found orthologous genes was calculated using BLASTn. Average nucleotide identities between Williamsia sp. ARP1 and reference genomes were calculated with JSpecies [65].
Genome properties
The Williamsia sp. ARP1 draft genome sequence contained a total of 4,745,080 bp distributed over 50 large contigs (≥1000 bp) with an average GC content of 68.63 %. Of the 4509 predicted genes, 4438 (98.42 %) were protein-coding, and 3505 (77.73 %) annotated with putative function. Pseudogenes were not detected. Genes not linked to a function were annotated as hypothetical or unknown function. Of these, 45 belonged to tRNA genes, 21 to ncRNA genes and five to rRNA genes (Table 3). One operon comprising a 16S rRNA, a 5S rRNA and a 23S rRNA gene was found. However two additional 5S rRNA genes suggest the presence of at least three rRNA operons. Functional assignments using COGs, a total of 2204 (59.59 %) of the coding sequences were classified into 23 different classes (Table 4, Fig. 3). Using TIGRFAM or Pfam, 793 (17.59 %) and 1330 (29.50 %) of the sequences could be classified (Table 3). For testing the genome completeness, a set of 111 essential gene markers was searched and 106 (=95.50 %) of them were present in Williamsia sp. ARP1. Except two marker genes (ribosomal proteins bS18 and bl28), all of them were found only once (Additional file 1). Within the RAST annotation, 1625 sequences were assigned to 402 metabolic subsystems. The highest ranking among the metabolic subsystems are linked to amino acids and derivatives (8.41 %), cofactors, vitamins and pigments (6.25 %), carbohydrates (5.77 %), protein metabolism (5.61 %), fatty acids, and lipids and isoprenoids (4.32 %) followed by stress response (2.86 %), (Fig. 4).
Table 3.
Genome statistics
| Attribute | Value | % of total |
|---|---|---|
| Genome size (bp) | 4,745,080 | 100.00 |
| DNA coding (bp) | 4,347,123 | 91.61 |
| DNA G+C (bp) | 3,256,678 | 68.63 |
| DNA scaffolds | 50 | |
| Total genes | 4509 | 100.00 |
| Protein coding genes | 4438 | 98.42 |
| RNA genes | 71 | 1.57 |
| tRNA genes | 45 | 1.00 |
| rRNA genes | 5 | 0.01 |
| rRNA operons | 1a | |
| Pseudo genes | 0 | 0.00 |
| Genes in internal clusters | NA | |
| Genes with function prediction | 3505 | 77.73 |
| Genes assigned to COGs | 2207 | 48.95 |
| Genes with Pfam domains | 1330 | 29.50 |
| Genes with TIGRFAM domains | 793 | 17.59 |
| Genes with signal peptides | 334 | 7.41 |
| Genes with transmembrane helices | 1140 | 25.28 |
| CRISPR repeats | 2 | 0.04 |
aOnly one RNA operon appears to be complete
Table 4.
Number of genes associated with general COG functional categories
| Code | Value | % age | Description |
|---|---|---|---|
| J | 143 | 3.17 | Translation, ribosomal structure, and biogenesis |
| A | 1 | 0.02 | RNA processing and modification |
| K | 183 | 4.06 | Transcription |
| L | 85 | 1.89 | Replication, recombination, and repair |
| B | 1 | 0.02 | Chromatin structure and dynamics |
| D | 0 | 0.00 | Cell cycle control, Cell division, chromosome partitioning |
| V | 31 | 0.69 | Defense mechanisms |
| T | 74 | 1.64 | Signal transduction mechanisms |
| M | 102 | 2.26 | Cell wall/membrane biogenesis |
| N | 11 | 0.24 | Cell motility |
| U | 18 | 0.40 | Intracellular trafficking and secretion |
| O | 79 | 1.75 | Posttranslational modification, protein turnover, chaperones |
| C | 184 | 4.08 | Energy production and conversion |
| G | 125 | 2.77 | Carbohydrate transport and metabolism |
| E | 226 | 5.01 | Amino acid transport and metabolism |
| F | 66 | 1.46 | Nucleotide transport and metabolism |
| H | 118 | 2.62 | Coenzyme transport and metabolism |
| I | 194 | 4.30 | Lipid transport and metabolism |
| P | 154 | 3.42 | Inorganic ion transport and metabolism |
| Q | 141 | 3.13 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 346 | 7.67 | General function prediction only |
| S | 184 | 4.08 | Function unknown |
| - | 2231 | 49.48 | Not in COGs |
The total is based on the total number of protein coding genes in the genome
Fig. 3.
