

Abstract
Inactivation of the ARID1A tumor suppressor gene is frequent in ovarian endometrioid (OEC) and clear cell carcinomas (OCCC), often in conjunction with mutations activating the PI3K/AKT and/or canonical Wnt signaling pathways. Prior work has shown that conditional bi-allelic inactivation of the Apc and Pten tumor suppressor genes in the mouse ovarian surface epithelium (OSE) promotes outgrowth of tumors that reflect the biological behavior and gene expression profiles of human OECs harboring comparable Wnt and PI3K/AKT pathway defects, though the mouse tumors are more poorly differentiated than their human tumor counterparts. We found that conditional inactivation of one or both Arid1a alleles in OSE concurrently with Apc and Pten inactivation unexpectedly prolonged survival of tumor-bearing mice and promoted striking epithelial differentiation of the cancer cells, resulting in morphological features akin to those in human OECs. Enhanced epithelial differentiation was linked to reduced expression of mesenchymal markers N-cadherin and vimentin, and increased expression of epithelial markers Crb3 and E-cadherin. Global gene expression profiling showed enrichment for genes associated with mesenchymal-to-epithelial transition in the Arid1a-deficient tumors. We also found that an activating (E545K) Pik3ca mutation, unlike Pten inactivation or Pik3ca H1047R mutation, cannot cooperate with Arid1a loss to promote ovarian cancer development in the mouse. Our results indicate the Arid1a tumor suppressor gene has a key role in regulating OEC differentiation, and paradoxically the mouse cancers with more initiating tumor suppressor gene defects had a less aggressive phenotype than cancers arising from fewer gene alterations.
Keywords: ovarian endometrioid carcinoma, mesenchymal-to-epithelial transition, mouse ovarian cancer model
Introduction
Ovarian cancer is the deadliest gynecological cancer for women in the United States [1]. The two endometriosis-associated subtypes of ovarian cancer, namely ovarian endometrioid carcinoma (OEC) and clear cell carcinoma (OCCC), together account for approximately 20% of ovarian carcinomas overall [2,3]. Two independent studies reported inactivating mutations in the ARID1A (AT-rich interactive domain 1A [SWI-like]) gene in 46-57% of OCCCs and 30% of OECs [4,5], and subsequent studies confirmed the findings [6,7]. ARID1A encodes the ARID1A/BAF250a protein, a key component of the eukaryotic multi-protein SWI/SNF-A (also known as BAF) chromatin-remodeling complex [8,9]. SWI/SNF complexes use ATP to mobilize nucleosomes, modulating the accessibility of promoters to activate or repress transcription of genes involved in many important cellular processes, including development, differentiation, proliferation, and DNA repair [10-12]. The mechanisms by which ARID1A functions as a tumor suppressor remain incompletely understood, and few studies have addressed ARID1A function in normal and neoplastic ovarian epithelial cells. Guan and colleagues showed that restoration of ARID1A expression in one OCCC-derived cell line with bi-allelic ARID1A inactivation suppressed growth of the cells in vitro, whereas knockdown of ARID1A expression in cells derived from normal ovarian surface epithelium increased cellular proliferation in vitro and enhanced tumorigenicity in nude mice [13]. The same group also provided evidence that ARID1A collaborates with p53 to activate CDKN1A and SMAD3 transcription and inhibit ovarian tumor cell growth. Several groups have reported findings suggesting that ARID1A mutations may exist concurrently with PI3K/AKT signaling pathway defects in human endometriosis-associated ovarian carcinomas, particularly OCCC [14-16]. Recently, Chandler and colleagues reported that bi-allelic inactivation of Arid1a and activation of mutant (H1047R) Pik3ca in the mouse ovarian surface epithelium (MOSE) lead to highly penetrant development of OCCCs in mice [17]. Similarly, concurrent bi-allelic inactivation of Arid1a and Pten in the MOSE results in the development of endometrioid and undifferentiated carcinomas [18].
In past efforts, we analyzed a large collection of human OECs for the presence or absence of several oncogene (CTNNB1, KRAS, PIK3CA) and tumor suppressor gene (PTEN, TP53) mutations and found that mutations predicted to activate the PI3K/AKT and canonical Wnt signaling pathways frequently co-occur in OECs [19]. We further showed that deregulation of these pathways in the MOSE by conditional inactivation of the Apc and Pten tumor suppressor genes promoted the outgrowth of tumors that reflect the biological behavior and gene expression profiles of human OECs with comparable signaling pathway defects. However, the Apc-;Pten- mouse tumors are more poorly differentiated than most human OECs and show a mixture of epithelial and mesenchymal differentiation features in the neoplastic cells. The genetically defined Apc/Pten-deficient OEC murine model provides an excellent setting in which to assess the effects of additional gene mutations that are frequently observed in human OECs on tumor development and progression. For instance, we have reported previously that the presence of mutant Trp53 or Pik3ca alleles in the Apc/Pten-deficient OEC model leads to a more aggressive tumor phenotype and shorter overall survival of tumor-bearing mice [20]. In this study, we assessed effects of mono- and bi-allelic Arid1a inactivation on the Apc/Pten-deficient OEC phenotype. We also tested whether activation of a Pik3ca “hotspot” mutation (E545K) affecting the helical domain of p110α rather than p110α’s kinase domain, can cooperate with Arid1a loss to generate OCCCs in the mouse.
MATERIALS AND METHODS
Mouse strains, tumor induction, and histopathology
Apcfl/fl;Ptenfl/fl and Pik3caLSL-E545K/+ mice and ovarian bursal delivery of replication-incompetent recombinant adenovirus expressing Cre recombinase (AdCre) have been previously described in detail [19-21]. Arid1afl/fl mice have also been reported previously [22]. For tumor induction, 5 × 107 plaque-forming units (p.f.u.) of AdCre (University of Michigan Vector Core) with 0.1% Evans Blue (Sigma-Aldrich Inc., St. Louis, MO) were injected into the right ovarian bursal cavities of 7-8-week-old female mice. In each mouse, the left ovarian bursa was not injected and served as control. All animal studies were performed under a protocol (PRO00006370) approved by the University of Michigan’s University Committee on Use and Care of Animals, the institutional committee that helps ensure the ethical and humane care, and justified use of animals for research. Mice were examined at necropsy for gross organ abnormalities. The genital system, liver, lungs, spleen, kidney, brain, and digestive tract were collected, fixed in 10% (v/v) buffered formalin, and embedded in paraffin for histopathological evaluation.
