
Figure 1.
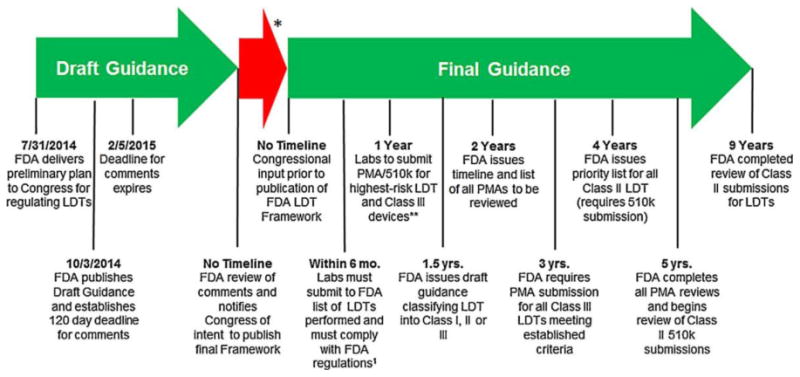
FDA timeline for implementation of oversight of laboratory-developed tests. *Time from FDA comment review and congressional input to the release of the final guidance is unknown. **PMA process is based on a per se demonstration of safety and effectiveness through “adequate and well-controlled” clinical trials. A successful PMA submission results in approval of the new device. The 510(k) process is somewhat analogous to the “generic” drug concept in that Premarket Notification is used to obtain marketing clearance for a device that is “substantially equivalent” in safety and effectiveness to another lawfully marketed device or to a standard recognized by the FDA when used for the same intended purpose(s).1 Federal regulations include medical device reporting as defined in the Code of Federal Register (CFR §803) and implementation of quality systems regulation (21 CFR §820).