

Abstract
AIM: To assess the effects of somatostatin on proliferation and apoptosis of activated rat hepatic stellate cells (HSCs).
METHODS: HSCs isolated from the livers of adult Sprague-Dawley rats (weighing 400-500 g) by in situ perfusion and purified by single-step density gradient centrifugation with Nycodenz, became activated after 10 days’ cultivation. Then the apoptotic rate of HSCs treated with different doses of somatostatin for 72 h, was assayed by acridine orange/ ethidium bromide fluorescent staining, terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling, transmission electron microscopy and flow cytometry, while the proliferation of HSCs was measured by MTT assay. Furthermore, the mechanisms of somatostatin were investigated by cytodynamic analysis.
RESULTS: Somatostatin at the concentration of 10-6-10-9 mol/L could decrease the proliferative rate, and promote the apoptosis of activated rat HSCs in a dose-dependent way. Its action was most significant when the concentration reached 10-6 mol/L or 10-7 mol/L (P < 0.05-0.01). An obvious cell-cycle arrest (G0/G1 arrest) was the important way for somatostatin to exert its action.
CONCLUSION: Antiproliferative and proapoptotic effects of low-dose somatostatin on activated rat HSCs can be obtained. These findings reveal its potential antifibrotic action.
INTRODUCTION
Hepatic stellate cells (HSCs), critical mesenchymal cells in producing hepatic fibrosis and hepatic cirrhosis[1-9], have been the major target of antifibrotic treatment for a long time, although success has been seldom achieved. Fortunately, progress in the research on inducing apoptosis of activated HSCs, together with inhibition of their proliferation, may provide us with a promising solution to fibrogenesis caused by different kinds of chronic hepatic diseases.
Recent researches have revealed the apoptosis-inductive and proliferation-inhibitory effects of somatostatin on various cells. Furthermore, somatostatin also has an inhibitory effect on the secretion of some cytokines such as epidermal growth factor (EGF), transforming growth factor-α (TGF-α) and the like, which are essential for the activation of HSCs. Thus we attempted to investigate the effect of somatostatin on proliferation and apoptosis of HSCs and their possible therapeutic potential in hepatic fibrosis.
MATERIALS AND METHODS
Isolation, culture and identification of rat HSCs
Adult male Sprague-Dawley rats (400-500 g) were employed in the experiment. After anesthesia, in situ serial infusions were performed via the portal vein with D-Hank’s solution and perfusion medium (Hank’s medium containing 0.5 g/L collagenase IV and 1 g/L pronase E). Then the liver was broken into pieces and redigested with collagenase IV and DNase. Finally, HSCs were separated from the cell suspension by single-step density gradient centrifugation with 180 g/L Nycodenz (Sigma,USA). After cultured in Dulbecco’s modified Eagle medium (DMEM) (Gibco, USA) containing 200 mL/L calf serum (Shishen, China), penicillin (100 IU/mL) and streptomycin (100 mg/mL), HSCs were inoculated into culture flasks and maintained at 37 °C in an atmosphere of 50 mL/L CO2.
The percentage of freshly isolated living HSCs was up to 95% as defined by trypan blue staining (Sigma, USA), while their purity was over 90% when assessed by light microscopic appearance and their characteristic autofluorescence, which reflected the droplet of vitamin A in cytoplasm (at 325-328 nm). Immunocytochemical staining of desmin (Boster, China) and α-smooth muscle actin (α-SMA) (Boster, China), as well as myofibroblast-like phenotype showed the overall activation of almost all HSCs on the 10th day.
Determination of proliferation of activated HSCs
Exponentially growing activated first-passage-HSCs seeded in 96 well plates at 1 × 104 cells per well, were divided into six groups at random, namely the control group and five somatostatin-treatment groups of 10-6 mol/L, 10-7 mol/L, 10-8 mol/L, 10-9 mol/L and 10-10 mol/L. Six duplicate wells were arranged in each group. They were incubated at 37 °C in 50 mL/L CO2 for five d, and then the following steps were taken. (a) Twenty micoliters of MTT (5 mg/mL) was added to each well and incubated for four h. (b) The supernatant was aspirated and discarded. (c) One hundred microliters of DMSO was dropped into each well and the plate was agitated for a few min. (d) The optical density (OD) was analysed on an ELISA reader at a test wavelength of 490 nm and a reference wavelength of 620 nm. The inhibition rate (IR) of cell proliferation was calculated according to the following equation. IR = [1-(OD of somatostatin well/OD of control well)] × 100%.