Immunohistochemical analysis of tumor tissues
Formalin-fixed, paraffin-embedded (FFPE) mouse tumor tissues were sectioned and subjected to routine immunohistochemical analysis. A human ovarian cancer FFPE tissue microarray including 20 OECs obtained from the University of Michigan surgical pathology archives has been previously described [23]. Use of these tissues was approved by our Institutional Review Board (protocols HUM00044063 and HUM00042103). Details of immunohistochemical staining are provided in the Supplementary Methods.
Gene expression profiling and data analysis
Total RNA from Apc-;Pten- and Apc-;Pten-;Arid1a- mouse tumors was isolated using Trizol (Invitrogen Life Technologies, Carlsbad, CA) according to the manufacturer’s protocol, aliquoted, and stored at −80°C until use. RNA was purified with RaNeasy columns (Qiagen, Valencia, CA), and RNA quality was assessed by an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA). Following biotinylated cRNA synthesis, labeled cRNAs were hybridized to Affymetrix Mouse Genome 430 2.0 arrays (Affymetrix, Santa Clara, CA) by the Microarray Core at the University of Michigan. Transcript abundances were estimated using the Robust Multi-array Average (RMA) method in the Bioconductor package of the R statistical programming language [24]. Probe set annotation was obtained from Affymetrix web downloads (version na33). We analyzed log2-transformed data. The raw and processed data, and statistical analysis, were deposited in NCBI’s Gene Expression Omnibus [25] and can be accessed with series accession number GSE67695. The following private link was created to permit review before release to the general public: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=wpkpgsgqhpoldsl&acc=GSE67695. We collapsed our selection of 1935 up and 1960 down probe-sets (of 45101 probe-sets total) to 20116 distinct mouse genes annotated with unique Entrez Gene IDs, of which 1424 were up and 1241 were down genes. These up and down gene lists were separately used for enrichment testing against 8445 gene sets, each set corresponding to a Gene Ontology (GO) molecular process term. We used one-sided Fisher Exact tests for this, which ask if the intersection of two gene lists is significantly larger than expected by chance. Mouse genes were mapped to 14729 distinct homologous human genes using build 65 of NCBI’s Homologene, using only 1-to-1 best homologs, which gave 1266 up and 1097 down human genes. These 2 lists were tested for enrichment in 3000 curated gene sets (collection C2) held in version 3 of the Molecular Signatures Database (MSigDB, http://www.broadinstitute.org/gsea/msigdb/index.jsp)[26], as well as to 232 gene sets for pathways from the Kyoto Encyclopedia of Genes and Genomes (KEGG). False discovery rates were estimated based on identical analysis of 100 data sets in which the gene identifiers were randomly permuted, and are less than 5% for all reported enrichments. The input and output files, as well as the code to perform the enrichment testing and estimate false discovery rates are provided in a supplementary download available in our GEO submission. Files and code to perform the permutation testing used to estimate false discovery rates for our probe-set selections described in Results are also available there.
RESULTS
Inactivation of Arid1a enhances epithelial differentiation in a murine model of OEC
Deregulation of the PI3K/AKT and canonical Wnt signaling pathways in the MOSE via conditional inactivation of the Apc and Pten tumor suppressor genes results in the development of OEC-like tumors in 100% of mice [19]. In this Apc/Pten-deficient OEC model, tumors show glandular epithelial differentiation elements (cytokeratin- and E-cadherin-positive), admixed with less differentiated, mesenchymal-appearing cells with vague spindle-cell morphology (vimentin-positive and cytokeratin- and E-cadherin-negative). In most tumors, the solid/spindle component exceeds 50% of the total tumor mass (equivalent to human FIGO grade 3). In order to explore effects of Arid1a inactivation on the Apc/Pten-deficient OEC phenotype, we crossed Apcfl/fl;Ptenfl/fl mice with mice allowing for conditional deletion of Arid1a exon 8, to generate Apcfl/fl;Ptenfl/fl;Arid1afl/+ and Apcfl/fl;Ptenfl/fl;Arid1afl/fl mice. We injected adenovirus expressing Cre recombinase (AdCre) into the right ovarian bursa to target the multiple tumor suppressor gene alleles in the MOSE. Mice were monitored for tumor formation and then euthanized based on health status, determined by criteria for humane endpoints and guidelines for the humane treatment of animals. We observed that Apc/Pten-deficient tumors that also had inactivation of one or both Arid1a alleles showed strikingly more epithelial differentiation than tumors arising in littermates in which both Arid1a alleles were intact (5-50% solid growth pattern, equivalent to human FIGO grade 2). The morphology of the murine Apc/Pten-deficient OEC tumors with Arid1a loss closely resembles that of human OECs (Figure 1A). Immunohistochemistry (IHC), immunofluorescence (IF) and/or Western blotting were used to evaluate expression of several markers of epithelial and mesenchymal differentiation in the murine OECs with intact Arid1a or with loss of one or both Arid1a alleles, including cytokeratin 8, cytokeratin 19, E-cadherin, N-cadherin, vimentin and the epithelial cell polarity marker Crb3 (Crumbs homolog 3). Using IHC and IF analyses, we observed diffuse positive staining for cytokeratins 8 and 19 (data not shown), and E-cadherin throughout most of the Apc-;Pten-;Arid1a-/- and Apc-;Pten-;Arid1a-/+ tumor regions studied. Tumors with deletion of one or both copies of Arid1a also showed prominent expression of Crb3, and expression of the mesenchymal markers N-cadherin and vimentin was decreased relative to that seen in tumors with intact Arid1a alleles (Figure 1B). These findings were confirmed by Western blotting (Figure 1C). We further analyzed these epithelial and mesenchymal markers in two mouse cell lines derived from Apc/Pten-deficient tumors with and one without Arid1a deletion. Striking elevation of E-cadherin and Crb3 expression was accompanied by marked reduction of N-cadherin and vimentin expression in the cells with Arid1a knockout compared to cells in which Arid1a is intact (Figure 2, left panel). The mouse cell line data strongly support the results found in primary mouse tumors. In order to assess effects of ARID1A loss in human OECs, we employed two independent shRNAs to stably knockdown ARID1A in TOV112D (OEC-derived cell line with CTNNB1 mutation and intact ARID1A alleles) and ES2 (OCC-derived cell line with intact ARID1A). ES2 cells with ARID1A knockdown showed modest reduction of N-cadherin, while TOV112D cells with ARID1A knockdown showed a slight increase in Crb3 expression (Figure 2, right panel). Other markers were unchanged by ARID1A knockdown or were expressed at levels too low to detect by Western blot. To determine whether human OECs with loss of ARID1A expression showed similar findings, we performed IHC staining for ARID1A, N-cadherin, vimentin, CK7 and Crb3 on 20 human OECs, using primary tumor tissue samples present in an ovarian cancer tissue microarray. Representative staining is shown in Supplementary Figure S1). As observed in the murine tumors, in the human samples we saw a significant correlation between loss of ARID1A expression and decreased vimentin expression (Spearman’s rho = 0.59, p = 0.006); correlations for the other markers studied were not statistically significant (Figure 3) perhaps due in part to small sample size. Collectively, the findings suggest that ARID1A may have an important role in regulating OEC differentiation, with ARID1A loss enhancing epithelial differentiation, especially so in the mouse OEC model system.
