

Abstract
We recently reported that the P2Y2 receptor (P2Y2R) is the predominant nucleotide receptor expressed in human coronary artery endothelial cells (HCAEC) and that P2Y2R activation by ATP or UTP induces dramatic up-regulation of tissue factor (TF), a key initiator of the coagulation cascade. However, the molecular mechanism of this P2Y2R-TF axis remains unclear. Here, we report the role of a newly identified AP-1 consensus sequence in the TF gene promoter and its original binding components in P2Y2R regulation of TF transcription. Using bioinformatics tools, we found that a novel AP-1 site at −1363 bp of the human TF promoter region is highly conserved across multiple species. Activation of P2Y2R increased TF promoter activity and mRNA expression in HCAEC. Truncation, deletion, and mutation of this distal AP-1 site all significantly suppressed TF promoter activity in response to P2Y2R activation. EMSA and ChIP assays further confirmed that upon P2Y2R activation, c-Jun, ATF-2, and Fra-1, but not the typical c-Fos, bound to the new AP-1 site. In addition, loss-of-function studies using siRNAs confirmed a positive transactivation role of c-Jun and ATF-2 but unexpectedly revealed a strong negative role of Fra-1 in P2Y2R-induced TF up-regulation. Furthermore, we found that P2Y2R activation promoted ERK1/2 phosphorylation through Src, leading to Fra-1 activation, whereas Rho/JNK mediated P2Y2R-induced activation of c-Jun and ATF-2. These findings reveal the molecular basis for P2Y G protein-coupled receptor control of endothelial TF expression and indicate that targeting the P2Y2R-Fra-1-TF pathway may be an attractive new strategy for controlling vascular inflammation and thrombogenicity associated with endothelial dysfunction.
Keywords: endothelial cell, G protein-coupled receptor, gene expression, gene transcription, tissue factor, vascular biology, Fra-1, P2Y2 nucleotide receptor
Introduction
Nucleotides matter more than being the universal currency of energy transaction and the building blocks of genes. They are also short term signaling molecules and long term trophic factors through P2X or P2Y receptors (1). Among the eight G protein-coupled P2Y nucleotide receptors, the ADP-preferring P2Y12 receptor on platelets is targeted by Plavix to prevent thrombosis in cardiovascular diseases, including percutaneous coronary intervention and atherosclerosis (2, 3). In addition, non-platelet P2Y1, P2Y2, and P2Y6 receptors are documented to mediate inflammatory response and plaque formation in animal models of atherosclerosis or restenosis (4–6). A potential role of the P2Y2 receptor (P2Y2R)2 in thrombosis is barely recognized, but it is already targeted in clinical drug development to treat cystic fibrosis (7) and dry eye disease (8). We recently reported that stimulation of human coronary artery endothelial cells (HCAEC) by ATP/UTP leads to dramatic up-regulation of tissue factor (TF) expression and activity through the P2Y2R (11). Although we have defined the post-P2Y2R signaling mechanism in control of TF expression, the exact transcriptional and post-transcriptional mechanisms remain to be determined.
TF is a membrane-bound transmembrane glycoprotein that is normally not exposed to the blood circulation. It facilitates both intrinsic and extrinsic pathways of coagulation and is a crucial protein in thrombotic diseases. In a normal blood vessel, TF is present mostly in tunica adventitia (fibroblasts), moderately in tunica media (smooth muscle cells), but virtually undetectable in endothelium (9). However, a diversity of stimuli can induce TF expression in endothelium. Some are endogenous molecules, such as TNF-α, interleukin-1β, CD40 ligand (10), or UTP/ATP (11); others are exogenous factors, including endotoxin (12) and caffeine (13). These inducers share some similar signal transduction pathways regulating TF induction in different cells. Among them, the MAPKs p38, p42/44 (ERK1/2), and c-Jun N-terminal kinase (JNK), and protein kinase C are commonly discussed as the positive regulatory pathways. In contrast, the PI3K/AKT pathway is shown to negatively regulate TF expression through an unknown mechanism (10). In this regard, our initial study already showed that endothelial P2Y2R activates ERK1/2, JNK, and p38 MAPK pathways without affecting the PI3K/AKT negative pathway (11). At the transcriptional level, TF was shown to be under concerted regulation of NF-κB, AP-1, and Sp-1 in porcine aortic endothelial cells by binding to sequences in an ∼300-bp region from the transcription starting site (14). Egr-1 is also involved in TF transcription in mononuclear cells and affects progression of diseases such as pulmonary fibrosis (15, 16). However, the transcriptional molecular mechanism in response to G protein-coupled receptor activation, i.e. P2Y2R, remains largely unknown.
Increased TF expression and activity are characterized in unstable atherosclerotic plaques and correlated with a higher risk of atherothrombosis, which leads to myocardial infarction and ischemic stroke (17). Elevated levels of TF are also characterized in patients with other cardiovascular complications such as hypertension, diabetes, and dyslipidemia (10). To understand how TF expression is up-regulated at the transcriptional level after P2Y2R activation, we have performed a bioinformatics search and found a new distal AP-1-binding site beyond the conventional ∼300-bp TF promoter region. Thus, the principal goal of this study was to determine whether this new AP-1 site is active in response to P2Y2R activation and, if so, to identify the exact AP-1 components controlled by P2Y2R signaling. Our findings demonstrate that in HCAEC, this new distal AP-1 site plays a significant role in P2Y2R-mediated TF transcription by recruiting c-Jun and a new positive regulator ATF-2. Furthermore, a negative regulator Fra-1 for the TF gene was also identified for the first time.
Experimental Procedures
Cell Culture
HCAEC were cultured in EBM-2 medium supplemented with VEGF, FGF, EGF, IGF, ascorbic acid, GA 1000 (Lonza), and 5% FBS at 37 °C in a humidified atmosphere of 5% CO2. HCAEC were used between the third and eighth passages. Before stimulation, cells were seeded to grow for 24 h and starved overnight. Where inhibitors were used, cells were pretreated with inhibitors for 1 h before stimulation.
RT-PCR Analysis
Total RNA and DNA were extracted from HCAEC using the RNeasy and DNeasy kits, respectively (Qiagen). On-column DNA digestion was carried out during RNA extraction. For the synthesis of the first strand of cDNA, 1 μg of total RNA after DNase (Ambion) treatment was reverse-transcribed using a cDNA synthesis kit (Applied Biosystems). The cDNA samples were then amplified by PCR using 2.5 units of TaqDNA polymerase (Qiagen). Real time PCR was performed on an iCycler iQ5 detection system (Bio-Rad) with SYBR Green reagents (Qiagen). The sequences of primers are as follows: TF mature mRNA: forward, 5′-ACGCTCCTGCTCGGCTGGGT-3′, and reverse, 5′-CGTCTGCTTCACATCCTTCA-3′; TF hnRNA (pre-mRNA): forward, 5′-CCCCTGGGTTGCTATGAGG-3′, reverse, 5′-CCTGGCTGTGGTGTTCTGTGC-3′; GAPDH: forward, 5′-TCAACAGCGACACCCACTCC-3′, and reverse, 5′-TGAGGTCCACCACCCTGTTG-3′.
Western Blotting Assay
After stimulation, cells were lysed, and standard Western blotting was performed as described previously (18–20). The individual primary antibodies used were anti-p-c-Jun, anti-p-c-Fos, anti-p-ATF-2, anti-c-Jun, anti-Fra-1, anti-ATF-2 (Cell Signaling Technology), and anti-tissue factor (10H10, Calbiochem). Equal protein loading was verified by stripping off the original antibodies and re-probing the membranes with the primary antibody anti-GAPDH, anti-β-tubulin, or anti-β-actin (Cell Signaling Technology).
Tissue Factor Promoter Constructs
Promoter constructs encompassing the region from −1427 to +207 relative to the transcription starting site of the human TF gene were amplified from human genomic DNA using specifically designed forward and reverse primers containing the EcoRV restriction enzyme site. The forward primer is 5′-GCTAGCCTCGAGGATATCCTACCTTCAATCCCAGAG-3′, and the reverse primer is AGGCCAGATCTTGATATCTCCATGTCTACCAGTTGGCG-3′. The promoter region was cloned into pGL4.12 vector (Promega) with In-Fusion HD EcoDry cloning kit (Clontech). Deleted, truncated, or mutated constructs were built with same method but with different primers as follows: deletion forward primer, 5′-CTCTTAGGGAAAAGGCTAGAGCCTGCATAAAAAGAG-3′, and deletion reverse primer, 5′-CCTTTTCCCTAAGAGATCTTCAGCTCCACCTGGGAT-3′; truncation forward primer, 5′-GCTAGCCTCGAGGATATCGGCTAGAGCCTGCATAAAAAG-3′, and truncation reverse primer is same as wild-type reverse primer; mutation forward primer, ′-GGAAAAGTCAGACGGC-TAGAGCCTGCATAAAAAGAG-3′, and mutation reverse primer, 5′-CCGTATGACTTTTCCCTAAGAGATCTTCAGCTCCAC-3′.