Apoptotic analysis of activated HSCs
Activated first-passage HSCs during their logarithmic growth period were inoculated on the surface of L-polylysine-covered-glass slides in six well plates. According to the same grouping method in the proliferation test, all these wells were divided into six groups for each item of experiment. The apoptotic rates of activated HSCs treated with different concentrations of somatostatin for 72 h, were measured by acridine orange (AO)/ethidium bromide (EB) fluorescent staining, terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL), transmission electron microscopy and flow cytometry, respectively.
Firstly, HSC suspension was prepared via trypsinized method and modulated to a density of 0.5 × 106 cells/mL. Then it was mixed with AO/EB solution (PBS medium containing 0.1 mg/mL AO and 0.1 mg/mL EB) at a proportion of 25:1. Apoptotic cells among 300 randomly selected HSCs were distinguished under high power fluorescent microscope.
Secondly, HSCs fixed with 40 g/L polyformaldehyde were processed in accordance with the instructions of the TUNEL kit (Boster, China): (a) treatment by 30 mL/L H2O2 and pronase K successively, (b) reaction with the reactive mixture consisting of 1 μL of TdT, 1 μL of digoxin-labeled dUTP, 18 μL of signal buffer and 20 μL of signal medium, in a wet box at 37 °C overnight, (c) incubation with blocking medium, anti-digoxin-biotin and SABC sequentially, (d) exposure to DAB for coloration. Thereafter, twenty high power fields under the microscope in each glass slide were chosen at random and positive cells with brown nuclei were counted. Apoptosis Index (AI) = (apoptotic cells/total cells) × 100%.
Thirdly, the following steps were taken for the digested HSCs: namely to rinse with PBS, to fix with cold ethanol at -20 °C, to incubate with 100 μL 10 g/L RNase at 37 °C for 15 min, and to stain with propidium iodide (PI) for 30 min in darkness. Then the apoptotic rate and cell cycle could be obtained by flow cytometry.
Finally, HSCs in culture flask were fixed in 20 g/L glutaraldehyde, centrifuged, embedded in glass and sliced in series. HSC ultrastructure was observed by transmission electron microscopy.
Statistical analysis
Results were expressed as mean ± SD. One way ANOVA and t test were applied for data analysis. P values less than 0.05 were considered statistically significant.
RESULTS
Effects of somatostatin on HSC proliferation
Data taken from colorimetric MTT assays showed that somatostatin (10-6 mol/L-10-9 mol/L) could significantly inhibit the proliferation of activated HSCs in a dose-dependent way (Table 1). As compared with the control group, the inhibitory rates of somatostatin treatment groups rose up to 30.53% and 15.81% when the concentrations of somatostatin reached 10-6 mol/L and 10-7 mol/L, respectively. But no obvious effect was found in 10-10 mol/L somatostatin treatment group.
Table 1.
Effects of somatostatin on HSC proliferation (mean ± SD)
| SST concentration (mol/L) | ODs | Inhibitory rates (%) |
| 10-6 | 0.19 ± 0.03b | 30.53 |
| 10-7 | 0.23 ± 0.02b | 15.81 |
| 10-8 | 0.25 ± 0.03 | 7.77 |
| 10-9 | 0.26 ± 0.03 | 4.48 |
| 10-10 | 0.27 ± 0.04 | 0 |
| 0 | 0.27 ± 0.04 | 0 |
P < 0.01 vs control group.
Apoptosis of HSCs examined by fluorescent staining
After 72 h’ treatment, round-shaped HSCs with shrunk nuclei and conglomerated granules became visible in all somatostatin treatment groups. These HSCs underwent early apoptosis and displayed stronger green fluorescence. The experimental results showed that the higher the concentration of somatostatin, the stronger its proapoptotic action (Figure 1). The apoptosis-inductive effect of somatostatin reached its maximum at the concentration of 10-6 mol/L-10-7 mol/L (P < 0.05).
Figure 1.