Figure 1. Deletion of one or both Arid1a alleles enhances epithelial differentiation in mouse OECs.
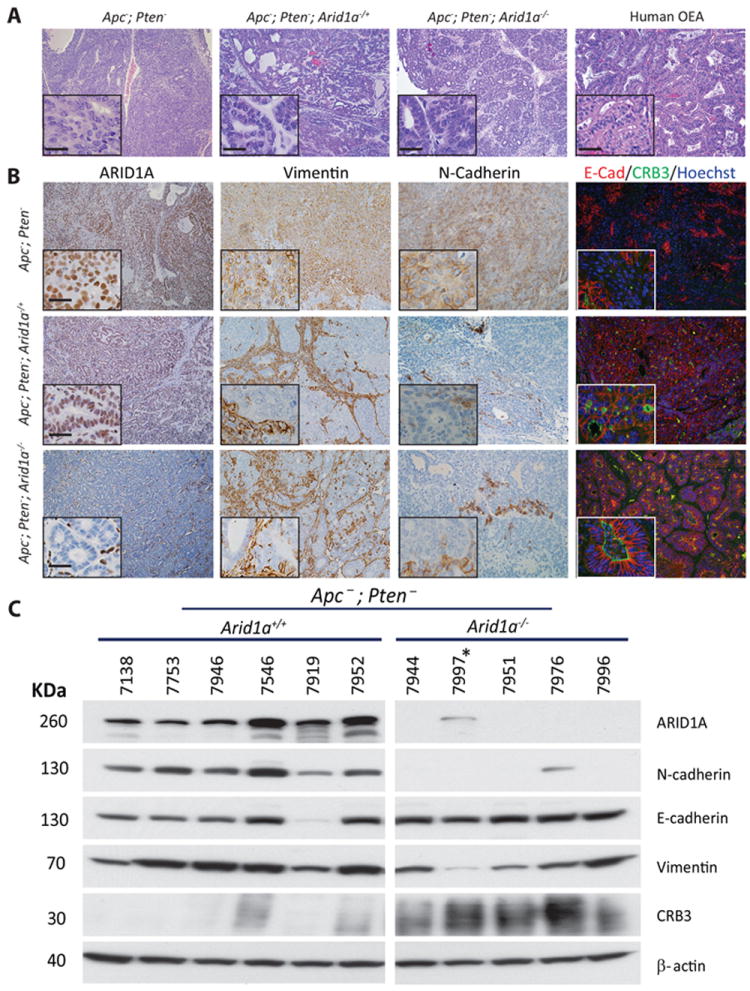
A) Hematoxylin and eosin stained sections of mouse tumors with two, one, or zero intact copies of Arid1a. Representative human OEC is shown for comparison. B) Immunohistochemical staining of ARID1A, vimentin, and N-cadherin and immunofluorescence staining of E-cadherin and Crb3 in mouse tumors with varying Arid1a copy number. Scale bar in insets represents 25 μm. C) Western blot analysis of ARID1A, N-cadherin, E-cadherin, Vimentin and Crb3 in mouse tumors with and without intact Arid1a. Mouse 7997* has the Apcfl/fl;Ptenfl/fl;Arid1afl/+ genotype.
Figure 2. Western blot analysis for EMT markers in human ovarian cancer cell lines with Arid1a knockdown and mouse tumor-derived cell lines.
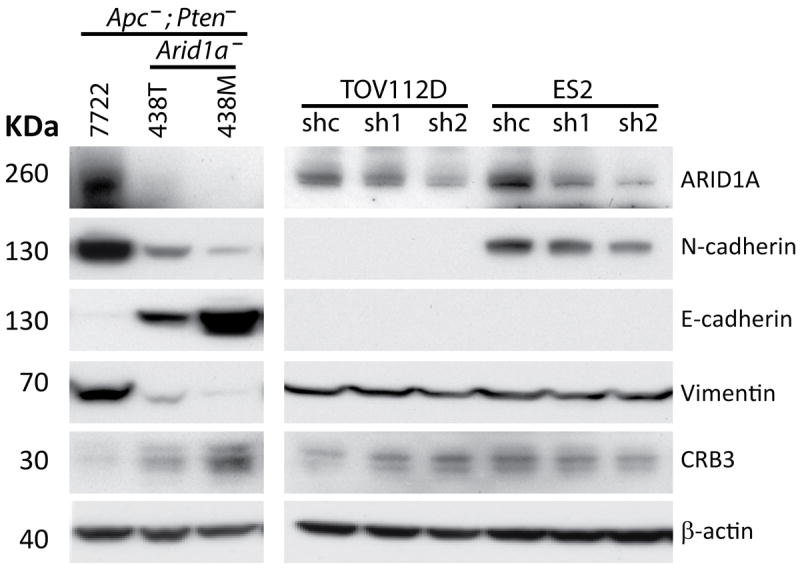
Left panel shows cell lines derived from Apc/Pten-deficient mouse tumors with intact (7772) or bi-allelic deletion (438T, 438M) of Arid1a analyzed by Western blot for ARID1A, N-cadherin, E-cadherin, Vimentin and Crb3. Right panel shows analysis of the same markers in TOV112D and ES2 cells with ARID1A knockdown using two independent shRNAs.