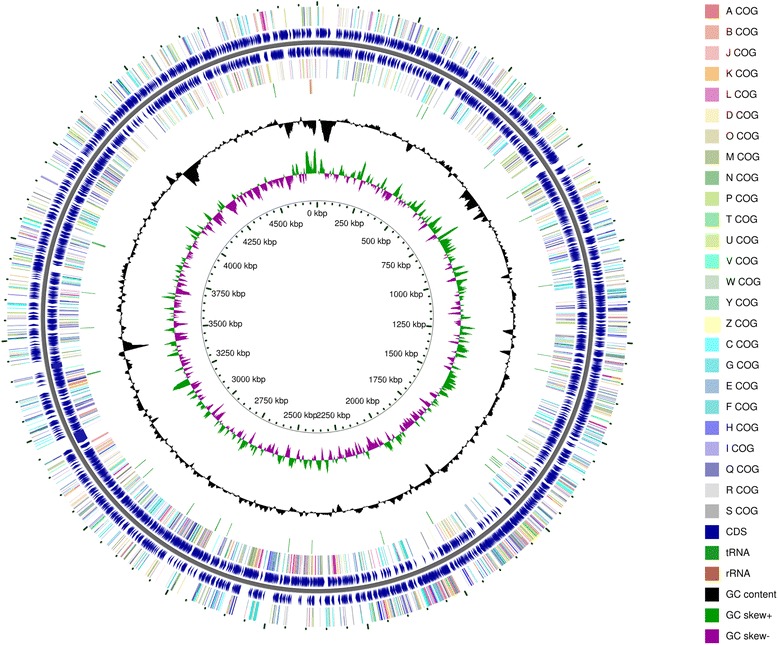
Graphical circular map of the Williamsia sp. ARP1 genome. Starting from the outmost circle and moving inwards, each ring of the circle contains information of the genome: genes on the forward strand (colored according to their COG categories), CDS on the forward strand (blue arrows), CDS on the reverse strand (blue arrows), genes on the reverse strand (colored according to their COG categories), tRNA and rRNA genes on both strands (green and orange), GC content (black), GC skew (green and purple) and genome region by kbp
Fig. 4.
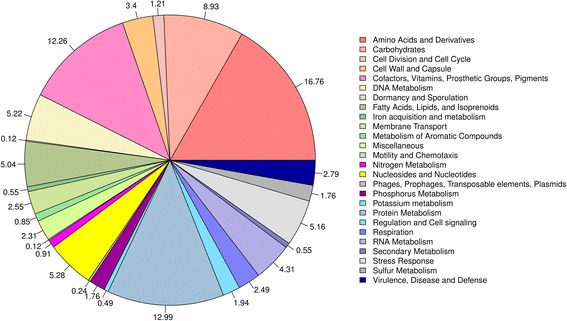
Metabolic subsystems of Williamsia sp. ARP1 annotated through the RAST webserver
Insights from the genome sequence
The genome of Williamsia sp. ARP1 was smaller but displayed a higher CG content (68.63 %) than its nearest relative genomes (Table 5), thus rendering this genome more similar to the G. bronchialis and G. polysoprenivorans VH2 (67.00 and 66.96 %) than to Williamsia sp. D3 (64.60 %) (Table 5). Considering the similarity between 16S rRNA sequences and its placement in the phylogenetic tree, strain ARP1 was however clearly assigned to the genus Williamsia (Fig. 1). With respect to orthologous genes, Williamsia sp. D3 was found to be the most similar strain to Williamsia sp. ARP1 with an average nucleotide identity of these orthologs of 75.53 %. Notably, the differences between Williamsia sp. ARP1 and the Gordonia strains and VH2 (75.17 and 74.84 % identity, respectively) is similar to the difference between the two Williamsia strains (75.53 %), (Additional file 2). Neither the clustering of COG classes nor the average nucleotide identities (ANI) were discriminative between the two genera (Fig. 5, Additional file 3). The ANI values are noticeably lower than the calculated cut-off values for species level identification (95) [66].