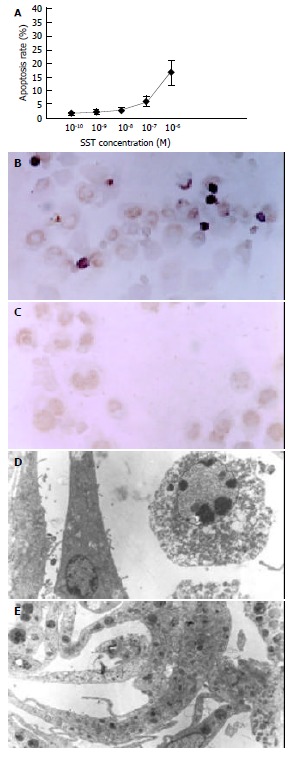
Effect of somatostatin on apoptosis of HSCs. A: Apoptosis rate of somatostatin-treated HSCs (fluorescent staining), B: Apoptosis of 10-6M somatostatin-treated HSCs dem-onstrated by TUNEL ( × 400), C: Apoptosis could not be detected in normal HSCs by TUNEL ( × 400), D: Apoptosis of 10-6M soma-tostatin-treated HSCs demonstrated by transmission electron mi-croscopy ( × 8000). E: Apoptosis could not be detected in normal HSCs by transmission electron microscopy ( × 8000).
Apoptosis of HSC examined by flow cytometry
As detected by flow cytometry, somatostatin of different concentrations could dose-dependently promote apoptosis of activated HSCs. In 10-6 mol/L and 10-7 mol/L somatostatin treatment groups, the apoptotic rate was significantly higher than that in the control group (P < 0.01) (Table 2).
Table 2.
Effects of somatostatin on HSC apoptosis (mean ± SD)
| Somatostatin concentration (mol/L) | Apoptosis rate (%) |
| 10-6 | 16.54 ± 4.59b |
| 10-7 | 6.06 ± 1.79b |
| 10-8 | 2.81 ± 0.72 |
| 10-9 | 2.25 ± 0.98 |
| 10-10 | 1.55 ± 0.64 |
| 0 | 1.53 ± 1.19 |
P < 0.01 vs control group.
Analysis of cytodynamics demonstrated that somatostatin could increase the percentage of HSCs in G0/G1 phase, and reduce it in S phase. However, there seemed to be no alternation in G2/M phase. Experimental findings also made it clear that the percentage of somatostatin-treated HSCs was 93.14 ± 6.69% (10-6 mol/L) and 90.65 ± 8.03% (10-7 mol/L) in G0/G1 phase, and 78.48 ± 4.43% in control group respectively. Meanwhile, the percentage of somatostatin treated HSCs were 1.75 ± 0.44% (10-6 mol/L), 3.87 ± 0.91% (10-7 mol/L) in S phase, and 12.50 ± 2.89% in control group, respectively. The difference between somatostatin treatment groups and control group was statistically significant (P < 0.01-0.05).
Apoptosis of HSC examined by TUNEL
The number of early apoptotic HSCs characterized by pyknosis of nuclear chromatin and condensation of cytoplasm, increased with somatostatin treatment in a dose-dependent manner (Figure 1). The apoptotic rates of different somatostatin treatment groups were 14.65 ± 3.86% (10-6 mol/L), 5.97 ± 1.04% (10-7 mol/L), 2.30 ± 0.62% (10-8 mol/L), 2.02 ± 0.81% (10-9 mol/L) and 1.65 ± 0.88% (10-10 mol/L), respectively. Statistical difference was found in the rates between two somatostatin treatment groups (10-6 mol/L, 10-7 mol/L) and the control group (P < 0.01).
Apoptosis of HSC examined by transmission electron microscopy
Typical apoptotic HSCs could be identified under transmission electron microscope after treatment of somatostatin for 72 h. They were characterized by shrinkage of cells with vacuoles in cytoplasm, swelling of mitochondria, dilated endoplasmic reticulum, irregular nuclei and pyknosis and conglomeration of chromatin ranging along inside of the nuclear membrane (Figure 1).
DISCUSSION
Somatostatin, an important peptide for inhibiting cellular proliferation and differentiation, could slow down the growth of various kinds of cells by blocking the synthesis and/or secretion of many important cytokines and hormones[10-14]. On the other hand, extensive physiological effects of somatostatin and its analogues are mainly mediated by five subtypes of somatostatin receptors (SSTR), including SSTR1, SSTR2 (SSTR2a and SSTR2b), SSTR3, SSTR4 and SSTR5. According to our knowledge, somatostatin-14 may be the main endogenous molecular form, although somatostatin-20, somatostatin-25 and somatostatin-28 also exist in the human body.