Figure 3. Expression of ARID1A and vimentin are inversely correlated in human OECs.
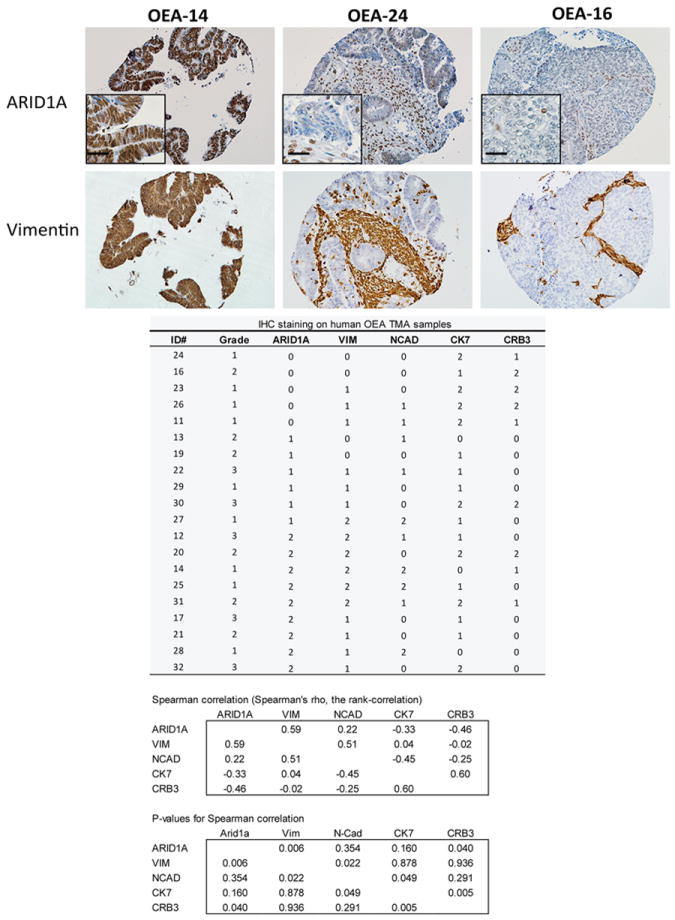
Photomicrographs of IHC staining for ARID1A and vimentin in representative OECs in a human ovarian cancer tissue microarray. FIGO grade and IHC score for each tumor and antibody and statistical analysis are also shown.
Gene expression profiling identifies signatures associated with mesenchymal to epithelial transition in OECs with Arid1a loss
We employed oligonucleotide microarrays to compare gene expression in Apc-;Pten- vs. Apc-;Pten-;Arid1a- mouse tumors (n=3 independent tumors for each of the two groups). Using log-transformed data on 45101 probe-sets, we selected those probe-sets for which two-sample T-tests gave p<.01 and the difference in means was at least 1.3-fold, to obtain 3895 differentially expressed probe-sets (1935 up and 1960 down in Arid1a-/- tumors). We similarly analyzed data-sets that were identical except that the sample labels were permuted, and obtained just 61.2 qualifying probe-sets on average, so that a simple estimate of the false discovery rate for this selection was 1.6% (= 61.2/3895). Figure 4 shows a small subset of these selected differences for which average changes were at least 10-fold. The complete dataset and analysis is publicly available in GEO. We collapsed our selections to distinct genes and performed enrichment testing against 3000 curated gene sets from MSigDB. The 5 top gene sets for lists of genes up- and down-regulated in tumors with bi-allelic Arid1a deletion are shown in Supplementary Table 1. Notably, our down-regulated gene list’s best enrichment (p=6E-15) was for a list of genes higher in luminal vs mesenchymal breast cell line samples [27], while our up-regulated gene list’s second best enrichment was to the corresponding list of genes higher in mesenchymal genes from the same study (p=1.8E-21). Similarly, when we assessed Gene Ontology (GO) biological process terms, the third best term for our down-regulated genes was “positive regulation of epithelial to mesenchymal transition” (10 of 20 genes, p=8E-8), including Axin2, Bmp2, Ctnnb1, Tgfb2 and Tgfb3, and our top hit was to “cell adhesion” (p=9E-12), which included Fn1, being down over 5-fold. For our down-regulated genes, “Wnt signaling pathway” was the second most significant enrichment in KEGG pathways and the ninth most significant in GO. Wnt5a was in these intersections and was decreased by 76-fold in our Arid1a-deficient mouse tumors. Levels of Axin2, an indicator of Wnt pathway activation and transcriptional target of β-catenin, were decreased approximately 2-fold.
Figure 4. Differential gene expression in OECs with and without knockout of Arid1a.
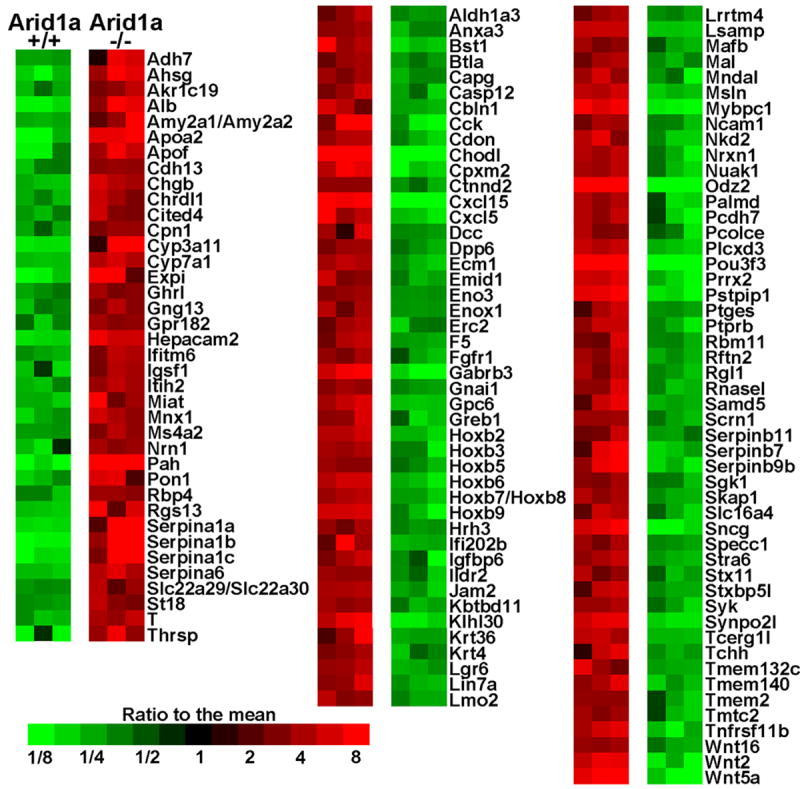
Heatmap shows genes with at least 10-fold average differences in gene expression and p<.01 by two-sample T-test. Only probe-sets with well-annotated genes are shown, and where several probe-sets for the same gene qualified, the one with the smallest p-value is shown.