Table 5.
Used actinomycete reference genomes in this study
| Species | Strain | Accession number | Genome Size [Mbp] | G+C content |
|---|---|---|---|---|
| Williamsia sp. | D3 | NZ_AYTE000000000.1 | 5.62 | 64.60 |
| Gordonia bronchialis | CP001802.1 | 5.21 | 67.00 | |
| G. polysoprenivorans | VH2 | NC_016906.1 | 5.67 | 66.96 |
Fig. 5.
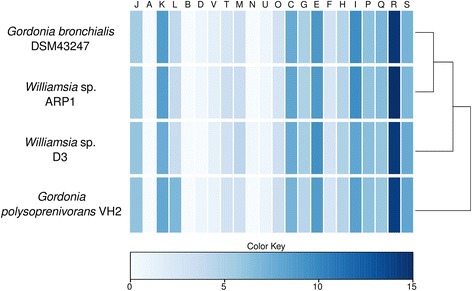
Comparison of COG classes between strain ARP1 and reference genomes. The color keys provide the relative percentage of each COG class per genome. The dendrogram is based on correlation analysis
Extended insights
UV radiation
UV radiation may impose stress on bacteria inhabiting plant leaves. In this context, a cluster of genes synthesizing mycosporins was found. These secondary metabolites are known to protect cells by absorbing UV light without generating reactive oxygen species (ROS) [67, 68]. Additionally, genes involved in the repair of UV-damaged DNA were found, which comprise DNA photolyases, the UvrABC endonuclease enzyme complex, and the DNA helicase II UvrD of the UvrABC system. The red color of Williamsia sp. ARP1 might protect it against photo-oxidative stress as pigmentation is known to be a common feature of phyllosphere colonizers [69]. All genes of the carotenoid biosynthetic pathway were found, consisting of a geranylgeranyl diphosphate synthase, a phytoene synthase, a phytoene desaturase, a carotene desaturase and a lycopene-β-cyclase. The products of this pathway are lycopene and β-carotene, both producing orange to red pigments.
Oxidative stress
Further adaptions to an epiphytic lifestyle are encoded on genes responding to reactive oxygen species (ROS; e.g. hydrogen peroxide, superoxide, hydroperoxil radical), which are products of the plant defense [70, 71]. Here, two genes encoding for glutathione peroxidases, two superoxide dismutases with copper/zinc or manganese as active site, two glutaredoxins, three thioredoxins, and one catalase were found.
Temperature shifts
Regarding temperature shifts, the heatshock chaperones DnaK, DnaJ and GrpE and the cold shock protein CspC were identified.
Uptake
ABC transporters for the uptake of carbohydrates such as ribose, glycerol or maltose, amino acids such as methionine, known plant photosynthates such as fructose, and enzymes for fructose utilization were identified. Also, genes mediating the uptake of choline and subsequent biosynthesis (choline dehydrogenase, betaine-aldehyde dehydrogenase) of the osmoprotectant betaine were found.
Desiccation
Trehalose is a compatible solute and known to prevent cells from desiccation and water loss [72]. Eight genes encoding for the biosynthesis pathway (Malto-oligosyltrehalose synthase, 1,4-alpha-glucan (glycogen) branching enzyme, GH-13-type trehalose-6-phosphate phosphatase, putative glucanase glgE, malto-oligosyltrehalose trehalohydrolase, glycogen debranching enzyme alpha, alpha-trehalose-phosphate synthase, glucoamylase) were identified.
Conclusions
The isolate ARP1 was isolated from the Arabidopsis thaliana phyllosphere. Phylogenetic analysis based on the 16S rRNA gene confirmed its affiliation to the genus Williamsia. However genomic properties also showed close similarities to Gordonia, as derived from GC content, COGs, and average nucleotide identities. Thus, an unequivocal delinearization based on the functional genomics level was not possible, which may be due to the underrepresentation of genomes from this genus. The genomic features of strain ARP1 would be consistent with a lifestyle within the phyllosphere, including putative adaptions to UV radiation, heat and cold shock, desiccation and oxidative stress. With this study, we provide novel genomic insights into the rarely sequenced genus Williamsia and discuss its putative adaptations to the phyllosphere habitat.