Plasmid Transfection and Luciferase Activity Assay
HCAEC were seeded in 48-well plates and transiently transfected with various pGL4.12 vector constructs with Xfect Transfection Reagent (Clontech) following the manufacturer's instruction. Transfection efficiency was corrected by co-transfection with a plasmid containing the β-galactosidase gene driven by the SV40 promoter (Promega). β-Galactosidase activity was measured with β-Galactosidase Enzyme Assay System (Promega). Luciferase activity was measured for reporter expression in triplicate according to the instructions provided in the Luciferase Reporter Assay System (Promega).
Chromatin Immunoprecipitation Assay
The chromatin immunoprecipitation (ChIP) assay was performed using the Agarose ChIP kit (Pierce) according to the manufacturer's instructions. HCAEC were treated with or without UTP stimulation, after which cells were cross-linked in formaldehyde. Micrococcal nuclease was used to digest the chromatin. Approximately 10% of the resulting sample was kept to serve as an input genomic DNA control, and the rest was incubated with candidate antibody overnight with rotation, followed by 1 h of incubation with protein A-agarose beads for ChIP. The precipitated chromatin-protein complexes were washed and eluted, and the cross-links were reversed. The genomic DNA fragments were amplified by real time quantitative PCR. For the sequence containing the new AP-1 site, the primer sequences were as follows: forward, 5′-CTACCTTCAATCCCAGAG-3′, and reverse, 5′-CCTTGTAGACTTTCCTTCC-3′.
Electrophoretic Mobility Shift Assay
DNA probe fragments were ordered from IDT and labeled with biotin at the 3′ end with the DNA 3′ end biotinylation kit (Pierce). Nuclear proteins were isolated from HCAEC with NE-PER nuclear and cytoplasmic extraction reagents (Pierce). Each binding reaction contained 10 μg of nuclear extract proteins. To distinguish specific from nonspecific protein-DNA complex formation, incubations were also performed in the presence of 200-fold excess unlabeled probes. After incubation, the reaction mixtures were electrophoresed on a pre-casted 6% TBE polyacrylamide gel (Lonza). The reactions were transferred to a nylon membrane and detected with LightShift chemiluminescent EMSA kit (Pierce). The sequence of the human TF promoter −1363 AP-1 site is as follows: 5′-GGAAAATGACTCAGGCTAGA-3′. Antibodies against the individual AP-1 subunits (Santa Cruz Biotechnology) and 10 μg of nuclear extracts were incubated overnight at 4 °C before adding biotin-labeled probe to the reaction. The binding reactions then follow the steps as described for EMSA. To confirm the specificity of the antibodies, two different amounts of antibodies were used, and corresponding IgG was used in place of antibodies as negative controls.
Co-immunoprecipitation
HCAEC were starved overnight and treated with or without UTP for 30 min, lysed, and sonicated on ice. Cell lysate was pre-cleared and then incubated with 1 μg of rabbit anti-c-Jun antibody (Santa Cruz Biotechnology) overnight at 4 °C. Protein A-agarose beads (Cell Signaling Technology) were added to the lysate/antibody mixture and incubated for another 2 h. The beads were then washed five times with cell lysis buffer and resuspended with 3× SDS sample buffer. Samples were heated to 95 °C, loaded on SDS-polyacrylamide gel, and analyzed by standard Western blotting.
Immunofluorescence Assay
HCAEC were seeded in 8-chamber glass slides (Nunc), starved overnight, and then treated with or without UTP. After a 30-min treatment, the medium was aspirated, and cells were fixed for 15 min in cold methanol. The fixed cells were washed with PBS three times and blocked with 5% horse serum for 1 h at room temperature. Then the cells were incubated with rabbit monoclonal antibodies to human p-c-Jun, p-Fra-1, and p-ATF-2 (1:50) (Cell Signaling Technology) or rabbit polyclonal antibodies to human total c-Jun, Fra-1, and ATF-2 (1:50) (Santa Cruz Biotechnology), overnight at 4 °C followed by incubation with FITC-conjugated anti-rabbit IgG (1:200) (Kirkegaard & Perry Laboratories) for 90 min at room temperature in darkness. For negative controls, cells were incubated with non-immune rabbit IgG in place of the specific primary antibody or only the FITC-conjugated secondary antibody. Cells were then washed with PBS three times and incubated with Alexa Fluor 555 phalloidin (Cell Signaling Technology) for 1 h. Finally, mounting medium containing DAPI was added to seal the slides. Images with fluorescent signals in random fields were acquired and captured using an AMG EVOS digital inverted multifunctional microscope.
Silencing of AP-1 Subunits with siRNA
To knock down the three AP-1 subunits, c-Jun, Fra-1, and ATF-2, HCAEC were transfected with the four sequence pool (ON-TARGET plus Human SMARTpool L-003268-00-0005; L-004341-00-0005; L-009871-00-0005, Dharmacon) using DharmaFECT 4 transfection reagent following the manufacturer's protocol. Briefly, HCAEC were seeded in 6-well plates at 80–90% confluence; the medium was replaced with complete EBM-2 without antibiotics before transfection. DharmaFECT 4 and siRNA products were incubated separately in EBM-2 at room temperature for 5 min. Mixtures were combined, incubated another 20 min, and added to cells at a final concentration of 2 μl/ml DharmaFECT 4 and 25 nm siRNAs. Cells in the control group were treated with DharmaFECT 4 plus a scramble control siRNA. For UTP stimulation of the cells, siRNA and transfection reagent were removed 24 h post-transfection, and complete culture medium was added. After overnight starvation, cells were stimulated by UTP for 30 min or not. Western blotting was employed to verify knockdown efficiency 24 h post-transfection. Real time quantitative RT-PCR was used to detect TF pre-mRNA level after knockdown of the individual AP-1 subunits.
Materials
DNA primers were purchased from Integrated DNA Technologies. EcoRV was purchased from Takara Bio Inc. Anti-c-Jun, anti-ATF-2, and anti-Fra1 antibodies used in ChIP assay were from Santa Cruz Biotechnology. TNF-α was purchased from R&D Systems. Actinomycin D, U0126, Y-27632, SB203580, SP600125, 2-thio-UTP tetrasodium salt were purchased from Tocris Bioscience. PP2 was from EMD. Purified UTP was obtained from Sigma.
Data Analysis
Data are expressed as the mean ± S.E. The means of two groups were compared using Student's t test (unpaired, two tailed), and one-way analysis of variance was used for comparison of more than two groups with p < 0.05 considered to be statistically significant. Unless otherwise indicated, all experiments were repeated at least three times.
Results
P2Y2R Activation Boosts TF Expression by Promoting Gene Transcription
Because our previous study demonstrated that P2Y2R is the only UTP-sensitive nucleotide receptor expressed in HCAEC (11), UTP was routinely used to activate the P2Y2R throughout this study. Two pairs of primers were designed to selectively amplify non-spliced TF pre-mRNA (also termed hnRNA) and mature TF mRNA, respectively. The pair for mature mRNA spans from exon 1 to exon 3 with two introns in the middle. The amplicon length is 233 bp for the TF mature mRNA after splicing (Fig. 1A). The pair of primers for pre-mRNA is located in the same intron (intron 2), which will be spliced away before forming the mature mRNA. This pair of primers produces a 244-bp amplicon from the non-spliced pre-mRNA pool (Fig. 1A). Fig. 1B shows that activation of HCAEC by TNF-α increased the TF expression levels of pre-mRNA and mature mRNA, which peaked at 30 min and 1 h, respectively. A similar pattern of time-dependent induction of both TF pre-mRNA and mature mRNA by UTP stimulation of the P2Y2R was observed (Fig. 1, C and D). To rule out possible DNA contamination in TF pre-mRNA amplification, we performed PCR using DNA samples with or without DNase treatment that we routinely used in total RNA isolation. Fig. 1E (lanes 2 and 3) shows that the PCR experiment using the DNA sample produced an expected band, but DNA sample treated with DNase generated no band, indicating the effectiveness of our protocol in DNA removal. Fig. 1E further shows that UTP stimulation significantly induced TF pre-mRNA expression (lane 6 versus lane 4), but omission of reverse transcriptase in our RT-PCR assay generated no amplicons (lane 7), suggesting no DNA contamination. These data indicate that TF pre-mRNA and mature mRNA are changed similarly over time by TNF-α and UTP stimulation and that HCAEC is an appropriate cell model for studying TF transcriptional mechanisms.
FIGURE 1.