Arid1a inactivation results in prolonged survival in the Apc/Pten-deficient OEC model
To date, no consistent relationship has been established between ARID1A mutation or loss of expression and prognosis in human cancer types [28]. Genetically engineered mouse models offer an opportunity to assess how Arid1a inactivation not only affects tumor differentiation state but also tumor aggressiveness, as reflected by overall survival of mice, in an in vivo model where the tumors differ chiefly in Arid1a genotype. We compared the overall survival of mice with Apc-;Pten- tumors that had two intact (n=18); one intact and one inactive (n=28), or two inactive (n=16) Arid1a alleles. Mice with Arid1a-deficient tumors had significantly longer survival than mice with tumors that retained ARID1A expression (Figure 5A). Interestingly, this effect was seen in mice bearing tumors with deletion of one or both copies of Arid1a, with longest survival observed in mice with tumors that were haploinsufficient for Arid1a. ARID1A expression in Arid1a haploinsufficient tumors was analyzed by qRT-PCR, immunohistochemistry and Western blot. We found a significant reduction, but not complete loss, of Arid1a transcripts in these tumors. Furthermore, at least some ARID1A protein expression was retained in the tumors. A few showed areas in which ARID1A expression was absent, suggesting the presence of sub-clones within the tumor that have deleted or silenced the wild-type Arid1a allele (Supplementary Figure S2). At necropsy, tumors with loss of one or both copies of Arid1a were larger than their counterparts with intact Arid1a (Figure 5B), and were more frequently associated with metastatic disease (Table 1), presumably because the mice with ARID1A-deficient tumors survived longer. Although tumors with intact Arid1a were smaller and not found to be associated with distant metastases, half of the mice with Arid1a-intact tumors died spontaneously and were found to have hemorrhagic ascites at necropsy. Others required euthanasia because they became visibly ill (reached humane endpoints). Interestingly, in prior work we found that ovarian tumors usually fail to develop unless both alleles of Apc and Pten are deleted in the MOSE [29]. In this study mice with at least one intact Apc or Pten allele were monitored for 40 or 60 weeks following ovarian bursal injection with AdCre. We found that deletion of one or both copies of Arid1a can promote outgrowth of cancer cells that still have intact function of one Apc or Pten allele (Table 1).
Figure 5. Deletion of one or both Arid1a alleles prolongs survival of mice with Apc-;Pten- OECs.
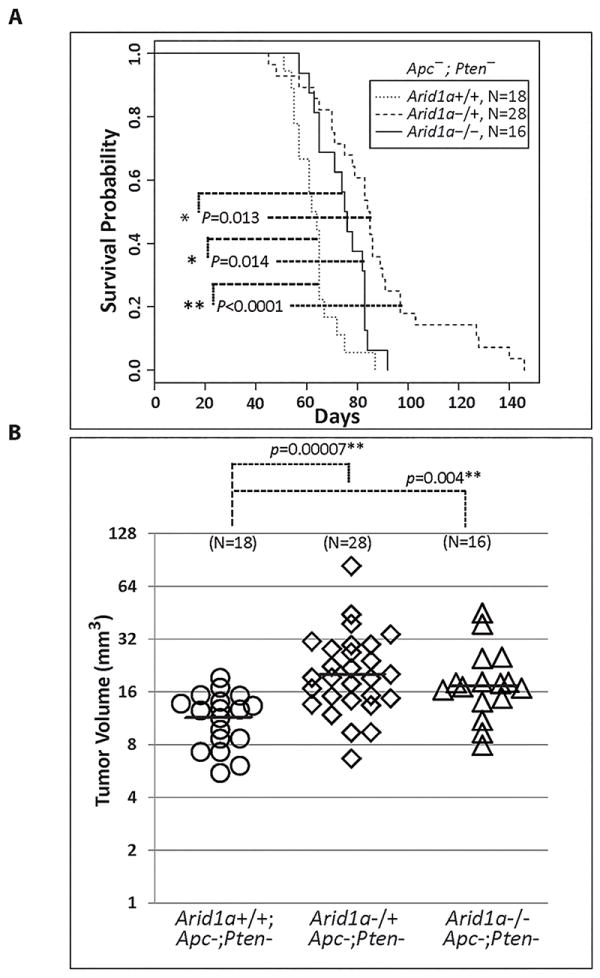
A) Kaplan-Meier survival curves of Apcfl/fl ;Ptenfl/fl ;Arid1afl/fl (n = 16), Apcfl/fl ;Ptenf/lfl ; Arid1afl/+ (n=28) and Apcfl/fl;Ptenfl/fl littermate controls (n = 18) following ovarian bursal injection of AdCre. Cox proportional hazards models showed that Arid1a+/+ mice died earlier than Arid1a-/- mice, with estimated hazard ratio (HR) of 2.40 for tumors with retained Arid1a expression (95% CI=1.19 - 4.83). Arid1a-/- mice died earlier than Arid1a+/- mice (HR=2.35, 95%CI = 1.19 - 4.61), and Arid1a+/+ mice died significantly earlier than Arid1a-/+ mice (HR=5.64, 95% CI = 2.82 - 11.26, p=9.6×10-7). Paired comparisons by Wald tests. B) Tumor volumes in the same groups of mice. Tumors with knockout of one or both Arid1a alleles were 1.8 and 1.6 fold larger on average than Arid1a+/+ tumors, respectively (p= 7×10-5 and .0044, ANOVA model with 3 means on log-transformed data).
Table 1.