Acknowledgements
We thank Dr. Eva E. Reisberg for original isolation of the strain, Christine Gernert and Srikkanth Balasubramanian for technical assistance, Wiebke Sickel for library preparation, the Department of Human Genetics for access to the MiSeq device, and Daniela Bunsen for help with the electron microscope (all University of Würzburg). This work was financially supported by the DFG Graduiertenkolleg GK1342 (TP A8).
Additional files
Identified essential genes in the Williamsia sp. ARP1 genome. (PDF 75 kb)
Orthologous gene comparison of Williamsia sp. ARP1 and three other actinomycete genomes. (PDF 58 kb)
Average nucleotide identities between Williamsia sp. ARP1 and nearest actinomycete genomes. (PDF 54 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HH designed the study, carried out the genome analysis, performed electron microscopy, phylogenetic analysis, and drafted the manuscript. AK carried out the sequencing and helped to draft the manuscript. UHi participated in the study design. PK performed laboratory experiments. MR conceived the study design and participated in its coordination. UHe conceived of the study, participated in its design, coordinated and drafted the manuscript. All authors read and approved the final manuscript.
References
- 1.Kämpfer P, Andersson MA, Rainey FA, Kroppenstedt RM, Salkinoja-Salonen M. Williamsia muralis gen. nov., sp. nov., isolated from the indoor environment of a children’s day care centre. Int J Syst Evol Microbiol. 1999;49:681–7. doi: 10.1099/00207713-49-2-681. [DOI] [PubMed] [Google Scholar]
- 2.Ludwig W, Euzéby J, Schumann P, Busse H-J, Trujillo M, Kämpfer P, et al. Bergey’s manual of systematic bacteriology. Vol. 5, The actinobacteria. New York: Springer; 2012. [Google Scholar]
- 3.Stackebrandt E, Rainey FA, WardRainey NL. Proposal for a new hierarchic classification system, Actinobacteria classis nov. Int J Syst Bacteriol. 1997;47(2):479–91. doi: 10.1099/00207713-47-2-479. [DOI] [Google Scholar]
- 4.Guerrero LD, Makhalanyane TP, Aislabie JM, Cowan DA. Draft genome sequence of Williamsia sp. strain D3, isolated from the Darwin Mountains, Antarctica. Genome Announc. 2014;2(1). doi:10.1128/genomeA.01230-13. [DOI] [PMC free article] [PubMed]
- 5.Yassin AF, Hupfer H. Williamsia deligens sp. nov., isolated from human blood. Int J Syst Evol Microbiol. 2006;56(Pt 1):193–7. doi: 10.1099/ijs.0.63856-0. [DOI] [PubMed] [Google Scholar]
- 6.Jones AL, Payne GD, Goodfellow M. Williamsia faeni sp. nov., an actinomycete isolated from a hay meadow. Int J Syst Evol Microbiol. 2010;60(Pt 11):2548–51. doi: 10.1099/ijs.0.015826-0. [DOI] [PubMed] [Google Scholar]
- 7.Sazak A, Sahin N. Williamsia limnetica sp. nov., isolated from a limnetic lake sediment. Int J Syst Evol Microbiol. 2012;62(6):1414–8. doi: 10.1099/ijs.0.032474-0. [DOI] [PubMed] [Google Scholar]
- 8.Pathom-Aree W, Nogi Y, Sutcliffe IC, Ward AC, Horikoshi K, Bull AT, et al. Williamsia marianensis sp. nov., a novel actinomycete isolated from the Mariana Trench. Int J Syst Evol Microbiol. 2006;56(Pt 5):1123–6. doi: 10.1099/ijs.0.64132-0. [DOI] [PubMed] [Google Scholar]
- 9.Stach JE, Maldonado LA, Ward AC, Bull AT, Goodfellow M. Williamsia maris sp. nov., a novel actinomycete isolated from the Sea of Japan. Int J Syst Evol Microbiol. 2004;54(Pt 1):191–4. doi: 10.1099/ijs.0.02767-0. [DOI] [PubMed] [Google Scholar]
- 10.Kämpfer P, Wellner S, Lohse K, Lodders N, Martin K. Williamsia phyllosphaerae sp. nov., isolated from the surface of Trifolium repens leaves. Int J Syst Evol Microbiol. 2011;61(Pt 11):2702–5. doi: 10.1099/ijs.0.029322-0. [DOI] [PubMed] [Google Scholar]
- 11.Yassin AF, Young CC, Lai WA, Hupfer H, Arun AB, Shen FT, et al. Williamsia serinedens sp. nov., isolated from an oil-contaminated soil. Int J Syst Evol Microbiol. 2007;57(Pt 3):558–61. doi: 10.1099/ijs.0.64691-0. [DOI] [PubMed] [Google Scholar]