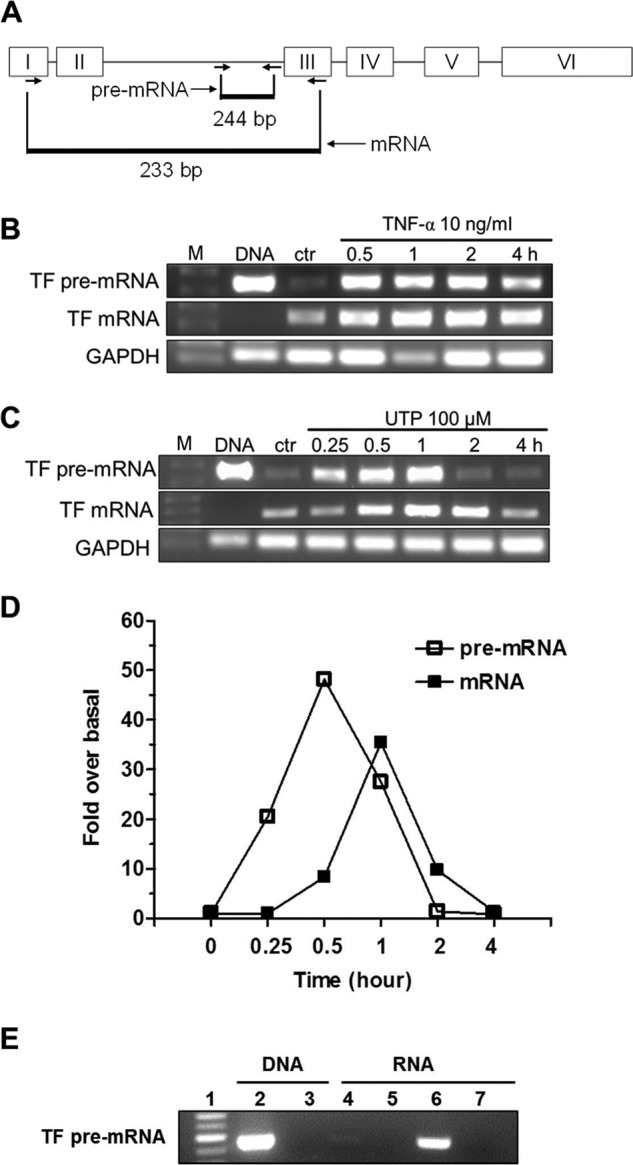
P2Y2R activation promotes TF gene transcription in HCAEC. Two pairs of primers were designed to quantify human TF mature mRNA and pre-mRNA (hnRNA) by RT-PCR, respectively. The relative position of primers to the TF gene is depicted in A. Representative RT-PCR gel images show time-dependent effect of TNF-α (B) or UTP (C) on the expression of TF pre-mRNA and mature mRNA. In both panels, the 1st lanes are DNA markers (M); 2nd lanes are PCR products of DNA as a positive control; 3rd lanes are from total RNA samples made from HCAEC without stimulation. The rest of the lanes reflect time course changes caused by TNF-α (B) and UTP (C), respectively. GAPDH was used as a control. Quantitative data on time course change of TF mRNA and pre-mRNA in response to UTP (100 μm) treatment were determined by real time RT-PCR (D). For clarity, error bars and markers for statistical analysis were omitted (n = 3). E, evidence of complete clearance of genomic DNA by DNase treatment in DNA or total RNA samples. Lane 1, standard DNA ladder 100 bp; lane 2, PCR product amplified from 200 ng of genomic DNA prepared from HCAEC; lane 3, PCR product amplified from 200 ng of genomic DNA treated with DNase; lane 4, product from RT-PCR of total RNA prepared from HCAEC without UTP stimulation; lane 5, PCR product amplified from non-reverse-transcribed total RNA prepared from HCAEC without UTP stimulation; lane 6, product from RT-PCR of total RNA prepared from HCAEC treated with 100 μm UTP for 1 h; lane 7, PCR product amplified from non-reverse-transcribed total RNA prepared from HCAEC treated with 100 μm UTP for 1 h (n = 3).
New AP-1-binding Site Regulates TF Gene Transcription
A classic proximate TF promoter model (up to −227 bp) has been established through the years and is consistently used to study TF transcription (Fig. 2A) (21). There are binding sites for four transcription factors in this region as follows: AP-1, NF-κB, Egr-1, and Sp1. Sp1 is required for constitutive TF gene transcription, although the other three are responsible for inducible TF gene transcription (21). Although Egr-1 is an immediate early gene product, our previous study showed that de novo protein synthesis is not required for P2Y2R-mediated TF induction (11). In addition, our prior study also excluded the involvement of the NF-κB pathway (11), leaving AP-1 as an attractive candidate. However, when we searched through TF promoter/enhancer region up to −5000 bp, we found another 100% consensus AP-1-binding site at −1363 bp (Fig. 2A). We also located the TF gene promoter in other mammalian species and found significant conservation of this new AP-1 site among them (Fig. 2B). To investigate this new AP-1-binding site, HCAEC were transfected with a luciferase construct and a plasmid containing β-galactosidase driven by the SV40 promoter to normalize the transfection efficiency. The TF promoter region up to −1427 bp containing the new AP-1 site or its deleted/truncated/mutated forms was cloned into a luciferase vector. 12 h after transfection, cells were starved overnight and then treated with 100 μm UTP for 4 h. Then luciferase and β-galactosidase activities were determined. Fig. 2C shows that UTP induced ∼8-fold of increase in luciferase activity over non-stimulated cells. Deletion, truncation, and mutation of this new AP-1 site all significantly decreased the promoter activity (Fig. 2C). In addition, treatment of HCAEC with the transcription inhibitor actinomycin D abolished UTP-induced TF promoter activity, suggesting transcription as the major mechanism (Fig. 2C). These data indicate that the new AP-1 site at −1363 bp has significant impact on TF gene transcription.
FIGURE 2.
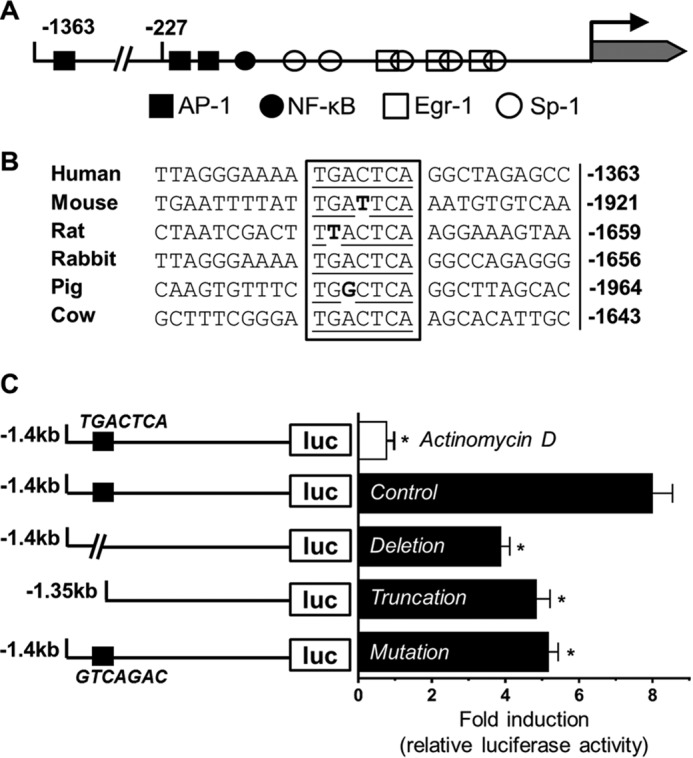
Critical role of a new distal AP-1-binding site in TF promoter. A, schematic description of human TF gene promoter and relevant transcription factor-binding sites. The new distal AP-1-binding site at −1363 bp is well conserved across multiple species (B). HCAEC were transfected with different luciferase plasmid constructs (C) and stimulated with UTP (100 μm) for 4 h, after which levels of luciferase activity relative to non-stimulated control were corrected for transfection efficiency, and summarized data are shown on the right. Top bar in white indicates result from co-treatment with UTP and actinomycin D (10 μg/ml). (n = 5, *, p < 0.05).
An EMSA was conducted with the biotin-labeled AP-1 probe equivalent to the region −1369 to −1350 bp in the human TF promoter to confirm our luciferase activity assay. We found that nuclear extract from HCAEC has higher occupancy over this TF AP-1 site after UTP stimulation of the cells for 30 min. In addition, excessive unlabeled probes competed against labeled ones and eliminated the signal, indicating the specificity of the protein-DNA complex. Moreover, TNF-α stimulation for 30 min caused the same change as UTP, which suggests that the same site may also be taken up in TNF-α-induced TF induction (Fig. 3A). Quantitative data indicate that both UTP and TNF-α treatment increased probe occupancy about 4-fold (Fig. 3B).
FIGURE 3.