Mouse ovarian tumor formation, progression and survival of mice with conditional knockout of Apc, Pten, and Arid1a
| Genotype | Tumor formation (n/N) * | Metastasis (n/N) | Median survival (days) |
|---|---|---|---|
| Apcfl/fl;Ptenfl/fl | 18/18 | 0/18 | 64.5 |
| Apcfl/fl;Ptenfl/fl;Arid1afl/+ | 28/28 | 5/28 | 86.5 |
| Apcfl/fl;Ptenfl/fl;Arid1afl/fl | 15/15 | 9/15 | 74.5 |
|
| |||
| Euthanized (weeks) | |||
|
| |||
| Apcfl/+;Ptenfl/fl;Arid1afl/fl | 3/8 | 40 | |
| Apcfl/fl;Ptenfl/+;Arid1afl/fl | 2/7 | 1/7 | 40 |
| Apcfl/fl;Ptenfl/+;Arid1afl/+ | 2/9 | 40 | |
| Apcfl/+;Ptenfl/fl;Arid1afl/+ | 2/8 | 1/8 | 40 |
| Apcfl/+;Ptenfl/+;Arid1afl/fl | 2/5 | 40 | |
| Apcfl/+;Ptenfl/+;Arid1afl/+ | 0/4 | 40 | |
| Apcfl/+;Ptenfl/fl | 1/20 | 1/20 | 60 |
| Apcfl/fl;Ptenfl/+ | 0/20 | 60 | |
n: number of mice that formed tumors or metastases;
N: total number of mice evaluated
Concurrent conditional inactivation of Arid1a and activation of mutant (E545K) Pik3ca in the MOSE are insufficient to generate ovarian clear cell or endometrioid carcinomas in mice
Several studies have reported that ARID1A mutations are found together with lesions activating the PI3K/AKT signaling pathway in human endometriosis-associated ovarian carcinomas, particularly OCCC. Conditional inactivation of Arid1a coupled with conditional activation of kinase-domain mutant (H1047R) Pik3ca resulting from ovarian bursal injection of AdCre was recently reported to promote rapid outgrowth of OCCC-like tumors in the mouse, with a median survival of only 7.5 weeks [17]. We sought to determine whether an oncogenic Pik3ca mutation (E545K) affecting the helical domain of p110α could similarly cooperate with Arid1a inactivation in outgrowth of OCCC-like tumors. Of note, we did not observe the development of OCCC- or OEC-like tumors following ovarian bursal injection of AdCre in Arid1afl/fl;Pik3caLSL-E545K mice (n=17), even though the mice were monitored for an average of 8.7 months (range 3-12 months). Cre-mediated recombination of Arid1a and Pik3ca alleles in the ovarian tissues was confirmed by PCR in representative mice (Supplementary Figure S3). The findings suggest that various mutant Pik3ca alleles may differ in their oncogenic potential in collaborating with Arid1a defects to promote OCCC-like tumors in the mouse.
DISCUSSION
We have previously reported that Apc/Pten-deficient OECs display more aggressive behavior (shorter overall survival and more widespread metastases) when mutant Trp53 or Pik3ca alleles are also present [20]. Our observations here that Arid1a loss prolongs survival and enhances epithelial differentiation in our Apc/Pten-deficient OEC model were unexpected, but perhaps not entirely surprising, given some other published data. For example, Jordan and colleagues reported that RNAi silencing of the SWI/SNF chromatin-remodeling factor Smarcd3/Baf60c led to a robust mesenchymal to epithelial transition (MET) in EpCam- negative breast cancer cells, whereas expression of Smarcd3/Baf60 in immortalized mammary epithelium induced an epithelial to mesenchymal transition (EMT) that could be reversed by inhibition of Wnt5a [30]. Smarcd3/Baf60c was shown to directly bind the promoters of WNT5A and CDH1 (E-cadherin), and to promote EMT through induction of Wnt5a signaling and repression of E-cadherin expression. These authors concluded that Smarcd3/Baf60 epigenetically regulates EMT by activating Wnt signaling. Interestingly, we observed that Wnt5a was remarkably (>70-fold) down-regulated in Arid1a-deficient, Apc/Pten-deficient OECs compared to Apc/Pten-deficient OECs in which both Arid1a alleles were intact. Others have also reported a role for the SWI/SNF chromatin remodeling complexes in regulating EMT/MET. For example, Huang et al. showed that ablation of BAF180, a component of the SWI/SNF-B (PBAF) chromatin remodeling complex, leads to impaired EMT [31].
In this study, we also showed that deletion of one or both Arid1a alleles leads to enhanced epithelial differentiation of Apc/Pten-deficient mouse tumors and tumor-derived cell lines. Compared to the poorly differentiated Apc/Pten-deficient tumors, the morphologic features of tumors that also have Arid1a deletion much more closely resemble human EOCs, most of which show overt gland formation. Recently, Guan et al reported that bi-allelic Arid1a and Pten inactivation in the MOSE led to the development of undifferentiated (62%) or OEC-like (38%) ovarian carcinomas in 13 of 22 mice during a 6-9 month surveillance period [18]. They did not explore the molecular basis underlying the different phenotypes. However, our collective data suggest that deregulation of Wnt signaling in the context of Pten and Arid1a loss leads to a more rapid and penetrant tumor phenotype and more consistent epithelial differentiation. Interestingly, in our model system, we also found that 3 out of 8 Apcfl/+;Ptenfl/fl;Arid1fl/fl mice developed ovarian tumors over a 10 month surveillance period, of which two were EOCs and one was an undifferentiated carcinoma.
Our observation that ARID1A loss is associated with loss of vimentin expression in human OECs is perhaps unexpected. Vimentin is widely accepted as a marker of mesenchymally-derived cells or cells undergoing epithelial-to-mesenchymal transition (EMT) during both normal development and neoplastic progression. However, human OECs (which typically display overt epithelial differentiation) often express vimentin [32]. More recently, Desouki and colleagues reported that vimentin is usually expressed by uterine endometrioid carcinomas, but is usually not expressed by primary OECs [33]. Our data suggest that the presence or absence of vimentin expression in human OECs may be influenced by the status of ARID1A.