In recent years, researches have revealed the involvement of somatostatin and its receptors in the differentiation of HSC, i.e., somatostatin-containing nerve fibers inside the liver lobule are in close contact with sinusoidal endothelium as well as HSC[15]. In 2001 Reynaert et al[16] demonstrated that HSCs expressed SSTR1, SSTR2 and SSTR3 during their activation. Therefore, it is reasonable to deduce that somatostatin may play a negative regulatory role in the activation of HSCs via paracrine route. Except for its actions through SSTRs, somatostatin exerts inhibitory effect on the secretion of mitogens, including insulin-like growth factor (IGF), EGF and TGF-α, which are essential for the continuous activation of HSCs. In addition, somatostatin can even reverse the action of EGF on EGFR. So somatostatin may be quite a potent antifibrotic agent for liver cirrhosis.
Being consistent with the hypothesis, somatostatin shows an obvious inhibitory effect on the proliferation of activated HSCs in a dose-dependent way. Its pharmacological action is most significant when its concentration reaches 10-6 mol/L or 10-7 mol/L. Once somatostatin binds to SSTRs on HSCs, the occurrence of cytostatic effect is due to: (a) inhibition of adenylate cyclase activity through Giα1, leading to a decrease in cAMP level in cells; (b) active induction of K+ channel via Giα3, whereas blockage of the voltage-operated Ca2+ channel through Goα so as to reduce intracellular Ca2+ concentration; (c) inhibition of mitogen-activated protein kinase (MAPK)-dependent signal transduction pathway on the basis of protein tyrosine phosphatase (PTP) activation and amino-dephosphorylation-induced deactivation of tyrosine kinase. Then the expression of c-fos, c-jun and c-myc was reduced significantly[17,18]. As for the action of somatostatin on cytodynamics, G0/G1 arrest was observed in our study, which was in accordance with the result of Sharma et al[19]. Cell-cycle-arrest may reflect the main mechanism of somatostatin.
In vivo and in vitro studies have illustrated that activated HSCs can be eliminated by different ways such as spontaneous apoptosis, stimulation of some membrane receptors, incubation with proapoptotic compounds[20-28]. Activation of SSTR3 by somatostatin may be relevant to this process. Binding of SSTR3 and its natural or synthetic ligands can trigger intracellular acidification, and cause a selective activation of cation-insensitive acidic endonuclease through PTP. Then DNA fragmentation caused by acidic endonuclease stimulates the overexpression of bax and wild-type p53, resulting in apoptosis of HSCs. Additionally, dephosphorylation of serine in wild-type p53 has been found to be another pathway towards apoptosis[29,30]. Our study has proved biochemically and ultrastructurally for the first time that somatostatin can dose-dependently promote the apoptosis of first-passage activated HSCs by fluorescent staining, TUNEL, flow cytometry and transmission electron microscopy. Furthermore, a significantly increased apoptosis rate could be obtained with low concentrations (10-6 mol/L-10-7 mol/L) of somatostatin.
In conclusion, somatostatin at a low dose may exert antiproliferative and proapoptotic actions on activated HSCs. This result may provide a basis for utilizing somatostatin and even its analogues to protect people from hepatic fibrosis and to treat those suffering from it. However, the underlying mechanisms need to be further studied.