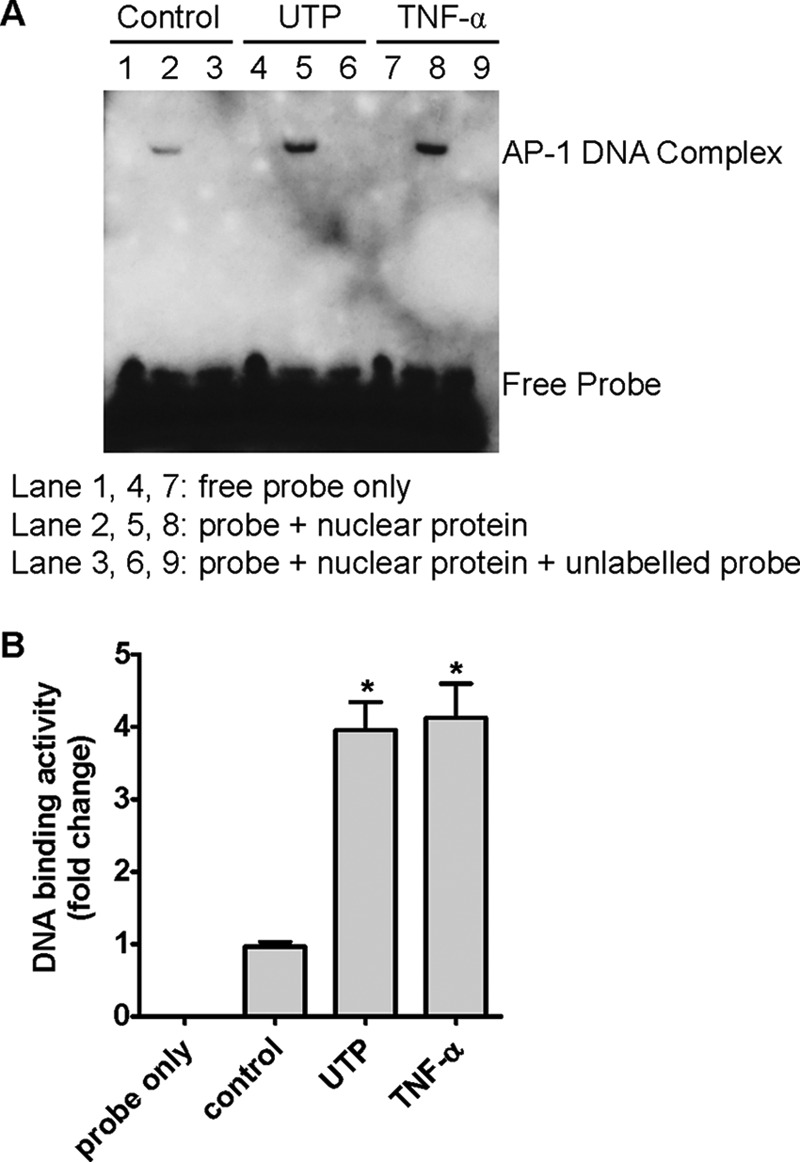
Evidence of nuclear protein binding to the new distal AP-1 site and its regulation by P2Y2R. A, HCAEC nuclear protein was isolated after treatment of the cells with UTP (lanes 4–6) or TNF-α (lanes 7–9) or vehicle (lanes 1–3) for 30 min, incubated with unlabeled and/or biotin-labeled probes, fractionated by electrophoresis, and visualized by ECL detection after the membrane was incubated with horseradish peroxidase-conjugated streptavidin. B, summarized data for band density of A were quantified with Quantity One. n = 4, *, p < 0.05.
P2Y2R Activation Has Differential Effects on AP-1 Subunits
AP-1 components bind to the DNA promoter in a dimerized form (23). The two subunits come from members of the Jun protein family (c-Jun, JunB, and JunD), Fos protein family (c-Fos, FosB, Fra-1, and Fra-2), and some members of the activating transcription factor (ATF) subfamily (ATFa, ATF-2, and ATF-3) (23). To determine the roles of different subunits, we first assessed the effect of P2Y2R activation on their phosphorylation status. Fig. 4, A–C and E–G, show that UTP or its analog 2-thio-UTP caused rapid phosphorylation of c-Jun, Fra-1, and ATF-2 in a dose- and time-dependent manner. However, UTP failed to induce phosphorylation of c-Fos, one of the most classic AP-1 subunits in a timely fashion (Fig. 4D). JunB and JunD, in contrast, were not detectable (data not shown). It is known that the Jun protein is mainly in charge of binding affinity, whereas the others rely on dimerizing with a Jun protein to influence the promoter activity (24). We furthered our study on subunits with a co-immunoprecipitation experiment to confirm that the phosphorylated Fra-1 and ATF-2 are c-Jun-bound so that they can recognize and attach to their targeting sites. Cell extracts from HCAEC treated with or without UTP were incubated with 1 μg of rabbit anti-c-Jun antibody, precipitated with protein A-agarose beads, and detected with antibodies specific for Fra-1, ATF-2, and c-Jun (as loading control). Fig. 4H shows that UTP treatment dramatically increased the amount of Fra-1 and ATF-2 bound to c-Jun, indicating their higher DNA-binding potential after forming a dimer with c-Jun.
FIGURE 4.
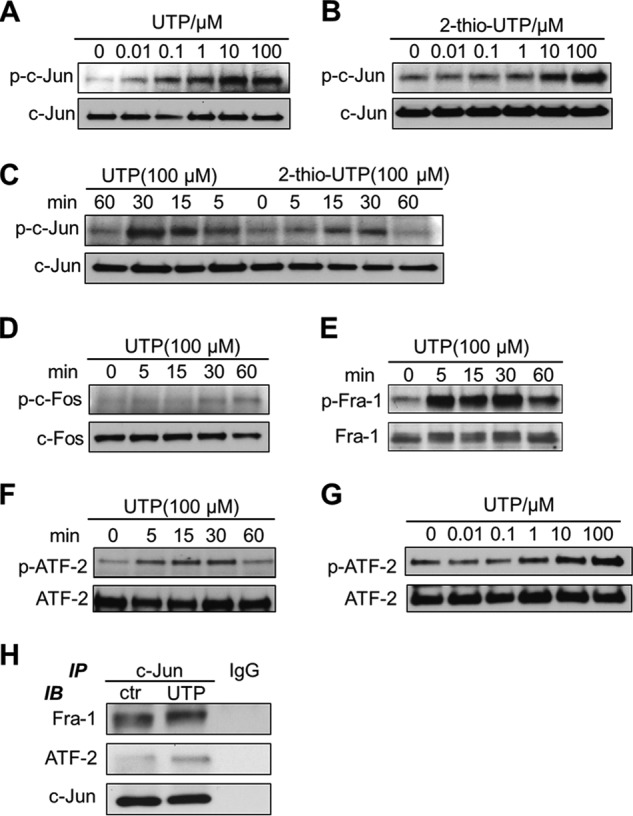
Differential effect of P2Y2R stimulation on AP-1 subunit activation. HCAEC were treated with UTP or its less degradable analog 2-thio-UTP and phosphorylation levels of c-Jun, c-Fos, Fra-1, and ATF-2 were detected by Western blotting. Concentration-dependent changes of phosphorylation levels after stimulation of the cells for 15 min are shown (A, B, and G). Time-dependent changes of phosphorylation levels after stimulation of the cells with 100 μm UTP or 2-thio-UTP are shown (C–F). Amount of Fra-1 and ATF-2 bound with c-Jun in HCAEC treated with or without UTP for 30 min was detected with co-immunoprecipitation and Western blotting (H). IgG was used as negative control. Data were representative of at least three independent experiments. IP, immunoprecipitation; IB, immunoblot.
For AP-1 subunits to be fully functional, they have to reside in the nucleus. The abundance of them in the nucleus correlates with their activity on the target (25).To define the location of phosphorylated c-Jun, Fra-1, and ATF-2, immunofluorescent images were compared between groups with or without UTP stimulation for 30 min. In control groups without stimulation, very little FITC signal was detected for any of the three phosphorylated subunits. In UTP-stimulated groups, however, intense green light from FITC indicates a high level of phosphorylated c-Jun, Fra-1, and ATF-2. Overlaying with DAPI and a phalloidin image shows that the majority of the phosphorylated subunits accumulated in the HCAEC nucleus, instead of the cytoplasm/cell skeleton (Fig. 5). Note that no positive staining was observed once p-c-Jun, p-Fra-1, and p-ATF-2 antibodies were replaced by its IgG control antibody or when only FITC-conjugated secondary antibody was used (Fig. 5). No significant change in FITC fluorescence was observed when using first antibodies against total c-Jun, total-Fra-1, or total ATF-2 in place of phosphorylated ones, respectively (data not shown).
FIGURE 5.