In our study, inactivation of one or both Arid1a alleles promoted epithelial differentiation in OECs and prolonged survival of tumor-bearing mice. The notion that there may be biological effects associated with Arid1a haploinsufficiency is consistent with some previous observations. For example, an early study reported that heterozygosity for Arid1a in mice leads to embryonic lethality (22). Further, only one ARID1A allele appears to be inactivated in roughly 70% of OCCCs with ARID1A mutations, although nearly three-quarters of OCCCs that retain one wild-type ARID1A allele lose expression of ARID1A protein by IHC [5]. In their review, Wu and Roberts suggest that ARID1A haploinsufficiency may promote transformation in some lineages, while homozygous inactivation is required in others [28]. They also argue that mouse models of disease will be critical for testing tissue and tumor type-specific effects of ARID1A insufficiency. Our studies provide strong evidence that inactivation of even one Arid1a allele has profound effects on the phenotype of OECs arising in the context of deregulated Wnt and PI3K/AKT pathway signaling.
We acknowledge that our study has some limitations. For example, deregulation of Wnt signaling in human OECs usually occurs as a result of activating CTNNB1 mutations, rather than bi-allelic APC inactivation as in our model. It is somewhat reassuring that Tanwar et al. have shown that combining activated β-catenin with homozygous Pten deletion in the murine OSE results in tumors similar to those in our model [34]. More importantly, our observations that Arid1a loss enhances epithelial differentiation and prolongs survival are interesting, but the mechanisms by which this occurs remain unclear. Functional testing of genes displaying marked differential expression in Arid1a-intact versus Arid1a-deficient tumors may yield new mechanistic insights, but identifying the specific genes that play key roles in these processes will be challenging.
Finally, our failure to observe ovarian tumor development in the context of conditional Arid1a inactivation and Pik3ca/p110α activation due to an E545K mutation, contrasts with the recent findings of Chandler et al. who found that conditional Arid1a inactivation coupled with activation of kinase-domain mutant (H1047R) p110α promoted outgrowth of OCCC-like tumors in the mouse [17]. We previously noted that different groups have observed variable effects of expressing mutant Pik3ca in MOSE, and the differences in effects observed may reflect at least in part the specific Pik3ca mutant allele tested [20]. Specifically, Kinross et al. and Liang et al. found that expression of activated p110α (H1047R or N-terminal myristoylated, respectively) resulted in hyperplasia of the MOSE, while expression of the E545K mutant did not cause epithelial proliferation in our model system [20,35,36]. The E545K mutation that we employed is in p110α’s helical domain, while the H1047R mutation affects the kinase domain. In their study of various PIK3CA mutants, Zhao and Vogt concluded that the helical and kinase domain mutations operate by two different and independent mechanisms [37]. Specifically, the functional consequences of helical domain mutations are not dependent on binding to p85 but require interaction with RAS-GTP, while function of the kinase domain mutants is dependent on the interaction with p85, but not RAS-GTP binding. In conjunction with the results of Chandler et al., our findings provide further evidence that Pik3ca mutations affecting the kinase vs. helical domains have different functional consequences in the MOSE, with the kinase domain mutants being more potent in promoting neoplastic transformation in mouse models. It is also possible that results obtained in different studies in part reflect differences in Pik3ca gene dosage. For example, Chandler et al. employed a conditional mutant (H1047R) Pik3ca allele knocked into the ROSA26 locus, while the mutant we used (E545K) was knocked into one of the endogenous Pik3ca loci. Obviously, in the case of human cancers where different mutant PIK3CA alleles arise and promote selective (clonal) outgrowth, both the PIK3CA E545K and H1047R alleles must have oncogenic potential by definition, though their functions and activity may be influenced by context, including cell differentiation state and other epigenetic and genetic alterations linked to transformation in a given cancer.
In closing, our work shows that inactivation of Arid1a enhances epithelial differentiation and prolongs survival in an Apc/Pten-deficient murine model of OEC. The mechanisms by which ARID1A interacts with its partner transcription factors in SWI/SNF-A chromatin remodeling complexes and modulates target gene expression remain incompletely understood. Further studies will likely substantially advance knowledge of the downstream factors and mechanisms through which ARID1A loss contributes to OEC and OCCC pathogenesis.
Supplementary Material
Acknowledgments
This work was supported in part by grants from the National Cancer Institute (R01CA94172, R01CA188516 and P30CA046592). The rabbit polyclonal anti-Crb3 antibody used for immunofluorescence staining of tumor tissues was a gift from Benjamin Margolis, University of Michigan Medical School.
Footnotes
STATEMENT OF AUTHOR CONTRIBUTIONS
YZ, RK, ZW, SJB, ERF, and KRC conceived experiments and analyzed data.
YZ, CT, RW, and MS carried out experiments.
All authors were involved in writing the paper and had final approval of the submitted version.
Microarray Data have been deposited in NCBI’s Gene Expression Omnibus (GSE67695)
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Seidman JD, Horkayne-Szakaly I, Haiba M, et al. The histologic type and stage distribution of ovarian carcinomas of surface epithelial origin. Int J Gynecol Pathol. 2004;23:41–44. doi: 10.1097/01.pgp.0000101080.35393.16. [DOI] [PubMed] [Google Scholar]
- 3.Kobel M, Kalloger SE, Lee S, et al. Biomarker-based ovarian carcinoma typing: a histologic investigation in the ovarian tumor tissue analysis consortium. Cancer Epidemiol Biomarkers Prev. 2013;22:1677–1686. doi: 10.1158/1055-9965.EPI-13-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones S, Wang TL, Shih Ie M, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228–231. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wiegand KC, Shah SP, Al-Agha OM, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363:1532–1543. doi: 10.1056/NEJMoa1008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maeda D, Mao TL, Fukayama M, et al. Clinicopathological Significance of Loss of ARID1A Immunoreactivity in Ovarian Clear Cell Carcinoma. International Journal of Molecular Sciences. 2010;11:5121–5129. doi: 10.3390/ijms11125120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McConechy MK, Ding J, Senz J, et al. Ovarian and endometrial endometrioid carcinomas have distinct CTNNB1 and PTEN mutation profiles. Mod Pathol. 2014;27:128–134. doi: 10.1038/modpathol.2013.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hargreaves DC, Crabtree GR. ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. 2011;21:396–420. doi: 10.1038/cr.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11:481–492. doi: 10.1038/nrc3068. [DOI] [PubMed] [Google Scholar]