Footnotes
Supported by the Scientific Development Programs of Science and Technology Commission Foundation of Shanghai, No. 004119047
Edited by Zhu LH and Wang XL Proofread by Xu FM
References
- 1.Liu WB, Yang CQ, Jiang W, Wang YQ, Guo JS, He BM, Wang JY. Inhibition on the production of collagen type I, III of activated hepatic stellate cells by antisense TIMP-1 recombinant plasmid. World J Gastroenterol. 2003;9:316–319. doi: 10.3748/wjg.v9.i2.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wei HS, Lu HM, Li DG, Zhan YT, Wang ZR, Huang X, Cheng JL, Xu QF. The regulatory role of AT 1 receptor on activated HSCs in hepatic fibrogenesis: effects of RAS inhibitors on hepatic fibrosis induced by CCl(4) World J Gastroenterol. 2000;6:824–828. doi: 10.3748/wjg.v6.i6.824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li X, Meng Y, Yang XS, Wu PS, Li SM, Lai WY. CYP11B2 expression in HSCs and its effect on hepatic fibrogenesis. World J Gastroenterol. 2000;6:885–887. doi: 10.3748/wjg.v6.i6.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liang ZW, Zhang G, Wang TC. Extracellular signal-regulated kinase in liver fibrogenesis of rat. Shijie Huaren Xiaohua Zazhi. 2003;11:730–732. [Google Scholar]
- 5.Wang JY, Zhang QS, Guo JS, Hu MY. Effects of glycyrrhetinic acid on collagen metabolism of hepatic stellate cells at different stages of liver fibrosis in rats. World J Gastroenterol. 2001;7:115–119. doi: 10.3748/wjg.v7.i1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen PS, Zhai WR, Zhou XM, Zhang JS, Zhang YE, Ling YQ, Gu YH. Effects of hypoxia, hyperoxia on the regulation of expression and activity of matrix metalloproteinase-2 in hepatic stellate cells. World J Gastroenterol. 2001;7:647–651. doi: 10.3748/wjg.v7.i5.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng ML, Wu J, Wang HQ, Xue LM, Tan YZ, Ping L, Li CX, Huang NH, Yao YM, Ren LZ, et al. Effect of Maotai liquor in inducing metallothioneins and on hepatic stellate cells. World J Gastroenterol. 2002;8:520–523. doi: 10.3748/wjg.v8.i3.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu XJ, Yang L, Mao YQ, Wang Q, Huang MH, Wang YP, Wu HB. Effects of the tyrosine protein kinase inhibitor genistein on the proliferation, activation of cultured rat hepatic stellate cells. World J Gastroenterol. 2002;8:739–745. doi: 10.3748/wjg.v8.i4.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang LT, Zhang B, Chen JJ. Effect of anti-fibrosis compound on collagen expression of hepatic cells in experimental liver fibrosis of rats. World J Gastroenterol. 2000;6:877–880. doi: 10.3748/wjg.v6.i6.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xia D, Zhao RQ, Wei XH, Xu QF, Chen J. Developmental patterns of GHr and SS mRNA expression in porcine gastric tissue. World J Gastroenterol. 2003;9:1058–1062. doi: 10.3748/wjg.v9.i5.1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo Y, Guo X, Yao XX. Changes of gastrointestinal hormones in chronic atrophic gastritis and their clinical significance. Shijie Huaren Xiaohua Zazhi. 2003;11:531–534. [Google Scholar]
- 12.Yao YL, Xu B, Zhang WD, Song YG. Gastrin, somatostatin, and experimental disturbance of the gastrointestinal tract in rats. World J Gastroenterol. 2001;7:399–402. doi: 10.3748/wjg.v7.i3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun FP, Song YG, Cheng W, Zhao T, Yao YL. Gastrin, somatostatin, G and D cells of gastric ulcer in rats. World J Gastroenterol. 2002;8:375–378. doi: 10.3748/wjg.v8.i2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li YY. Mechanisms for regulation of gastrin and somatostatin release from isolated rat stomach during gastric distention. World J Gastroenterol. 2003;9:129–133. doi: 10.3748/wjg.v9.i1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stoyanova II, Gulubova MV. Immunocytochemical study on the liver innervation in patients with cirrhosis. Acta Histochem. 2000;102:391–402. doi: 10.1078/0065-1281-00568. [DOI] [PubMed] [Google Scholar]