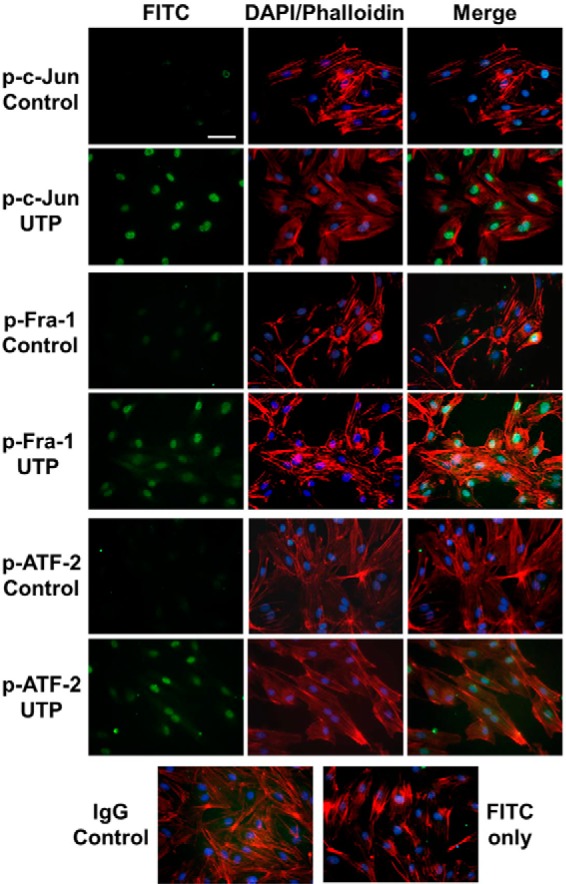
Nuclear accumulation of activated AP-1 subunits after P2Y2R stimulation in HCAEC. Locations of p-c-Jun, p-Fra-1, and p-ATF-2 were indicated by FITC staining (green). HCAEC were stimulated with vehicle (control) or 100 μm UTP for 30 min before fixing for standard immunofluorescence assays. Cell nuclei were counterstained with DAPI (blue). Cytoskeleton was stained with phalloidin (red). Isotype-matched primary antibody (IgG) and FITC-conjugated secondary antibody were used for negative controls. Scale bar, 20 μm. Data were representative of at least three independent experiments.
New AP-1 Site Is Responsible for Endogenous c-Jun, Fra-1, and ATF-2 Binding to TF Promoter
To confirm the binding specificity of the AP-1 components to the new AP-1-binding site, we further performed ChIP and EMSA studies using specific c-Jun, Fra-1, and ATF-2 antibodies and cell lysates with or without P2Y2R activation. With primers amplifying a PCR product spanning from −1427 to −1217 bp of TF promoter, which includes the AP-1 site, a 210-bp DNA fragment was amplified from anti-c-Jun, anti-Fra-1, and anti-ATF-2 chromatin precipitates by real time PCR (Fig. 6, A–C). All three precipitates from UTP-treated HCAEC resulted in a dramatic increment in DNA amplification with expected size and sequence. In addition to the ChIP assay, EMSA supershift assays were performed with labeled wild-type AP-1 site probe. Different AP-1 protein antibodies and control IgG were added to the nuclear extracts before electrophoresis to detect further decreases in electrophoretic mobility. The addition of c-Jun, Fra-1, and ATF-2 antibodies to the EMSA reaction mixture resulted in the inhibition of specific complex formation (Fig. 6, D–F), indicating that all three subunits are the proteins binding to this site in HCAEC after P2Y2R activation. A higher amount (2 μg) of antibodies caused more inhibition compared with a lower amount (1 μg) of the same antibody. Control IgG (2 μg) did not change the binding of the labeled probe with the AP-1 components. These data demonstrate that P2Y2R activation induces specific binding of c-Jun, Fra-1, and ATF-2 to the −1363-bp new AP-1 site of the TF gene in HCAEC.
FIGURE 6.

Identification of AP-1 components binding to the new distal AP-1 element within the TF promoter. Chromatin immunoprecipitation assay and electrophoretic mobility shift assay demonstrate specific binding of c-Jun, Fra-1, and ATF-2 to the new AP-1 site in response to P2Y2R activation in HCAEC. A–C, ChIP assay of the new AP-1 site in the TF promoter with anti-c-Jun, Fra-1, and ATF-2 antibodies using HCAEC chromatin. Histograms were acquired from four independent real time PCR experiments followed by electrophoresis gel imaging of amplified PCR products. D–F, confirmation of the roles of c-Jun, Fra-1, and ATF-2 binding to the new AP-1 site with EMSA experiments. 1 and 2 μg of antibody were used in binding reactions or 2 μg of IgG was used as negative controls. Location of arrows indicate band of interest. Data were representative of three independent experiments. IP, immunoprecipitation.
Knockdown of c-Jun, Fra-1, or ATF-2 Has Differential Roles in TF Transcription Following P2Y2R Activation
To determine the roles of the three AP-1 subunits in control of TF transcription, we knocked down c-Jun, Fra-1, and ATF-2, respectively, by an siRNA approach. Western blotting assay indicated a complete elimination of ATF-2 and a significant decrease of c-Jun and Fra-1 on their total protein levels in HCAEC (Fig. 7, A–C). The siRNA-transfected HCAEC were exposed to UTP for 30 min or not before TF pre-mRNA was quantified by real time PCR. Transfection with different siRNA pools did not change the basal levels of TF pre-mRNA without UTP treatment. Upon UTP treatment, however, TF pre-mRNA markedly decreased in HCAEC in the absence of c-Jun and ATF-2. Surprisingly, however, TF transcription level was significantly increased in cells with Fra-1 knockdown as compared with control scrambled siRNA-transfected ones (Fig. 7D). The same pattern was observed on the protein level of tissue factor expression after 4 h of UTP stimulation (Fig. 7E). Collectively, these data suggest that Fra-1 affects TF transcription negatively in response to P2Y2R activation in HCAEC, although the other two AP-1 subunits involved, c-Jun and ATF-2, are positive regulators.
FIGURE 7.
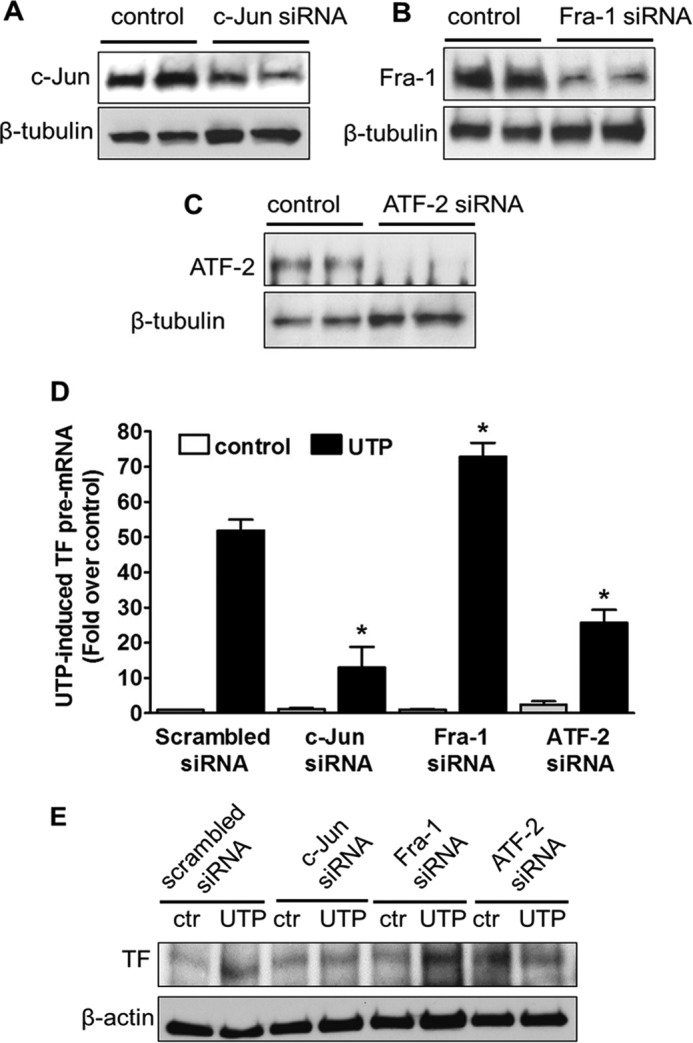
Differential roles of AP-1 subunits in P2Y2R-mediated TF gene transcription. A--C, c-Jun, Fra-1, and ATF-2 protein levels were determined by Western blotting after HCAEC were transfected with their respective siRNA for 48 h. UTP-induced TF pre-mRNA was evaluated after c-Jun, Fra-1, or ATF-2 was knocked down by siRNA in HCAEC. *, p < 0.05 versus UTP-stimulated cells were transfected with scrambled siRNA (D). n = 5. UTP-induced TF protein expression under the same siRNA intervention was determined by Western blotting (E). Data were representative of three independent experiments. ctr, control.