- 10.Sif S, Saurin AJ, Imbalzano AN, et al. Purification and characterization of mSin3A-containing Brg1 and hBrm chromatin remodeling complexes. Genes Dev. 2001;15:603–618. doi: 10.1101/gad.872801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang W, Xue Y, Zhou S, et al. Diversity and specialization of mammalian SWI/SNF complexes. Genes Dev. 1996;10:2117–2130. doi: 10.1101/gad.10.17.2117. [DOI] [PubMed] [Google Scholar]
- 12.Wang X, Haswell JR, Roberts CW. Molecular Pathways: SWI/SNF (BAF) Complexes Are Frequently Mutated in Cancer--Mechanisms and Potential Therapeutic Insights. Clin Cancer Res. 2014;20:21–27. doi: 10.1158/1078-0432.CCR-13-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guan B, Wang TL, Shih Ie M. ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Research. 2011;71:6718–6727. doi: 10.1158/0008-5472.CAN-11-1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Samartzis EP, Noske A, Dedes KJ, et al. ARID1A mutations and PI3K/AKT pathway alterations in endometriosis and endometriosis-associated ovarian carcinomas. International journal of molecular sciences. 2013;14:18824–18849. doi: 10.3390/ijms140918824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamamoto S, Tsuda H, Takano M, et al. Loss of ARID1A protein expression occurs as an early event in ovarian clear-cell carcinoma development and frequently coexists with PIK3CA mutations. Mod Pathol. 2012;25:615–624. doi: 10.1038/modpathol.2011.189. [DOI] [PubMed] [Google Scholar]
- 16.Yamamoto S, Tsuda H, Takano M, et al. PIK3CA mutations and loss of ARID1A protein expression are early events in the development of cystic ovarian clear cell adenocarcinoma. Virchows Arch. 2012;460:77–87. doi: 10.1007/s00428-011-1169-8. [DOI] [PubMed] [Google Scholar]
- 17.Chandler RL, Damrauer JS, Raab JR, et al. Coexistent ARID1A-PIK3CA mutations promote ovarian clear-cell tumorigenesis through pro-tumorigenic inflammatory cytokine signalling. Nature communications. 2015;6:6118. doi: 10.1038/ncomms7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guan B, Rahmanto YS, Wu RC, et al. Roles of deletion of Arid1a, a tumor suppressor, in mouse ovarian tumorigenesis. J Natl Cancer Inst. 2014;106 doi: 10.1093/jnci/dju146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu R, Hendrix-Lucas N, Kuick R, et al. Mouse model of human ovarian endometrioid adenocarcinoma based on somatic defects in the Wnt/B-catanin and PI3K/Pten signaling pathways. Cancer Cell. 2007;11:321–333. doi: 10.1016/j.ccr.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 20.Wu R, Baker SJ, Hu TC, et al. Type I to Type II Ovarian Carcinoma Progression: Mutant Trp53 or Pik3ca Confers a More Aggressive Tumor Phenotype in a Mouse Model of Ovarian Cancer. Am J Pathol. 2013;182:1391–1399. doi: 10.1016/j.ajpath.2012.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robinson G, Parker M, Kranenburg TA, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488:43–48. doi: 10.1038/nature11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao X, Tate P, Hu P, et al. ES cell pluripotency and germ-layer formation require the SWI/SNF chromatin remodeling component BAF250a. Proc Natl Acad Sci U S A. 2008;105:6656–6661. doi: 10.1073/pnas.0801802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwartz DR, Wu R, Kardia SL, et al. Novel candidate targets of beta-catenin/T-cell factor signaling identified by gene expression profiling of ovarian endometrioid adenocarcinomas. Cancer Res. 2003;63:2913–2922. [PubMed] [Google Scholar]
- 24.Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 25.Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 2013;41:D991–995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Charafe-Jauffret E, Ginestier C, Monville F, et al. Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene. 2006;25:2273–2284. doi: 10.1038/sj.onc.1209254. [DOI] [PubMed] [Google Scholar]
- 28.Wu JN, Roberts CW. ARID1A Mutations in Cancer: Another Epigenetic Tumor Suppressor? Cancer Discov. 2013;3:35–43. doi: 10.1158/2159-8290.CD-12-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu R, Hendrix-Lucas N, Kuick R, et al. Mouse model of human ovarian endometrioid adenocarcinoma based on somatic defects in the Wnt/beta-catenin and PI3K/Pten signaling pathways. Cancer Cell. 2007;11:321–333. doi: 10.1016/j.ccr.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 30.Jordan NV, Prat A, Abell AN, et al. SWI/SNF chromatin-remodeling factor Smarcd3/Baf60c controls epithelial-mesenchymal transition by inducing Wnt5a signaling. Mol Cell Biol. 2013;33:3011–3025. doi: 10.1128/MCB.01443-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang X, Gao X, Diaz-Trelles R, et al. Coronary development is regulated by ATP-dependent SWI/SNF chromatin remodeling component BAF180. Dev Biol. 2008;319:258–266. doi: 10.1016/j.ydbio.2008.04.020. [DOI] [PubMed] [Google Scholar]
- 32.Dabbs DJ, Sturtz K, Zaino RJ. The immunohistochemical discrimination of endometrioid adenocarcinomas. Hum Pathol. 1996;27:172–177. doi: 10.1016/s0046-8177(96)90371-8. [DOI] [PubMed] [Google Scholar]
- 33.Desouki MM, Kallas SJ, Khabele D, et al. Differential vimentin expression in ovarian and uterine corpus endometrioid adenocarcinomas: diagnostic utility in distinguishing double primaries from metastatic tumors. Int J Gynecol Pathol. 2014;33:274–281. doi: 10.1097/PGP.0b013e31829040b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanwar PS, Zhang L, Kaneko-Tarui T, et al. Mammalian target of rapamycin is a therapeutic target for murine ovarian endometrioid adenocarcinomas with dysregulated Wnt/beta-catenin and PTEN. PLoS One. 2011;6:e20715. doi: 10.1371/journal.pone.0020715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kinross KM, Montgomery KG, Kleinschmidt M, et al. An activating Pik3ca mutation coupled with Pten loss is sufficient to initiate ovarian tumorigenesis in mice. J Clin Invest. 2012;122:553–557. doi: 10.1172/JCI59309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liang S, Yang N, Pan Y, et al. Expression of activated PIK3CA in ovarian surface epithelium results in hyperplasia but not tumor formation. PLoS One. 2009;4:e4295. doi: 10.1371/journal.pone.0004295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao L, Vogt PK. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci U S A. 2008;105:2652–2657. doi: 10.1073/pnas.0712169105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.