- 16.Reynaert H, Vaeyens F, Qin H, Hellemans K, Chatterjee N, Winand D, Quartier E, Schuit F, Urbain D, Kumar U, et al. Somatostatin suppresses endothelin-1-induced rat hepatic stellate cell contraction via somatostatin receptor subtype 1. Gastroenterology. 2001;121:915–930. doi: 10.1053/gast.2001.27971. [DOI] [PubMed] [Google Scholar]
- 17.Ferjoux G, Bousquet C, Cordelier P, Benali N, Lopez F, Rochaix P, Buscail L, Susini C. Signal transduction of somatostatin receptors negatively controlling cell proliferation. J Physiol Paris. 2000;94:205–210. doi: 10.1016/s0928-4257(00)00206-0. [DOI] [PubMed] [Google Scholar]
- 18.Feng DY, Zheng H, Tan Y, Cheng RX. Effect of phosphorylation of MAPK and Stat3 and expression of c-fos and c-jun proteins on hepatocarcinogenesis and their clinical significance. World J Gastroenterol. 2001;7:33–36. doi: 10.3748/wjg.v7.i1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharma K, Patel YC, Srikant CB. C-terminal region of human somatostatin receptor 5 is required for induction of Rb and G1 cell cycle arrest. Mol Endocrinol. 1999;13:82–90. doi: 10.1210/mend.13.1.0220. [DOI] [PubMed] [Google Scholar]
- 20.Zhao WX, Zhao J, Liang CL, Zhao B, Pang RQ, Pan XH. Effect of caffeic acid phenethyl ester on proliferation and apoptosis of hepatic stellate cells in vitro. World J Gastroenterol. 2003;9:1278–1281. doi: 10.3748/wjg.v9.i6.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yao XX, Tang YW, Yao DM, Xiu HM. Effects of Yigan Decoction on proliferation and apoptosis of hepatic stellate cells. World J Gastroenterol. 2002;8:511–514. doi: 10.3748/wjg.v8.i3.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang XL, Liu L, Jiang HQ. Salvia miltiorrhiza monomer IH764-3 induces hepatic stellate cell apoptosis via caspase-3 activation. World J Gastroenterol. 2002;8:515–519. doi: 10.3748/wjg.v8.i3.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu XJ, Yang L, Wu HB, Qiang O, Huang MH, Wang YP. Apoptosis of rat hepatic stellate cells induced by anti-focal adhesion kinase antibody. World J Gastroenterol. 2002;8:734–738. doi: 10.3748/wjg.v8.i4.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Issa R, Williams E, Trim N, Kendall T, Arthur MJ, Reichen J, Benyon RC, Iredale JP. Apoptosis of hepatic stellate cells: involvement in resolution of biliary fibrosis and regulation by soluble growth factors. Gut. 2001;48:548–557. doi: 10.1136/gut.48.4.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wright MC, Issa R, Smart DE, Trim N, Murray GI, Primrose JN, Arthur MJ, Iredale JP, Mann DA. Gliotoxin stimulates the apoptosis of human and rat hepatic stellate cells and enhances the resolution of liver fibrosis in rats. Gastroenterology. 2001;121:685–698. doi: 10.1053/gast.2001.27188. [DOI] [PubMed] [Google Scholar]
- 26.Fischer R, Schmitt M, Bode JG, Häussinger D. Expression of the peripheral-type benzodiazepine receptor and apoptosis induction in hepatic stellate cells. Gastroenterology. 2001;120:1212–1226. doi: 10.1053/gast.2001.23260. [DOI] [PubMed] [Google Scholar]
- 27.Trim N, Morgan S, Evans M, Issa R, Fine D, Afford S, Wilkins B, Iredale J. Hepatic stellate cells express the low affinity nerve growth factor receptor p75 and undergo apoptosis in response to nerve growth factor stimulation. Am J Pathol. 2000;156:1235–1243. doi: 10.1016/S0002-9440(10)64994-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lang A, Schoonhoven R, Tuvia S, Brenner DA, Rippe RA. Nuclear factor kappaB in proliferation, activation, and apoptosis in rat hepatic stellate cells. J Hepatol. 2000;33:49–58. doi: 10.1016/s0168-8278(00)80159-2. [DOI] [PubMed] [Google Scholar]
- 29.Liu D, Martino G, Thangaraju M, Sharma M, Halwani F, Shen SH, Patel YC, Srikant CB. Caspase-8-mediated intracellular acidification precedes mitochondrial dysfunction in somatostatin-induced apoptosis. J Biol Chem. 2000;275:9244–9250. doi: 10.1074/jbc.275.13.9244. [DOI] [PubMed] [Google Scholar]
- 30.Teijeiro R, Rios R, Costoya JA, Castro R, Bello JL, Devesa J, Arce VM. Activation of human somatostatin receptor 2 promotes apoptosis through a mechanism that is independent from induction of p53. Cell Physiol Biochem. 2002;12:31–38. doi: 10.1159/000047824. [DOI] [PubMed] [Google Scholar]