Effect of Different Post-P2Y2R Signaling Pathways on AP-1 Subunits and TF Gene Transcription
We then further explored which post-P2Y2R signaling pathways are responsible for regulating the three AP-1 subunits and net TF transcription. As shown in Fig. 8A, treatment of the cells with SP60012, a well established JNK inhibitor, abolished c-Jun phosphorylation completely after P2Y2R activation. Similarly, phosphorylation of ATF-2 was changed in exactly the same way as c-Jun after JNK inhibition (Fig. 8A). However, Fra-1 phosphorylation is resistant to JNK pathway inhibition (Fig. 8A). Afterward, we used the classic ERK1/2 inhibitor U0126 to study the effect of ERK1/2 on c-Jun, Fra-1, and ATF-2. As shown in Fig. 8B, U0126 dose-dependently inhibited UTP-induced Fra-1 phosphorylation, whereas the phosphorylation level of c-Jun and ATF-2 were insensitive. Given that Rho kinase is the upstream regulator of JNK as we reported previously (11), we then hypothesized that Rho may induce c-Jun and ATF-2 activation through JNK. To test this hypothesis, we examined the effect of the Rho kinase inhibitor Y27632 on c-Jun and ATF-2 phosphorylation. Preincubation of HCAEC with Y27632 at 3 μm inhibited UTP-induced c-Jun and ATF-2 phosphorylation (Fig. 8C). Similarly, we verified whether the other kinase activated by UTP treatment, Src (11), can act on ERK1/2 and its downstream Fra-1, because there is a well established connection between Src and ERK1/2 (26). Fig. 8D shows that increasing the dosage of the Src inhibitor PP2 gradually suppressed phosphorylation of Fra-1. These data clearly indicate that P2Y2R utilizes different signaling pathways to control the activity of the three AP-1 components.
FIGURE 8.

P2Y2R signaling mechanism in control of AP-1 subunits and TF gene transcription in HCAEC. Phosphorylation levels of AP-1 subunits in response to P2Y2R activation were determined with Western blotting after HCAEC were pretreated for 1 h with the individual inhibitor for JNK (A), ERK1/2 (B), Rho kinase (C), and Src (D). TF pre-mRNA levels in response to UTP stimulation (100 μm for 30 min) were analyzed with real time PCR after HCAEC were pretreated for 1 h with the individual inhibitor for JNK, ERK1/2, Rho kinase, and Src. *, p < 0.05 versus UTP-stimulated cells; **, p < 0.01 versus UTP-stimulated cells (E). n = 5. A systematic diagram shows a new two-way fine-tuning mechanism underlying P2Y2R-controlled cell signaling, AP-1 activation, and TF gene transcription in HCAEC (F).
Finally, we validated the impact of the above pathways on TF transcription. Because Src and ERK1/2 are in control of Fra-1, they are expected to have the same effect on TF transcription as Fra-1 does. Likewise, Rho and JNK should possess the opposite effect as compared with Src/ERK1/2. In line with our assumption, ERK1/2 inhibitor U0126 and Src inhibitor PP2 mimicked the effect of Fra-1 knockdown and boosted TF transcription. In contrast, the JNK inhibitor SP60012 and Rho inhibitor Y27632 act in the same manner as c-Jun and ATF-2 knockdown and caused a loss in TF transcription (Fig. 8E). As summarized in Fig. 8F, under P2Y2R activation, Src/ERK/Fra-1 pathway negatively regulates TF transcription, whereas Rho/JNK/c-Jun/ATF-2 pathway has a positive impact.
Discussion
In this study, we show for the first time that activation of P2Y2R in HCAEC stimulates TF transcription through a new distal AP-1-binding site. We also show that in addition to c-Jun, ATF-2 is a new positive regulator of TF transcription in response to P2Y2R signaling to the JNK pathway. Furthermore, we have identified Fra-1 as a first negative regulator fine-tuning the overall transcription activity of TF gene promoter, which is controlled by P2Y2R signaling to the ERK1/2 pathway.
Aside from cell apoptosis and necrosis, nucleotide release also occurs in different cells, including platelets, monocytes/macrophages, and vascular endothelial cells (27). These cells release nucleotides, including ATP and UTP, under various mechanical and other stimuli, such as shear stress, hypotonic swelling, hypoxia, as well as in response to thrombin and other Ca2+-mobilizing receptor agonists (27). Released extracellular nucleotides have been established as important mediators of vascular inflammation (28), thrombosis (29), and atherosclerosis (30) by acting on various platelet, endothelial, and leukocyte P2X and P2Y receptors. P2Y2R, one of the eight G protein-coupled P2Y nucleotide receptors (P2Y1, -2, -4, -6, and -11–14), has been shown to induce VCAM-1 expression in HCAEC (31) and ICAM-1 expression in vascular smooth muscle cells (32), suggesting its potential role in control of leukocyte interaction with the vascular walls.
Initially we showed that the P2Y2R is up-regulated in response to coronary artery stenting (33), and recently we also reported that activation of the human coronary artery P2Y2R induces a dramatic up-regulation of TF, the initiator of the coagulation cascade (11). This original finding has been extended to mouse macrophages by other groups showing a role of P2X7 receptor in LPS-induced TF up-regulation (34, 35). Together, these findings suggest that non-platelet nucleotide receptors, i.e. P2Y2R, may have a role in the initiation and/or progression of thrombogenesis. However, the exact molecular mechanisms underlying P2X or P2Y receptor-controlled TF expression remained largely unknown. In this study, we found that activation of P2Y2R in HCAEC increased not only the mature TF mRNA level but also the TF pre-mRNA level, consistent with our previous study that blocking cellular transcription by actinomycin D suppressed UTP-induced TF mRNA accumulation (11), indicating a transcriptional mechanism responsible for P2Y2R-mediated TF up-regulation in HCAEC, although additional mechanisms at a post-transcriptional level cannot be excluded.
Studies on TF transcription have been limited to a short promoter region up to −227 bp from the transcription start site (21). Binding sites for four types of transcription factors are located in this area. Of these four factors, Sp-1 is in charge of constitutive TF transcription, whereas Egr-1, NF-κB, and AP-1 are responsible for induced TF transcription (21). It is well known that Egr-1 needs to be synthesized de novo (22), suggesting that it may not play a significant role in our testing system, because our previous report showed that de novo protein synthesis is not required for P2Y2R-mediated TF up-regulation (11). Also, in our prior study, no change in IκBα and p65 phosphorylation was observed in response to P2Y2R activation in HCAEC, excluding the involvement of the NF-κB pathway (11). Although the classical AP-1 sites are worth studying, we were more intrigued by our original finding through bioinformatics search on a much wider region of the TF promoter. Our in silico search identified a new distal AP-1-binding site that matches 100% to the AP-1 consensus sequence and also is well conserved across multiple species. In addition, we found that this new site is located at −1363 bp in the human TF promoter and that the deletion, truncation, or mutation of which all significantly diminished UTP-induced TF transcription, suggesting that this new distal AP-1 site plays a significant role in regulating TF gene transcription in response to membrane receptor activation in HCAEC. This notion is strongly supported by our EMSA study showing that both UTP and TNF-α treatments all significantly increased AP-1 probe occupancy.
To date, very few studies systemically reflect how nucleotide receptor activation acts on transcription factors and their target genes. P2Y2R is reported to mediate sustained phosphorylation of transcription factor cAMP-response element-binding protein on sensory neurons (36). Signaling through P2X7 receptor is reported to activate AP-1 (c-Jun/c-Fos) in human T cells (37), although others found that P2X7 receptor activates the FosB·AP-1 complex in monocytic and osteoblastic cells (38). In this study, we found that three AP-1 subunits and their binding affinity were under the control of P2Y2R in HCAEC, namely c-Jun, Fra-1, and ATF-2. Unexpectedly, we ruled out c-Fos, although it is the most classic partner of c-Jun and has been reported in TF induction (39, 40). Both Jun and ATF members can form homodimers by themselves and have different preferences for DNA-binding sites (41). Jun and Fos subunits favor the TPA-response element heptamer (TGAC/GTCA), whereas ATF members prefer the cAMP-response element octamer (TGACGTCA). It has been reported that when Jun dimerizes with ATF, the complex has higher affinity to cAMP-response element (42). Herein we demonstrated that transcription factor complexes containing ATF-2 could also recognize and bind to AP-1 consensus site as evidenced by our ChIP and EMSA studies showing that ATF-2-specific monoclonal antibody not only precipitated a TF promoter fragment containing the new distal AP-1 site, but also significantly decreased AP-1 probe binding capacity. In addition, we found that siRNA knockdown of ATF-2 also significantly suppressed UTP-induced TF gene transcription. This c-Jun/ATF-2 combination is new to both TF gene transcription and nucleotide receptor signaling.