- 12.Fang XM, Su J, Wang H, Wei YZ, Zhang T, Zhao LL, et al. Williamsia sterculiae sp. nov., isolated from a Chinese medicinal plant. Int J Syst Evol Microbiol. 2013;63(Pt 11):4158–62. doi: 10.1099/ijs.0.052688-0. [DOI] [PubMed] [Google Scholar]
- 13.Andeer P, Stahl DA, Lillis L, Strand SE. Identification of microbial populations assimilating nitrogen from RDX in munitions contaminated military training range soils by high sensitivity stable isotope probing. Environ Sci Technol. 2013;47(18):10356–63. doi: 10.1021/es401729c. [DOI] [PubMed] [Google Scholar]
- 14.Kato H, Saito M, Nagahata Y, Katayama Y. Degradation of ambient carbonyl sulfide by Mycobacterium spp. in soil. Microbiology. 2008;154(Pt 1):249–55. doi: 10.1099/mic.0.2007/011213-0. [DOI] [PubMed] [Google Scholar]
- 15.Leigh MB, Prouzova P, Mackova M, Macek T, Nagle DP, Fletcher JS. Polychlorinated biphenyl (PCB)-degrading bacteria associated with trees in a PCB-contaminated site. Appl Environ Microbiol. 2006;72(4):2331–42. doi: 10.1128/AEM.72.4.2331-2342.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.del Mar Tomas M, Moure R, Saez Nieto JA, Fojon S, Fernandez A, Diaz M, et al. Williamsia muralis pulmonary infection. Emerg Infect Dis. 2005;11(8):1324–5. doi: 10.3201/eid1108.050439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miteva VI, Sheridan PP, Brenchley JE. Phylogenetic and physiological diversity of microorganisms isolated from a deep greenland glacier ice core. Appl Environ Microbiol. 2004;70(1):202–13. doi: 10.1128/AEM.70.1.202-213.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shade A, Hogan CS, Klimowicz AK, Linske M, McManus PS, Handelsman J. Culturing captures members of the soil rare biosphere. Environ Microbiol. 2012;14(9):2247–52. doi: 10.1111/j.1462-2920.2012.02817.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaewkla O, Franco CM. Rational approaches to improving the isolation of endophytic actinobacteria from Australian native trees. Microb Ecol. 2013;65(2):384–93. doi: 10.1007/s00248-012-0113-z. [DOI] [PubMed] [Google Scholar]
- 20.Stiefel P, Zambelli T, Vorholt JA. Isolation of optically targeted single bacteria by application of fluidic force microscopy to aerobic anoxygenic phototrophs from the phyllosphere. Appl Environ Microbiol. 2013;79(16):4895–905. doi: 10.1128/AEM.01087-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vorholt JA. Microbial life in the phyllosphere. Nat Rev Microbiol. 2012;10(12):828–40. doi: 10.1038/nrmicro2910. [DOI] [PubMed] [Google Scholar]
- 22.Knief C, Delmotte N, Vorholt JA. Bacterial adaptation to life in association with plants–A proteomic perspective from culture to in situ conditions. Proteomics. 2011;11(15):3086–105. doi: 10.1002/pmic.201000818. [DOI] [PubMed] [Google Scholar]
- 23.Leveau JH, Lindow SE. Appetite of an epiphyte: quantitative monitoring of bacterial sugar consumption in the phyllosphere. Proc Natl Acad Sci U S A. 2001;98(6):3446–53. doi: 10.1073/pnas.061629598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindow SE, Brandl MT. Microbiology of the phyllosphere. Appl Env Microbiol. 2003;69(4):1875–83. doi: 10.1128/AEM.69.4.1875-1883.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Newton AC, Gravouil C, Fountaine JM. Managing the ecology of foliar pathogens: ecological tolerance in crops. Ann Appl Biol. 2010;157(3):343–59. doi: 10.1111/j.1744-7348.2010.00437.x. [DOI] [Google Scholar]
- 26.Marcell LM, Beattie GA. Effect of leaf surface waxes on leaf colonization by Pantoea agglomerans and Clavibacter michiganensis. Mol Plant Microbe Interact. 2002;15(12):1236–44. doi: 10.1094/MPMI.2002.15.12.1236. [DOI] [PubMed] [Google Scholar]
- 27.Castellani A, Chalmers AJ. Manual of tropical medicine. 1919. [Google Scholar]
- 28.Zhi XY, Li WJ, Stackebrandt E. An update of the structure and 16S rRNA gene sequence-based definition of higher ranks of the class Actinobacteria, with the proposal of two new suborders and four new families and emended descriptions of the existing higher taxa. Int J Syst Evol Microbiol. 2009;59(3):589–608. doi: 10.1099/ijs.0.65780-0. [DOI] [PubMed] [Google Scholar]
- 29.Goodfellow M, Isik K, Yates E. Actinomycete systematics: an unfinished synthesis. Nova Acta Leopold. 1999. [Google Scholar]
- 30.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 31.Pruesse E, Peplies J, Glockner FO. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics. 2012;28(14):1823–9. doi: 10.1093/bioinformatics/bts252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–21. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 33.Reisberg EE. Der Einfluss von Trichomen und kutikulären Lipiden auf die bakterielle Besiedelung von Arabidopsis thaliana Blättern. PhD thesis: University of Wuerzburg. 2013. [Google Scholar]
- 34.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26(5):541–7. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reisberg EE, Hildebrandt U, Riederer M, Hentschel U. Phyllosphere bacterial communities of trichome-bearing and trichomeless Arabidopsis thaliana leaves. Antonie Van Leeuwenhoek. 2012;101(3):551–60. doi: 10.1007/s10482-011-9669-8. [DOI] [PubMed] [Google Scholar]
- 36.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmieder R, Edwards R. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS One. 2011;6(3) doi: 10.1371/journal.pone.0017288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coil D, Jospin G, Darling AE. A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics. 2014 doi: 10.1093/bioinformatics/btu661. [DOI] [PubMed] [Google Scholar]
- 39.Peng Y, Leung HC, Yiu SM, Chin FY. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28(11):1420–8. doi: 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- 40.Zimin AV, Marcais G, Puiu D, Roberts M, Salzberg SL, Yorke JA. The MaSuRCA genome assembler. Bioinformatics. 2013;29(21):2669–77. doi: 10.1093/bioinformatics/btt476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comp Biol. 2012;19(5):455–77. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zerbino DR. Using the Velvet de novo assembler for short-read sequencing technologies. Curr Protoc Bioinformatics. 2010;Chapter 11:Unit 11 5. doi: 10.1002/0471250953.bi1105s31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29(8):1072–5. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hunt M, Kikuchi T, Sanders M, Newbold C, Berriman M, Otto TD. REAPR: a universal tool for genome assembly evaluation. Genome Biol. 2013;14(5):R47. doi: 10.1186/gb-2013-14-5-r47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clark SC, Egan R, Frazier PI, Wang Z. ALE: a generic assembly likelihood evaluation framework for assessing the accuracy of genome and metagenome assemblies. Bioinformatics. 2013;29(4):435–43. doi: 10.1093/bioinformatics/bts723. [DOI] [PubMed] [Google Scholar]
- 46.Vezzi F, Narzisi G, Mishra B. Reevaluating assembly evaluations with feature response curves: GAGE and assemblathons. PLoS One. 2012;7(12) doi: 10.1371/journal.pone.0052210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5(6):e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marchler-Bauer A, Anderson JB, Derbyshire MK, DeWeese-Scott C, Gonzales NR, Gwadz M, et al. CDD: a conserved domain database for interactive domain family analysis. Nucleic Acids Res. 2007;35(Database issue):D237–40. doi: 10.1093/nar/gkl951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30(9):1236–40. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Markowitz VM, Mavromatis K, Ivanova NN, Chen IM, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics. 2009;25(17):2271–8. doi: 10.1093/bioinformatics/btp393. [DOI] [PubMed] [Google Scholar]