Transcription activity of different AP-1 subunits has been uncanny on different genes under various conditions. Taking c-Jun for example, although usually a positive regulator, it was reported to suppress transcription of the PLA2G4A gene (43). Existing reports on Fra-1 are even more controversial (44, 45). Negative effect of Fra-1 was reported in chemotherapy resistance and tumor progression (46). Although TF transcription was up-regulated immensely under phosphorylation of all three subunits, our current loss-of-function study yielded more truth of their differential impact. To the best of our knowledge, this study is the first to identify Fra-1 as a negative regulator in control of TF transcription, whereas c-Jun and ATF-2 are strong positive ones in line with most studies. This is supported by our unexpected finding that knockdown of Fra-1 boosted TF gene transcription in response to P2Y2R activation in HCAEC. In addition, we found that c-Jun is slightly more effective than ATF-2 in TF transcription up-regulation. The overall effect of the three covered up the fact that Fra-1 is in the opposite direction as compared with c-Jun and ATF-2. These results indicate that at least in HCAEC, the TF gene is under fine-tuning by P2Y2R signaling.
Our previous report has documented that P2Y2R is the predominant P2Y nucleotide receptor subtype expressed in HCAEC and that activation of this receptor stimulates all the three MAPK pathways, including ERK1/2, JNK, and p38 (11). In addition, we found that inhibition of JNK or p38, but not ERK1/2, all attenuated UTP-induced TF protein expression (11), suggesting that both JNK and p38 play an essential positive role in TF protein up-regulation. However, whether such an MAPK signaling mechanism also applies to P2Y2R-controlled TF gene transcription was unknown. In this study, we found that inhibition of JNK suppressed UTP-induced phosphorylation of c-Jun and ATF-2, but not of Fra-1, indicating that JNK is an upstream kinase in control of the two positive regulators c-Jun and ATF-2 but not the negative regulator Fra-1. Because our prior study also demonstrated that P2Y2R activates JNK through the Rho kinase pathway (11), we reasoned that blocking Rho kinase should also inhibit c-Jun and ATF-2. Indeed, this notion is supported by our observation that UTP-induced c-Jun and ATF-2 phosphorylation was inhibited by Y27632, a well known selective Rho kinase inhibitor. However, although both ERK5 and ERK1/2 have been shown to activate Fra-1 in different cells (47), we did not find any evidence that P2Y2R activates ERK5 (data not shown). Instead, our findings indicate a new P2Y2R/Src/ERK1/2/Fra-1 pathway that negatively regulates TF gene transcription. Several lines of evidence support this view. 1) We found that UTP-induced Fra-1 phosphorylation was inhibited by U0126, a highly selective ERK1/2 kinase inhibitor that did not have any effect on ATF-2 and c-Jun phosphorylation. 2) We also found that UTP-induced Fra-1 phosphorylation was dose-dependently inhibited by PP2, a selective Src kinase inhibitor, which is consistent with our previous report showing that Src is required for P2Y2R signaling in HCAEC (11). 3) Importantly, blocking either ERK1/2 or Src kinase all boosted TF pre-mRNA transcription in response to P2Y2R activation.
In summary, we have identified a new active distal AP-1-binding site in the TF gene promoter, which plays an essential role in P2Y2R-mediated TF gene transcription in human coronary artery endothelial cell. In addition, we revealed a new positive regulatory pathway for TF gene transcription, namely P2Y2R-Rho-JNK-c-Jun/ATF-2. Furthermore, for the first time, we discovered an original repressor Fra-1 for the TF gene, which is controlled by P2Y2R signaling via Src and ERK1/2. These findings indicate that the TF gene is under complex two-way fine-tuning by membrane receptor signaling. Overall, our findings highlight that coronary artery endothelial P2Y2R is an intriguing intersection of extracellular nucleotide signaling, vascular inflammation, and thrombogenesis. Future study should focus on the in vivo significance of the P2Y2R-TF axis and whether P2Y2R antagonist or Fra-1-selective chemical activator would be effective new pharmacotherapy options for the prevention and/or treatment of TF-related thrombotic and inflammatory diseases.
Author Contributions
Y. L. conducted most of the experiments, analyzed the results, and wrote most of the paper. L. Z., C. W., and S. R. performed some parts of the study. J. S. conceived the main idea for the project, analyzed part of the data with Y. L., and wrote the paper with Y. L.
This work was supported in part by National Scientist Development Award 12SDG8850011 from the American Heart Association (to J. S.) and by Auburn University Intramural Grant Program IGP150301 (to J. S.). The authors declare that they have no conflicts of interest with the contents of this article.
This article was selected as a Paper of the Week.
- P2Y2R
- P2Y2 receptor
- HCAEC
- human coronary artery endothelial cell
- TF
- tissue factor
- hnRNA
- heterogeneous nuclear RNA
- ATF
- activating transcription factor.
References
- 1. Burnstock G. (2011) Introductory overview of purinergic signalling. Front. Biosci. 3, 896–900 [DOI] [PubMed] [Google Scholar]
- 2. Jarvis B., and Simpson K. (2000) Clopidogrel: a review of its use in the prevention of atherothrombosis. Drugs 60, 347–377 [DOI] [PubMed] [Google Scholar]
- 3. Verheugt F. W. (2013) Antithrombotic therapy during and after percutaneous coronary intervention in patients with atrial fibrillation. Circulation 128, 2058–2061 [DOI] [PubMed] [Google Scholar]
- 4. Guns P. J., Hendrickx J., Van Assche T., Fransen P., and Bult H. (2010) P2Y receptors and atherosclerosis in apolipoprotein E-deficient mice. Br. J. Pharmacol. 159, 326–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hechler B., Freund M., Ravanat C., Magnenat S., Cazenave J. P., and Gachet C. (2008) Reduced atherosclerotic lesions in P2Y1/apolipoprotein E double-knockout mice: the contribution of non-hematopoietic-derived P2Y1 receptors. Circulation 118, 754–763 [DOI] [PubMed] [Google Scholar]
- 6. Seye C. I., Kong Q., Yu N., Gonzalez F. A., Erb L., and Weisman G. A. (2007) P2 receptors in atherosclerosis and postangioplasty restenosis. Purinergic Signal. 3, 153–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kellerman D., Rossi Mospan A., Engels J., Schaberg A., Gorden J., and Smiley L. (2008) Denufosol: a review of studies with inhaled P2Y(2) agonists that led to Phase 3. Pulm. Pharmacol. Ther. 21, 600–607 [DOI] [PubMed] [Google Scholar]
- 8. Lau O. C., Samarawickrama C., and Skalicky S. E. (2014) P2Y2 receptor agonists for the treatment of dry eye disease: a review. Clin. Ophthalmol. 8, 327–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wilcox J. N., Smith K. M., Schwartz S. M., and Gordon D. (1989) Localization of tissue factor in the normal vessel wall and in the atherosclerotic plaque. Proc. Natl. Acad. Sci. U.S.A. 86, 2839–2843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Steffel J., Lüscher T. F., and Tanner F. C. (2006) Tissue factor in cardiovascular diseases: molecular mechanisms and clinical implications. Circulation 113, 722–731 [DOI] [PubMed] [Google Scholar]
- 11. Ding L., Ma W., Littmann T., Camp R., and Shen J. (2011) The P2Y(2) nucleotide receptor mediates tissue factor expression in human coronary artery endothelial cells. J. Biol. Chem. 286, 27027–27038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Colucci M., Balconi G., Lorenzet R., Pietra A., Locati D., Donati M. B., and Semeraro N. (1983) Cultured human endothelial cells generate tissue factor in response to endotoxin. J. Clin. Invest. 71, 1893–1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gebhard C., Holy E. W., Camici G. G., Akhmedov A., Stämpfli S. F., Stähli B. E., von Rickenbach B., Breitenstein A., Greutert H., Yang Z., Lüscher T. F., and Tanner F. C. (2012) Caffeine induces endothelial tissue factor expression via phosphatidylinositol 3-kinase inhibition. Thromb. Haemost. 107, 884–894 [DOI] [PubMed] [Google Scholar]
- 14. Moll T., Czyz M., Holzmüller H., Hofer-Warbinek R., Wagner E., Winkler H., Bach F. H., and Hofer E. (1995) Regulation of the tissue factor promoter in endothelial cells. Binding of NFκB-, AP-1-, and Sp1-like transcription factors. J. Biol. Chem. 270, 3849–3857 [DOI] [PubMed] [Google Scholar]