- 52.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST) Nucleic Acids Res. 2014;42(Database issue):D206–14. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–64. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35(9):3100–8. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29(22):2933–5. doi: 10.1093/bioinformatics/btt509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–9. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 58.Sonnhammer EL, von Heijne G, Krogh A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc Int Conf Intell Syst Mol Biol. 1998;6:175–82. [PubMed] [Google Scholar]
- 59.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8(10):785–6. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 60.Edgar RC. PILER-CR: fast and accurate identification of CRISPR repeats. BMC Bioinformatics. 2007;8:18. doi: 10.1186/1471-2105-8-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E, et al. antiSMASH 2.0–a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013;41(Web Server issue):W204–12. doi: 10.1093/nar/gkt449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol. 2013;31(6):533–8. doi: 10.1038/nbt.2579. [DOI] [PubMed] [Google Scholar]
- 63.Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14(9):755–63. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 64.Remm M, Storm CE, Sonnhammer EL. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J Mol Biol. 2001;314(5):1041–52. doi: 10.1006/jmbi.2000.5197. [DOI] [PubMed] [Google Scholar]
- 65.Richter M, Rossello-Mora R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A. 2009;106(45):19126–31. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57(Pt 1):81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 67.Gao Q, Garcia-Pichel F. An ATP-grasp ligase involved in the last biosynthetic step of the iminomycosporine shinorine in Nostoc punctiforme ATCC 29133. J Bacteriol. 2011;193(21):5923–8. doi: 10.1128/JB.05730-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gao Q, Garcia-Pichel F. Microbial ultraviolet sunscreens. Nat Rev Microbiol. 2011;9(11):791–802. doi: 10.1038/nrmicro2649. [DOI] [PubMed] [Google Scholar]
- 69.Jacobs JL, Carroll TL, Sundin GW. The role of pigmentation, ultraviolet radiation tolerance, and leaf colonization strategies in the epiphytic survival of phyllosphere bacteria. Microb Ecol. 2005;49(1):104–13. doi: 10.1007/s00248-003-1061-4. [DOI] [PubMed] [Google Scholar]
- 70.Liu X, Williams CE, Nemacheck JA, Wang H, Subramanyam S, Zheng C, et al. Reactive oxygen species are involved in plant defense against a gall midge. Plant Physiol. 2010;152(2):985–99. doi: 10.1104/pp.109.150656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hammond-Kosack KE, Jones JD. Resistance gene-dependent plant defense responses. Plant Cell. 1996;8(10):1773–91. doi: 10.1105/tpc.8.10.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brown AD. Microbial water stress. Bacteriol Rev. 1976;40(4):803–46. doi: 10.1128/br.40.4.803-846.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87(12):4576–9. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Garrity GM, Holt JG. The road map to the manual. In: Boone D, Castenholz R, Garrity G, editors. Bergey’s manual® of systematic bacteriology. New York: Springer; 2001. pp. 119–66. [Google Scholar]
- 75.Buchanan RE. Studies in the nomenclature and classification of the bacteria: II. The primary subdivisions of the schizomycetes. J Bacteriol. 1917;2(2):155–64. doi: 10.1128/jb.2.2.155-164.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Skerman VBD, Mcgowan V, Sneath PHA. Approved lists of bacterial names. Int J Syst Bacteriol. 1980;30(1):225–420. doi: 10.1099/00207713-30-1-225. [DOI] [Google Scholar]
- 77.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]