- 15. Yan S. F., Zou Y. S., Gao Y., Zhai C., Mackman N., Lee S. L., Milbrandt J., Pinsky D., Kisiel W., and Stern D. (1998) Tissue factor transcription driven by Egr-1 is a critical mechanism of murine pulmonary fibrin deposition in hypoxia. Proc. Natl. Acad. Sci. U.S.A. 95, 8298–8303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aljada A., Ghanim H., Mohanty P., Kapur N., and Dandona P. (2002) Insulin inhibits the pro-inflammatory transcription factor early growth response gene-1 (Egr)-1 expression in mononuclear cells (MNC) and reduces plasma tissue factor (TF) and plasminogen activator inhibitor-1 (PAI-1) concentrations. J. Clin. Endocrinol. Metab. 87, 1419–1422 [DOI] [PubMed] [Google Scholar]
- 17. Moons A. H., Levi M., and Peters R. J. (2002) Tissue factor and coronary artery disease. Cardiovasc. Res. 53, 313–325 [DOI] [PubMed] [Google Scholar]
- 18. Ma W., Liu Y., Ellison N., and Shen J. (2013) Induction of C-X-C chemokine receptor type 7 (CXCR7) switches stromal cell-derived factor-1 (SDF-1) signaling and phagocytic activity in macrophages linked to atherosclerosis. J. Biol. Chem. 288, 15481–15494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shen J., Chandrasekharan U. M., Ashraf M. Z., Long E., Morton R. E., Liu Y., Smith J. D., and DiCorleto P. E. (2010) Lack of mitogen-activated protein kinase phosphatase-1 protects ApoE-null mice against atherosclerosis. Circ. Res. 106, 902–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shen J., and DiCorleto P. E. (2008) ADP stimulates human endothelial cell migration via P2Y1 nucleotide receptor-mediated mitogen-activated protein kinase pathways. Circ. Res. 102, 448–456 [DOI] [PubMed] [Google Scholar]
- 21. Li Y. D., Ye B. Q., Zheng S. X., Wang J. T., Wang J. G., Chen M., Liu J. G., Pei X. H., Wang L. J., Lin Z. X., Gupta K., Mackman N., Slungaard A., Key N. S., and Geng J. G. (2009) NF-κB transcription factor p50 critically regulates tissue factor in deep vein thrombosis. J. Biol. Chem. 284, 4473–4483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cui M. Z., Parry G. C., Oeth P., Larson H., Smith M., Huang R. P., Adamson E. D., and Mackman N. (1996) Transcriptional regulation of the tissue factor gene in human epithelial cells is mediated by Sp1 and EGR-1. J. Biol. Chem. 271, 2731–2739 [DOI] [PubMed] [Google Scholar]
- 23. Glover J. N., and Harrison S. C. (1995) Crystal structure of the heterodimeric bZIP transcription factor c-Fos-c-Jun bound to DNA. Nature 373, 257–261 [DOI] [PubMed] [Google Scholar]
- 24. Hess J., Angel P., and Schorpp-Kistner M. (2004) AP-1 subunits: quarrel and harmony among siblings. J. Cell Sci. 117, 5965–5973 [DOI] [PubMed] [Google Scholar]
- 25. Schreck I., Al-Rawi M., Mingot J. M., Scholl C., Diefenbacher M. E., O'Donnell P., Bohmann D., and Weiss C. (2011) c-Jun localizes to the nucleus independent of its phosphorylation by and interaction with JNK and vice versa promotes nuclear accumulation of JNK. Biochem. Biophys. Res. Commun. 407, 735–740 [DOI] [PubMed] [Google Scholar]
- 26. Arany I., Megyesi J. K., Kaneto H., Price P. M., and Safirstein R. L. (2004) Cisplatin-induced cell death is EGFR/src/ERK signaling dependent in mouse proximal tubule cells. Am. J. Physiol. Renal Physiol. 287, F543–F549 [DOI] [PubMed] [Google Scholar]
- 27. Yegutkin G. G. (2008) Nucleotide-and nucleoside-converting ectoenzymes: important modulators of purinergic signalling cascade. Biochim. Biophys. Acta 1783, 673–694 [DOI] [PubMed] [Google Scholar]
- 28. Deaglio S., and Robson S. C. (2011) Ectonucleotidases as regulators of purinergic signaling in thrombosis, inflammation, and immunity. Adv. Pharmacol. 61, 301–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Robson S. C., Wu Y., Sun X., Knosalla C., Dwyer K., and Enjyoji K. (2005) Ectonucleotidases of CD39 family modulate vascular inflammation and thrombosis in transplantation. Semin. Thromb. Hemost. 31, 217–233 [DOI] [PubMed] [Google Scholar]
- 30. Di Virgilio F., and Solini A. (2002) P2 receptors: new potential players in atherosclerosis. Br. J. Pharmacol. 135, 831–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Seye C. I., Yu N., Jain R., Kong Q., Minor T., Newton J., Erb L., González F. A., and Weisman G. A. (2003) The P2Y2 nucleotide receptor mediates UTP-induced vascular cell adhesion molecule-1 expression in coronary artery endothelial cells. J. Biol. Chem. 278, 24960–24965 [DOI] [PubMed] [Google Scholar]
- 32. Seye C. I., Agca Y., Agca C., and Derbigny W. (2012) P2Y2 receptor-mediated lymphotoxin-α secretion regulates intercellular cell adhesion molecule-1 expression in vascular smooth muscle cells. J. Biol. Chem. 287, 10535–10543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shen J., Seye C. I., Wang M., Weisman G. A., Wilden P. A., and Sturek M. (2004) Cloning, up-regulation, and mitogenic role of porcine P2Y2 receptor in coronary artery smooth muscle cells. Mol. Pharmacol. 66, 1265–1274 [DOI] [PubMed] [Google Scholar]
- 34. Lee R., Williams J. C., and Mackman N. (2012) P2X7 regulation of macrophage tissue factor activity and microparticle generation. J. Thromb. Haemost. 10, 1965–1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Furlan-Freguia C., Marchese P., Gruber A., Ruggeri Z. M., and Ruf W. (2011) P2X7 receptor signaling contributes to tissue factor-dependent thrombosis in mice. J. Clin. Invest. 121, 2932–2944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Molliver D. C., Cook S. P., Carlsten J. A., Wright D. E., and McCleskey E. W. (2002) ATP and UTP excite sensory neurons and induce CREB phosphorylation through the metabotropic receptor, P2Y2. Eur. J. Neurosci. 16, 1850–1860 [DOI] [PubMed] [Google Scholar]
- 37. Budagian V., Bulanova E., Brovko L., Orinska Z., Fayad R., Paus R., and Bulfone-Paus S. (2003) Signaling through P2X7 receptor in human T cells involves p56lck, MAP kinases, and transcription factors AP-1 and NF-κB. J. Biol. Chem. 278, 1549–1560 [DOI] [PubMed] [Google Scholar]
- 38. Gavala M. L., Hill L. M., Lenertz L. Y., Karta M. R., and Bertics P. J. (2010) Activation of the transcription factor FosB/activating protein-1 (AP-1) is a prominent downstream signal of the extracellular nucleotide receptor P2RX7 in monocytic and osteoblastic cells. J. Biol. Chem. 285, 34288–34298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Parry G. C., and Mackman N. (1995) Transcriptional regulation of tissue factor expression in human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 15, 612–621 [DOI] [PubMed] [Google Scholar]
- 40. Oeth P., Parry G. C., and Mackman N. (1997) Regulation of the tissue factor gene in human monocytic cells. Role of AP-1, NF-κB/Rel, and Sp1 proteins in uninduced and lipopolysaccharide-induced expression. Arterioscler. Thromb. Vasc. Biol. 17, 365–374 [DOI] [PubMed] [Google Scholar]
- 41. van Dam H., and Castellazzi M. (2001) Distinct roles of Jun: Fos and Jun: ATF dimers in oncogenesis. Oncogene 20, 2453–2464 [DOI] [PubMed] [Google Scholar]
- 42. Benbrook D. M., and Jones N. C. (1990) Heterodimer formation between CREB and JUN proteins. Oncogene 5, 295–302 [PubMed] [Google Scholar]
- 43. Bickford J. S., Beachy D. E., Newsom K. J., Barilovits S. J., Herlihy J. D., Qiu X., Walters J. N., Li N., and Nick H. S. (2013) A distal enhancer controls cytokine-dependent human cPLA2α gene expression. J. Lipid Res. 54, 1915–1926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rajasekaran S., Vaz M., and Reddy S. P. (2012) Fra-1/AP-1 transcription factor negatively regulates pulmonary fibrosis in vivo. PLoS ONE 7, e41611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Belguise K., Kersual N., Galtier F., and Chalbos D. (2005) FRA-1 expression level regulates proliferation and invasiveness of breast cancer cells. Oncogene 24, 1434–1444 [DOI] [PubMed] [Google Scholar]
- 46. Obenauf A. C., Zou Y., Ji A. L., Vanharanta S., Shu W., Shi H., Kong X., Bosenberg M. C., Wiesner T., Rosen N., Lo R. S., and Massagué J. (2015) Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 520, 368–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Terasawa K., Okazaki K., and Nishida E. (2003) Regulation of c-Fos and Fra-1 by the MEK5-ERK5 pathway. Genes Cells 8, 263–273 [DOI] [PubMed] [Google Scholar]